
Abstract
Wastewater-based epidemiology (WBE) has been previously used as a tool for pathogen identification within communities. After the SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) outbreak, in 2020, Daughton proposed the implementation of a wastewater surveillance strategy that could determine the incidence of COVID-19 (coronavirus disease 2019) nationally. Individuals in various stages of SARS-CoV-2 infection, including presymptomatic, asymptomatic and symptomatic patients, can be identified as carriers of the virus in their urine, saliva, stool and other bodily secretions. Studies using this method were conducted to monitor the prevalence of the virus in high-density populations, such as cities but also in smaller communities, such as schools and college campuses. The aim of this pilot study was to assess the feasibility and effectiveness of wastewater surveillance in Bucharest, Romania, and wastewater samples were collected weekly from seven locations between July and September 2023. RNA (ribonucleic acid) extraction, followed by dPCR (digital polymerase chain reaction) analysis, was performed to detect viral genetic material. Additionally, NGS (next generation sequencing) technology was used to identify the circulating variants within the wastewater of Bucharest, Romania. Preliminary results indicate the successful detection of SARS-CoV-2 RNA in wastewater, providing valuable insights into the circulation of the virus within the community.
Similar content being viewed by others

Introduction
The COVID-19 global pandemic that started in 2019 and caused severe acute respiratory syndrome has been considered a health emergency that needs international attention1. It is important to note that individuals in various stages of infection with COVID-19, including presymptomatic, asymptomatic, and symptomatic patients, have been found to carry a viral load in their faeces2,3. For this reason, monitoring the virus through wastewater surveillance has become a tool to support clinical studies. This method of detection provides a broader perspective on the prevalence of the virus within a population, aiding early detection and efforts to mitigate the spread of COVID-194,5,6,7. The Centers for Disease Control and Prevention (CDC) implemented a National Wastewater Surveillance System (NWSS) in September 2020, followed by the European Commission, which established a list of recommendations for wastewater monitoring to track COVID-19 and its variants according to the Commission Recommendation8 (2021). Considering that SARS-CoV-2 sewage surveillance is a vital component of early detection and is critical for mitigating the spread of COVID-19 within communities, all EU (European Union) member states, including Romania, have taken action. WBE has been applied to measure the prevalence of various human viruses, including measles9, enteroviruses10, hepatitis A and E11,12, noroviruses13,14 and SARS-CoV-215. After Daughton16 (2020) suggested that this type of approach would allow the early identification of SARS-CoV-2 nationwide, various methods have been developed for monitoring viral presence in sewage systems. Therefore, WBE has proven to be a promising approach that has provided information on outbreaks within populations and the efficacy of measures taken by public health officials17,18. Wastewater surveillance involves the collection of samples from the sewage system, followed by pathogen detection analysis19. Wastewater-based epidemiology, unlike clinical surveillance, offers an overview of the circulating virus in a population by analysing sewage samples. In this way, early detection of outbreaks and gathering of data on asymptomatic individuals are possible20. Therefore, WBE is carried out using a non-invasive sampling method that can complement clinical surveillance and can help obtain a comprehensive picture of SARS-CoV-2 prevalence21,22,23,24.
WBE for COVID-19 has been implemented in many countries at a high rate since the start of the pandemic, and more than 70 countries have reported using this surveillance method today according to COVIDPoops1925. The detection of SARS-CoV-2 in wastewater has been reported in countries such as Australia, the Czech Republic, Japan, India, Italy, the Netherlands, the United States, Spain and China15,17,26,27,28,29,30,31,32,33. Among these studies, the workflow was similar and involved the following main steps: sample collection, the addition of a recovery control to determine how a related virus will behave during processing, determination of the concentration of the wastewater samples, RNA extraction, and RNA quantification through polymerase chain reaction (PCR) and sequencing28. Studies conducted in Pakistan34, Germany35, Spain36,37 and China38 have reported significant results by using NGS analysis of wastewater samples, offering relevant information on the emerging lineages and variants across different regions.
These findings prove that wastewater surveillance can serve as an early warning system for COVID-19 outbreaks and provide data about newly occurring mutations. However, wastewater surveillance faces challenges such as environmental RNA degradation and uneven genome coverage in regard to sequencing39,40. Other factors that may affect the quality of the analysis are low concentrations of SARS-CoV-2, PCR inhibitors, and the composition of sewage water that contains genetic material from humans, bacteria and other viruses41,42. Additionally, another problem with processing wastewater is the abundance of organic matter and solid suspensions that affect RNA structure43. Wastewater samples can have variable RNA concentrations, which may affect PCR analysis. Additionally, inhibitors present in the samples may interfere with the reaction mixture and result in false-negative results44. For this reason, we chose to use Digital PCR for viral detection, as it has been previously considered a more sensitive approach than qPCR (quantitative polymerase chain reaction) and is more tolerant to PCR inhibitors44,45. Sequencing viral genomes has provided a guide for understanding the spread and genetic diversity of SARS-CoV-2 during the pandemic and additional information on pathogen evolution, transmission and spread in the community. NGS has been a crucial tool for the characterization of circulating SARS-CoV-2 variants in a population. NGS of positive samples can reveal the diversity of SARS-CoV-2 variants circulating in a population; this method simplifies and accelerates genotyping compared to other historical methods, such as Sanger sequencing23,24. By sequencing viral RNA from wastewater, researchers can now obtain information on viral lineages and introduction events in a population and detect low-frequency variants or variants of concern41,46. In the Netherlands, for example, a study using NGS compared variants (single nucleotide polymorphisms) between sewage and clinical samples and determined the diversity of the circulating SARS-CoV-2 variants in the region23. Similar studies were conducted in South Africa, Canada and Greece, where variants of concern were successfully detected24,40,47. The results of these studies contributed to establishing the prevalence of specific SARS-CoV-2 lineages and variants in different regions around the globe, highlighting the importance of sequencing wastewater samples.
Here, we show that valuable data can be obtained via NGS technology using sewage samples collected from seven locations in Bucharest from July 2023 to September 2023. The aim of this study was to assess the viral concentrations collected from different locations in Bucharest, to measure the recovery rate for Bovine Coronavirus strain “S379 Riems” (EVAg 015 V-02282) and determine the prevalent variants through sequencing.
Materials and methods
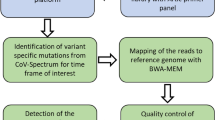
According to Eurostat48 (2022), Bucharest, a densely populated city in Romania, has an area of approximately 239 \(\:\text{k}{\text{m}}^{\text{2}}\) and almost 2.3 million residents. Therefore, this study was initially planned to evaluate the feasibility of a wastewater-based epidemiological tool in a crowded area of a Romanian municipality. The percentage population of Bucharest served by the sampling locations (Fig. 1h) was distributed as follows: location A is discharge collector A2 in A0 and serves 2%, location B is discharge collector A3 in cassette and serves 46%, location C is discharge collector B2-B3 and serves 6%, location D is discharge collector B5 in cassette and serves 5%, location E is discharge collector B6 in cassette and serves 10% and location F is discharge collector B7 in cassette and serves 31%. Location G is a wastewater treatment plant serving Bucharest and several neighboring localities. The study was carried out by implementing a wastewater surveillance system that used the bovine coronavirus strain S379 Riems (EVAg 015 V-02282) according to the method described by Deák42 (2023) and an automated extraction method through QIAcube Connect. For the detection of SARS-CoV-2, dPCR was chosen because it has proven to be more sensitive for these types of samples than other PCR methods44. To obtain the SARS-CoV-2 datasets of the circulating variants and strains in Bucharest, we used amplicon-based NGS via MiSeq (Illumina). Moreover, the development of a preconcentration system was patented with publishing no. 137,598 A049. Our workflow, as previously mentioned, included sample collection, primary processing, RNA extraction, RNA amplification, NGS and data analysis. According to the Commission Recommendation8 (2021), all samples should be analysed at least in duplicate to avoid false-positive or false-negative results; therefore, in this study, samples were processed in triplicate.
Sample collection
The samples were collected from six sewage collectors and one wastewater treatment plant located in Bucharest. Wastewater from the sewage system was collected weekly from July 19, 2023 until September 27, 2023. For this purpose, a separate entity was assigned to ensure sample collection and transportation. No precipitation was recorded before sampling at least 48 h. Samples were grab sampled and placed in 1 L containers. Sample collection was carried out weekly between 7:30 am and 10:50 am. After collection, the samples were transported to the laboratory under refrigerated conditions and received the same day at 11:20 am at the latest, followed by a verification step in which the pH and temperature of the samples were measured. The samples were then stored in a refrigerator at 4 °C until the next day for primary processing and RNA extraction.
Concentration and nucleic acid extraction
Sample concentration is an important step in the process because wastewater is a complex matrix that needs to be filtered. For this reason, in our study, we chose to use ultrafiltration using Millipore Vivaspin 20 tubes with 30 kDa filter membranes (#UFC903024). Ultrafiltration has proven to be efficient because it relies on excluding particles based on size and has the advantage of maintaining a stable viral load, unlike other methods that rely on precipitation50. Before concentration, 40 mL of the wastewater samples were transferred to conical tubes and mixed with BCoV working solution provided by the European Virus Archive – GLOBAL (EVAg 015 V-02282). The bovine coronavirus was chosen as a recovery control to observe viral loss during the whole process because of its high similarity with SARS-CoV-251. Before adding BCoV to the samples, the viral material had to be reconstituted, diluted, aliquoted and stored at -70 to -90 ºC. The quantity of added BCoV varied as follows: from week 29 to 36 (sample 1–56), 200 µL of BCoV were added per sample, while from week 37 to week 39 (sample 57–77) the amount was increased to 400 µL. The volume of added recovery control was adjusted to enhance the viral load of BCoV, ensuring it remained well above the detection limit. Samples were concentrated, RNA extracted, and the recovered BCoV was measured using the GT-Digital SARS-CoV-2 Wastewater Surveillance Kit for QIAcuity®. The ultrafiltration stage was performed by transferring 14 mL of supernatant into the previously mentioned Millipore filter tubes. All tubes were centrifuged at 5500 × g for 15 min until the whole volume was filtered through the tube membranes. After this step, the concentrated samples were recovered with a pipette and transferred to 2 mL Eppendorf tubes. Viral RNA was extracted using 200 µL of viral concentrate with the AllPrep PowerViral DNA/RNA Kit following the manufacturer’s protocol52. To remove potential inhibitors, a purification step with inhibitor removal solution (IRS) was done. The IRS solution has an important role in removing contaminants that would affect the PCR reaction. Automated RNA extraction was performed with a QIAcube Connect to obtain 80 µL of each eluted sample. The final RNA extract was stored at -80 °C for future applications.
Quantification of the N1 and N2 genes by digital PCR
For quantification, we used the GT-Digital SARS-CoV-2 Wastewater Surveillance Assay for QIAcuity® from GT-Molecular, which contains primer sets for the N1, N2 and BCoV genes53. A summary of the kits used for sample preparation can be found in Table 1. The Master Mix for each sample was prepared using 10 µL of QIAGEN 4X One-Step Viral RT‒PCR Master Mix, 0.4 µL of QIAGEN 100X Multiplex Reverse Transcription Mix, 2.0 µL of 20x N1, N2, and BCoV and 7.6 µL of RNase/DNase-free water. The volume per sample was multiplied by the number of analysed replicates. For quantification, we added 20 µL of prepared master mix and 20 µL of our extracted RNA samples to PCR strip tubes. After homogenization, the mixture was loaded and sealed in a QIAcuity Nanoplate 26k 24-well plate. For each nanoplate, a non-template control (NTC) containing RNase-Free Water and an N1-N2-BCoV positive control provided by GT Molecular was added. dPCR was performed according to the manufacturer’s instructions using the QIAcuity® Software Suite. The targeted regions were read on three different channels at different intensities. The green and yellow channels corresponding to N2 and BCoV, respectively, had an exposure duration of 300 ms. For the N1 gene, the red channel had an exposure duration of 600 ms. The dPCR parameters were set as follows: 1 cycle at 50 °C for 30 min, 1 cycle at 95 °C for 2 min and 45 cycles at 95 °C for 10 s and 55 °C for 30 s each. The QIAcuity Software Suite was used for automatic analysis of the raw data. If all three replicates had an average equal to or greater than 3 positive partitions, they were considered positive for SARS-CoV-2. Subsequently, to further define the samples as negative or positive, a statistical analysis of the obtained data was performed with Minitab® 21 Statistical Software54. This analysis helped assess which gene concentrations for N1 and N2 (copies/µl) corresponded to negative or positive samples. A range of values for characterizing the negative or positive samples was established using a graphical control chart model that plotted the average values +/- the standard deviation for each series of N1 and N2 concentrations. Thus, lower and upper limits were established for each of the series of N1 and N2 concentrations attributed to positive and negative SARS-CoV-2 samples.
Amplicon-based library preparation and genome sequencing
For library preparation, 48 samples were chosen according to the number of positive partitions and the concentrations of the N1 and N2 genes generated by dPCR. The libraries were prepared manually according to the manufacturer’s instructions using 8.5 µL of previously extracted RNA. Amplicon-based library preparation was performed using the Illumina® COVIDSeq™ Assay following the protocol from the Illumina COVIDSeq RUO Kit Reference Guide55. Before sequencing, the library had to be quantified using a Qubit dsDNA HS Assay and diluted to a 4 nM normalized concentration. The final step was to load 600 µL of the obtained 10 pM library on an Illumina MiSeq platform using the MiSeq Reagent Kit v2 for 300 cycles. The samples were sequenced on a MiSeq flow cell. Before running the protocol for SARS-CoV-2, the equipment was tested with a PhiX Control v3 Library. The run parameters showed a Q30 score of 71.6%, an alignment of 99.28% for Read 1 and 97.72% on Read 2 with error rates of 3.65% and 6.22% respectively.
Data analysis
The raw sequencing data were obtained from Illumina’s cloud-based BaseSpace Sequence Hub, which generated FASTQ files for each sample. Each consensus sequence, in FASTA file format, was then analysed using the DRAGEN COVID Lineage App (pipeline and lineage tool)55 to identify SARS-CoV-2 variants and annotate mutations. Information on the lineages of our sequenced samples was obtained through PANGOLIN (https://cov-lineages.org/resources/pangolin.html)56. For information on how our sequences were compared to the reference genome (Wuhan-Hu-1/2019) (NC_045512.2) and for clade assignment, the Nextclade tool (https://clades.nextstrain.org/) was used57.
Phylogenetic Analysis
For phylogenetic analysis, we obtained one dataset from the National Center for Biotechnology Information (NCBI) SARS-CoV-2 Data Hub, which is representative of SARS-CoV-2 whole-genome sequences across Europe and to which we added data from our samples. One of the criteria for selecting strains was ensuring they had a complete genome so the alignment would be successful. The selected sequences were derived from clinical samples and were chosen so the clades would be properly represented in the phylogenetic tree. All these parameters could be established by applying the following filters from the database: Pango lineage, geographic region, nucleotide completeness and host. Sequences were chosen depending on their lineage to obtain a representative tree of all existing clades for SARS-CoV-2. These sequences were aligned in MEGA11 software (Molecular Evolutionary Genetics Analysis Version 11) using MUSCLE (Multiple Sequence Comparison by Log Expectation), from which a maximum likelihood (ML) tree was constructed using 100 bootstrap replicates. Then, the condensed tree option was selected with a 50-cut-off value. The final dataset was displayed using the Interactive Tree of Life tool58. Clades were assigned using the Nextclade tool57.
Results
During the three-month period (19th July 2023–27th September 2023) of this study, a total of 77 wastewater samples were collected and tested for SARS-CoV-2, of which 56 tested positive for the N1 and N2 genes using the SARS-CoV-2 dPCR assay. Among the SARS-CoV-2-positive samples, 48 were chosen for sequencing analysis based on their partition number to determine their variants (Table 2).
Dynamics of SARS-CoV-2 concentrations in wastewater samples based on the N1, N2 and BCoV
Based on the dPCR analysis of wastewater samples, a graphic representation was generated showing the positivity trend for the seven locations from July to September 2023. After obtaining the average concentrations for N1 and N2 expressed in copies/µl, we calculated the average concentrations for the targeted genes in copies/L considering all of the dilutions that were made during laboratory processing. The graphic was made using the concentrations of N1 and N2 genes expressed in copies/L and calculated with log10. All NTCs were negative for N1, N2, and BCoV. According to the Minitab® statistical data, concentrations lower than 0.169 copies/µl (equivalent to 3.36 copies/L log10) were considered negative for SARS-CoV-2, and concentrations higher than 0.207 copies/µl (equivalent to 3.45 copies/L log10) were considered positive for SARS-CoV-2. Samples were considered positive if their concentration levels were above 3.36 copies/L log10 for one of the two targeted regions. As shown in Fig. 1, positive detection of SARS-CoV-2 was observed in both gene assays (N1 and N2) among the seven study sites, with the highest peak of 4.74 copies/L log10 for Location E in week 38. Virus presence varied between locations for the first seven weeks but started showing higher concentrations in September for all locations than in the previous months.

Graphic representation of SARS-CoV-2 positivity trends for the seven locations where (a)–(g) correspond to the locations on the Bucharest h map as follows: (a) – discharge collector A2 in A0, (b) – discharge collector A3 in cassette, (c) – discharge collector B2-B3, d – discharge collector B5 in cassette, e – discharge collector B6 in cassette, (f) – discharge collector B7 in cassette, (g) – water treatment plant entrance, (i) – correlation between the number of clinical cases of COVID-19, the positivity of wastewater samples and the percentage of the population associated with positive samples in weeks 29 to 39 of 2023.
For location A (Fig. 1a), the targeted genes were undetected until week 31, when they started to show higher values for SARS-CoV-2-targeted regions. Location B (Fig. 1b) tested positive from week 33 until the last date. The concentration for location C (Fig. 1c) fluctuated between positive and negative detection for the first weeks, showing lower concentrations compared to the other locations. Samples collected from site D (Fig. 1d) had high concentrations of our targeted genes, ranging between 3.40 and 4.41 copies/L log10 on all dates except week 33, when the values were lower than the selected dPCR threshold. Location E (Fig. 1e) exhibited constant concentrations for the entire timeline, with a peak of 4.74 copies/L log10 during week 38. For location F (Fig. 1f), samples started to show detectable concentrations after week 34, and they continued to rise until the last week of sample collection. The concentrations for location G (Fig. 1g), fluctuated between weeks, showing more visible results in week 33 and from week 35 to week 39, when the peak was approximately 3.90 copies/L log10. During this study, it was noticed that our results had a higher viral RNA recovery and improved sequencing rates for samples collected between 7:30 am and 7:50 am, even though these samples represent a smaller percentage of the population.
A correlation analysis was conducted between the number of COVID-19 cases reported by the Romanian Ministry of Health59 and the positivity percentage in wastewater samples for weeks 29–39 of 2023. Figure 1i shows a significant increase in clinical cases, with the number rising approximately 28-fold by week 39 compared to week 29, indicating a clear upward trend. Similarly, the positivity percentage in wastewater samples increased from 29% in week 29 to 100% by week 35, and remained at 100% through week 39. This strong correlation suggests that wastewater monitoring can effectively anticipate the rise in clinical cases, providing an early indication of the pandemic’s progression. A Pearson correlation analysis (Minitab®54, p = 0.814, p-value = 0.002) further supports the close correlation between reported clinical cases and the positivity percentage of samples from the corresponding sewage system population. The number of reported cases in the population served by the analysed sewage system increased exponentially between weeks 29 and 34, reaching a positivity level of 100% at approximately 500 clinical cases by week 35. Additionally, the positivity percentages of the samples were correlated with the percentages of the population served by the sewage system. Sample positivity ranging from 29 to 71% corresponded to a population coverage interval between 15% and 94%. The prediction model used in our analysis determined the lag period through cross-correlation analysis in Minitab®54. This approach, which has been used by several researchers in similar studies, identified a 2-week time delay (lag) between the detection of viral RNA in wastewater and the subsequent rise in clinical cases.
The samples analysed via sequencing were preselected based on dPCR concentrations ranging from 0.29 to 4.78 copies/µl for the N1 gene and 0.36 to 4.59 copies/µl for the N2 gene. From the 48 samples presented in Table 2, it was observed that samples 4 C (PP550127), 25 C (PP550858), 65 A (PP550929) and 68 A (PP551225) had sufficient sequencing data to identify their variants. Samples 4 C (PP550127), 25 C (PP550858), 65 A (PP550929) and 68 A (PP551225) exhibited values of 1.65, 1.92, 2.21 and 4.78 copies/µl, respectively, for the N1 gene and 1.60, 1.89, 1.87 and 4.59 copies/µl, respectively, for the N2 gene. Although there were samples with higher concentrations of the N1 and N2 genes, they were not fully analysed for variant identification. Based on this finding, we concluded that concentrations were not enough to serve as an indicator for sample preselection when performing sequencing. Unlike RT‒qPCR (quantitative reverse transcription polymerase chain reaction), which has predefined conditions for sample preselection for sequencing (Ct value 35–37)60, the conditions may vary based on specific assay requirements or inhibitory factors present in wastewater samples for dPCR wastewater analysis61.
The recovery percentage of the bovine coronavirus strain S379 Riems (EVAg 015 V-02282) was calculated as a ratio between the viral concentration obtained in the presence of a matrix and the viral concentration obtained in ultrapure water, that can be found as Supplementary Table S1, follows the formula: R% = (CBCoVmatrix/CBCoVwater) × 100, where Cmatrix is the viral concentration obtained in the presence of matrix and Cwater is the viral concentration obtained in ultrapure water. Recovery percentage values vary between 0.54 and 174.23. The concentration values for BCoV viral RNA in the presence of the matrix vary between 0.44 and 20.36 copies/µl. A recovery percentage between 0.54 and 43.96 was obtained for 71.79% of the samples from Supplementary Table S1 and approximately 18% of the samples were above 87.4% recovery. Also, the load of the analysed samples was comparable, therefore exogenous BCoV RNA could not be suspected. Regarding the BCoV positive control, values varied between 65,84 copies/µl and 88,02 copies/µl, having a relative standard deviation (RSD) of 0.143. The RSD value shows that the detection was precise and uniform. These results can be found as Supplementary Table S3.
Sequencing analysis
To obtain data for SARS-CoV-2, we sequenced 2 sample sets of 24 samples each using the Illumina COVIDSeq protocol. Among the 48 samples, 8.3% were fully analysed, and 91.7% had insufficient concentration. For this method, ARCTIC V4 primers were used because they have better genome recovery than previous versions55,62. We observed that the samples that were successfully sequenced had at least 81 detected amplicons out of 99 that were introduced. The minimum percentage of non-N bases (Coverage ≥ 10x) was 79.33%, and the minimum Coverage ≥ 30x was 72.89%. After the analysis, two types of graphics were generated, representing the median amplicon coverage (Fig. 2a) and the coverage depth (Fig. 2b) for samples 4 C (PP550127), 25 C (PP550858), 65 A (PP550929) and 68 A (PP551225).
According to Fig. 2, the samples had different median read depths: sample 4 C (PP550127) – 1001, sample 25 C (PP550858) – 1237, sample 65 A (PP550929) – 225 and sample 68 A (PP551225) – 378. These median amplicon coverage values showed that samples with greater read depths (4 C, 25 C) contained 82 and 87 detected amplicons out of 99. Samples with low read depths (65 A, 68 A) had more gaps in the genome, but 84 and 81 amplicons were detected. Sequencing low viral load has been previously noted to be challenging because of the heavily fragmented RNA and PCR inhibitors, which lead to poor sequencing coverage, making the analysis of genomic information from wastewater samples more difficult39. In our case, the poorly covered regions varied between samples, which suggests that this might have been due to their quality63. Another reason for the low coverage is that viral genomes from wastewater are a mixture of multiple genome origins, which makes specific identification of strains more difficult. Compared to clinical sampling, the amplicons obtained from different individuals cannot be associated with a single variant in wastewater sampling64. However, specific mutations corresponding to particular strains can still be detected, which provides a more comprehensive background for clinical data65,66 (Fig. 3).

Graphical representation of viral amplicon median coverage – (a) and coverage depth of the sequenced samples - (b) (sample PP550127, sample PP550858, sample PP550929 and sample PP551225).

Maximum likelihood phylogenetic tree of currently available SARS-CoV-2 genomes across Europe and the samples used for the present study where sample 4 C corresponds to PP550127, sample 25 C to PP550858, sample 65 A to PP550929 and sample 68 A to PP551225.
Phylogenetic tree
For the phylogenetic analysis, we used sequences across Europe available on NCBI to construct a maximum likelihood (ML) tree of the known clades and to determine the phylogeny of our samples. All sequences were evaluated against the reference genome NC_045512.2-Wuhan-Hu-1. By this analysis, we could obtain information about four of our samples, as they had enough sequencing information to be aligned with the NCBI dataset. Additionally, sample sequences were assigned to lineages using the Pangolin COVID-19 Lineage Assigner (Pangolin, 2023) to validate our findings and DRAGEN COVID Lineage56.
As shown in the phylogenetic tree in Fig. 3, our samples were integrated into different clades as follows: sample RO4C (PP550127) was grouped in clade 23D, lineage XBB.1.9.1, and samples RO25C (PP550858) and RO65A (PP550929) were grouped in clade 23B, lineage XBB1.16.1, and XBB.16, respectively, and sample RO68A (PP551225) was grouped in clade 23E, lineage XBB.2.3. The origin for all of our strains was clade 22 F, corresponding to the XBB PANGOLIN lineage. According to the Pangolin COVID-19 Lineage Assigner 4.3, v1.22 assignment, sample RO4C (PP550127) was assigned to lineage FL.14 (1/1), RO25C (PP550858) to XBB.1.16.1 (1/1), RO65A (PP550929) to XBB.1.22(1/3), XBB.1.42.2(1/3), XBB.1.9.1(1/3) and RO68A (PP551225) to XBB.2.3 (1/1)67. The DRAGEN COVID Lineage tool showed that sequence 4 C (PP550127.1) was assigned to lineage BA.2.10.1 (1/1), sequence 25 C (PP550858.1) to XBB.1.16.1 (1/1), sequence 65 A (PP550929.1) to XBB.1 (2/4), XBB.1.22 (1/4), XBB.1.9.1 (1/4), and sequence 68A (PP551225.1) to XBB (1/1). Our results matched with the PANGOLIN lineage assignment. Notably, FL.14 was used as an alias for XBB.1.9.1.14, which is consistent with the findings presented in Fig. 3, as sample RO4C (PP550127) was grouped with XBB.1.9.168. However, sample RO65A (PP550929) had a different assignment in PANGOLIN, which may be due to the assignment conflict of 0.66666 that was shown by the tool.
Discussion
Several studies have already demonstrated the significant role of wastewater surveillance information for emerging infectious pathogens such as SARS-CoV-2 and thus the regional variation dynamics could be tracked using advanced molecular biological techniques for measuring viral RNA concentration. Previous studies have also shown that WBE monitoring is an effective and non-invasive testing strategy for mass surveillance, especially when individual testing is limited, but it has its own limitations. Sensitivity of detection techniques is one of these. The potential of using dPCR for the detection of SARS-CoV-2 viral RNA in wastewater has been previously demonstrated by several studies32,34,69. Similar studies regarding the SARS-CoV-2 concentration in wastewater have been conducted using detection methods such as RT–qPCR and RT‒ddPCR (Droplet Digital PCR), which have shown different results depending on the location and epidemiologic status70,71. In general, N1 and N2 assays were frequently chosen for this type of research, as they were reported to be more sensitive for detection in wastewater72,73,74. However, N3 and E gene assays were also used for the same purpose75. In northern Italy, for example, a qPCR analysis of sewage samples from the N1 and N3 regions revealed an average of 3.46 × 102 to 9.10 × 103 gc/L for the N1 gene assay and 2.92 × 102 to 2.90 × 104 gc/L for the N3 gene assay75. A study conducted in Japan on municipal wastewater reported concentrations ranging from 3.30 to 4.15 log10 copies/L for SARS-CoV-264. Another study that compared the CDC_N1N2 assay with NIID_N2 obtained better values with the CDC_N1N2 set with concentrations of 1.6 × 102–1.3 × 104 gene copies/L for both targeted regions69. Randazzo31 (2020), who quantified SARS-CoV-2 RNA by qPCR with an N1 or N2 primer mixture, reported average values of 5.1 ± 0.3 and 5.5 ± 0.2 log10 gc/L, respectively31. Isanovic70 (2022) who targeted the N1 and N2 regions via ddPCR, reported low viral RNA concentrations in wastewater influent (3.95 and 3.76 log10 RNA copies per litre, respectively)70.
To the best of our knowledge, this is the first SARS-CoV-2 surveillance study of the virus in wastewater from Romania. For this study, 7 representative locations in Bucharest were identified and sampled. The results of our study demonstrate the detection of SARS-CoV-2 viral RNA from wastewater using dPCR. By comparing the values obtained in this study and those presented in Fig. 1, it can be noted that N1 tends to have higher concentrations than N274,76. Additionally, it was noted that the concentration method has an impact on the levels of SARS-CoV-269. These observations underscore the heterogeneity in viral loads across distinct geographic locations, thereby showing the importance of systematic surveillance for more effective management of virus prevalence in sewage77. However, detection methods need to be further optimized for better sensitivity, as previously noted by Ahmed1 (2020) and Sherchan32 (2020).
Wastewater-based epidemiology (WBE) studies on SARS-CoV-2 have utilized various process controls, such as MHV, transmissible gastroenteritis virus, HCoV-229E, alphacoronavirus and BCoV78,79,80. After adding BCoV in our samples, the recovery efficiency was measured and we obtained values that varied between 0.54 and 43.96 for 71.79% of the samples from Supplementary Table S2. The importance of the viral recovery rate of wastewater samples was outlined in several studies, even if the recovery efficiency of process control viruses differs from one study to another due to variations in the characteristics of the wastewater studied, but also for reasons related to the volume used in sample processing or the methods of virus concentration81. González80 (2020) reported that in some cases the BCoV82 recovery rate was higher in samples that were not concentrated before extraction, with values reaching 59% (± 14%), compared to the recovery rate results that showed 5.5% (± 2.1%) where InnovaPrep was used and 4.8% (± 2.8%) after electronegative filtration. The study of Pasha83 (2024) evaluated the recovery rates of BCoV (bovine coronavirus) across various wastewater samples. The research observed that MHP samples (dozed before freezing) had the highest BCoV recovery at 54 ± 20%. In contrast, neighbourhood pumping stations showed recovery rates of less than 1% before freezing, though this improved 24–17% after filtration for Neighbourhoods A and B, respectively. Hospital wastewater, however, had a modest improvement from 2 ± 0.89% to 5 ± 3.61% after filtration and dosing83. Other studies, like Randazzo31 (2020), reported lower recovery rates for process controls, such as PEDV (porcine coronavirus) and MgV (mengovirus), in raw influents and final effluents, reporting the following values: 10 ± 3.5% and 10 ± 2.1%; 3.3 ± 1.6% and 6.2 ± 1.0%, respectively31. Duroobi84 (2023) noted that raw influent samples exhibited the highest mean recovery at 25.7 ± 12.5%, followed by the primary effluent with 20.2 ± 17.0%84. Depending on the recovery control and the method used in sample processing, studies showed different results: Qiu85 (2022) reported values that ranged from 11.6 to 73.4% (recovery rate) for hCoV-229E, results varying because of the extraction method85; Jafferali86 (2021) results were influenced by the concentration method, observing higher recovery rates for BCoV87 when using ultrafiltration instead of electronegative membrane filtration; and other multi-laboratory studies reported recoveries for the surrogate viruses that ranged from 0.08 to 66%. These fluctuations are caused by factors such as composition of wastewater, its inhibitors, the recovery control and the methods that were chosen for sample processing (e.g. concentration, extraction)81. The variability highlights the need to improve wastewater surveillance practices and create more standardized approaches for accurate detection and quantification.
According to the World Health Organization (WHO), the current circulating variants are the variants of interest (VOIs) XBB.1.5 (clade 23 A), XBB.1.16 (clade 23B), and EG.5 (unassigned) and the variants under monitoring (VUMs) DV.7, XBB, XBB.1.9.1 and XBB.2.388. Additional information was found in the European Centre for Disease Prevention and Control (ECDC) reports, which indicates that XBB.1.5, BA.2, BA.2.75 and XBB are the prevalent variants in Europe89. According to the reports on the National Institute of Public Health from Romania90 website, between week 29 and 33 of 2023, the prevailing variants were XBB and XBB.1.5. After week 35, variants such as XBB.1.16, XBB.2.3, XBB.1.9.1, EG.1, GE.1 and others started to be detected, the first two being the most prevalent90. Our sequencing data, shows that XBB.1.9.1 and XBB.1.16.1 variants were already present in wastewater during week 29 and week 32, while XBB.1.16 and XBB.2.3 started to show up during week 38. Therefore, it can be concluded that two variants (XBB.1.9.1, XBB.1.16.1) started to show up in wastewater samples before clinical cases were confirmed. This finding suggests that the phylogenetic analysis in the present study showed that our results are consistent with the data on circulating variants mentioned by the WHO and with the reported cases by National Institute of Public Health from Romania.
The correlation between rising clinical cases and significant increases in viral RNA detection in wastewater has been well-documented across various studies, reinforcing the utility of wastewater surveillance as a predictive tool in public health. For instance, D’Aoust91 (2022) highlighted that during periods of restricted clinical testing, such as when testing was limited to appointment-only, the wastewater-to-case ratio significantly increased. This suggested that the true infection rate in the community was higher than what was captured by clinical testing alone, due to reduced testing availability and lower daily test numbers, despite rising viral signals in wastewater91. Similarly, Zhu92 (2022) found a strong correlation between the positive detection rate of SARS-CoV-2 in wastewater and reported clinical cases, supporting the notion that wastewater monitoring can serve as a reliable predictive tool, even though directly estimating epidemic size remains challenging92. Additionally, Galani93 (2022) demonstrated that by averaging RNA copy numbers in wastewater over an 8-day period, it is possible to predict positive clinical cases approximately 5 days in advance. This method also allowed for predictions of new hospital and ICU admissions 8 and 9 days ahead, respectively. These findings underscore the potential of wastewater surveillance as an early warning system that can significantly aid in healthcare planning and resource allocation93. Other studies, such as those by Bagutti94 (2022), have observed a consistent 14-day correlation between wastewater measurements and clinical case data, further confirming the value of wastewater surveillance in tracking epidemiological trends over time. The sensitivity of this method is able to detect infections even in presymptomatic and asymptomatic patients, therefore, enhancing the early detection and response capabilities of public health systems94.
Our results are consistent with these findings, as we could effectively predict clinical cases within 2 weeks windows according to the statistical demonstrating model that was also used by Rezaeitavabe95 (2024). This aligns with the broader body of evidence supporting the predictive value of wastewater data in epidemiological surveillance.
We summarize that the detection of nucleic acids of relevance in wastewater by PCR techniques used in combination with NGS could serve as an early and complementary warning system that provides opportunities for community-based pathogen surveillance. Our future directions include extending the monitoring area and improving the current method as well as standardizing the protocol and obtaining a better recovery rate for the virus.
Conclusions
In conclusion, this is the first study to report the detection of SARS-CoV-2 in Romanian wastewater using a dPCR assay. Of the 77 samples analysed, 72.7% were positive for SARS-CoV-2 according to the established lower limit of 3.45 copies/L log10. In addition, it was possible to perform whole-genome sequencing of SARS-CoV-2 using amplicon-based NGS technology. As a result, 85.7% of the positive samples were preselected for sequencing, and of these, 8.3% were analysed in their entirety, while 91.7% had insufficient concentration. The minimum concentration for preselected samples was 0.29 copies/µl. Our results show that dPCR is a viable method for detecting low concentrations of SARS-CoV-2 isolated from complex environmental samples such as wastewater. In addition, NGS technology has proven successful in sequencing these samples and generating sufficient data to determine the variants that are present. WBE provides valuable population-level insights into local virus dynamics and enhances public health intervention opportunities. Immediate wastewater results offer significant advance notice of infection trends. The detection of SARS-CoV-2 in wastewater provides critical early warning signals that can guide public health responses, particularly in identifying infection surges before they become apparent in clinical data. This makes wastewater surveillance an indispensable tool for managing public health.
Data availability
All SARS-CoV-2 variant sequences are currently available at NCBI GenBank under the following accession numbers: PP550127, PP550858, PP550929 and PP551225.
References
Ahmed, W. et al. First confirmed detection of SARS-CoV-2 in untreated wastewater in Australia: a proof of concept for the wastewater surveillance of COVID-19 in the community. Sci. Total Environ.18, 138764. https://doi.org/10.1016/j.scitotenv.2020.138764 (2020).
Foladori, P. et al. SARS-CoV-2 from faeces to wastewater treatment: what do we know? A review. Sci. Total Environ.743, 140444. https://doi.org/10.1016/j.scitotenv.2020.140444 (2020).
Tiwari, A. et al. Persistence and occurrence of SARS-CoV-2 in water and wastewater environments: a review of the current literature. Environ. Sci. Pollut. Res.29, 85658–85668. https://doi.org/10.1007/s11356-021-16919-3 (2021).
Jones, D. L. et al. Shedding of SARS-CoV-2 in feces and urine and its potential role in person-to-person transmission and the environment-based spread of COVID-19. Sci. Total Environ.749, 141364. https://doi.org/10.1016/j.scitotenv.2020.141364 (2020).
Dhama, K. et al. SARS-CoV-2 existence in sewage and wastewater: a global public health concern? J. Environ. Manage.280, 111825. https://doi.org/10.1016/j.jenvman.2020.111825 (2021).
Wannigama, D. L. et al. Tracking COVID-19 with wastewater to understand asymptomatic transmission. J. Infect. Dis.108, 296–299. https://doi.org/10.1016/j.ijid.2021.05.005 (2021).
Soller, J. A. et al. Modeling infection from SARS-CoV-2 wastewater concentrations: promise, limitations, and future directions. J. Water Health. 20, 1197–1211. https://doi.org/10.2166/wh.2022.094 (2022).
Commission Recommendation (EU). 2021/472 of 17 March 2021 on a common approach to the establishment of a systematic surveillance of SARS-CoV-2 and its variants in EU waste water (OJ L 98, 19.3.2021, p. 3 ). (2021). https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32021H0472
Benschop, K. et al. Polio and measles down the drain: environmental enterovirus surveillance in the Netherlands, 2005 to 2015. Appl. Environ. Microbiol.83https://doi.org/10.1128/AEM.00558-17 (2017).
Brinkman, N. E., Fout, G. S. & Keely, S. P. Retrospective surveillance of wastewater to examine seasonal dynamics of enterovirus infections. MSphere83 (2017). https://doi.org/10.1128/msphere.00099-17
Alfonsi, V. et al. Hepatitis E in Italy: 5 years of national epidemiological, virological and environmental surveillance, 2012 to 2016. Euro. Surveill. 23https://doi.org/10.2807/1560-7917.ES.2018.23.41.1700517 (2018).
McCall, C., Wu, H., O’Brien, E. & Xagoraraki, I. Assessment of enteric viruses during a hepatitis outbreak in Detroit MI using wastewater surveillance and metagenomic analysis. J. Appl. Microbiol.131, 1539–1554. https://doi.org/10.1111/jam.15027 (2021).
Hellmér, M. et al. Detection of pathogenic viruses in sewage provided early warnings of hepatitis a virus and norovirus outbreaks. Appl. Environ. Microbiol.80, 6771–6781. https://doi.org/10.1128/AEM.01981-14 (2014).
Lu, L. et al. Changing predominance of norovirus strains in children with acute gastroenteritis in Shanghai, 2018–2021. Virol. Sin. 38, 671–679. https://doi.org/10.1016/j.virs.2023.08.005 (2023).
La Rosa, G., Bonadonna, L., Lucentini, L., Kenmoe, S. & Suffredini, E. Coronavirus in water environments: occurrence, persistence and concentration methods - a scoping review. Water Res.179, 115899. https://doi.org/10.1016/j.watres.2020.115899 (2020).
Daughton, C. G. The international imperative to rapidly and inexpensively monitor community-wide Covid-19 infection status and trends. Sci. Total Environ.726, 138149. https://doi.org/10.1016/j.scitotenv.2020.138149 (2020).
Kumar, M. et al. First proof of the capability of wastewater surveillance for COVID-19 in India through detection of genetic material of SARS-CoV-2. Sci. Total Environ.746, 141326. https://doi.org/10.1016/j.scitotenv.2020.141326 (2020).
Ferraro, G. B. et al. A state-of-the-art scoping review on SARS-COV-2 in sewage focusing on the potential of wastewater surveillance for the monitoring of the COVID-19 pandemic. Food Environ. Virol.14, 315–354. https://doi.org/10.1007/s12560-021-09498-6 (2021).
B Diamond, M. et al. Wastewater surveillance of pathogens can inform public health responses. Nat. Med.28, 1992–1995. https://doi.org/10.1038/s41591-022-01940-x (2022).
Wani, H. et al. Wastewater-based epidemiology of SARS-CoV-2: assessing prevalence and correlation with clinical cases. Food Environ. Virol.15, 131–143. https://doi.org/10.1007/s12560-023-09555-2 (2023).
Mao, K. et al. The potential of wastewater-based epidemiology as surveillance and early warning of infectious disease outbreaks. Curr. Opin. Environ. Sci. Health. 17, 1–7. https://doi.org/10.1016/j.coesh.2020.04.006 (2020).
Shrestha, S. et al. Wastewater-based epidemiology for cost-effective Mass Surveillance of COVID-19 in low- and Middle-Income countries: challenges and opportunities. Water. 13, 2897. https://doi.org/10.3390/w13202897 (2021).
Izquierdo-Lara, R. et al. Monitoring SARS-CoV-2 circulation and diversity through Community Wastewater sequencing, the Netherlands and Belgium. Emerg. Infect. Dis.27, 1405–1415. https://doi.org/10.3201/eid2705.204410 (2021).
Yousif, M. et al. SARS-CoV-2 genomic surveillance in wastewater as a model for monitoring evolution of endemic viruses. Nat. Commun.14https://doi.org/10.1038/s41467-023-41369-5 (2023).
COVIDPoops19. Summary of Global SARS-CoV-2 Wastewater Monitoring efforts by UC Merced Researchers. ArcGIS Dashboards (2024). https://www.arcgis.com/apps/dashboards/c778145ea5bb4daeb58d31afee389082
Arora, S. et al. Sewage surveillance for the presence of SARS-CoV-2 genome as a useful wastewater based epidemiology (WBE) tracking tool in India. Water Sci. Technol.82, 2823–2836. https://doi.org/10.2166/wst.2020.540 (2020).
Haramoto, E., Malla, B., Thakali, O. & Kitajima, M. First environmental surveillance for the presence of SARS-CoV-2 RNA in wastewater and river water in Japan. Sci. Total Environ.737, 140405. https://doi.org/10.1016/j.scitotenv.2020.140405 (2020).
Medema, G., Heijnen, L., Elsinga, G., Italiaander, R. & Brouwer, A. Presence of SARS-Coronavirus-2 RNA in sewage and correlation with reported COVID-19 prevalence in the early stage of the Epidemic in the Netherlands. Environ. Sci. Technol. Lett.7, 511–516. https://doi.org/10.1021/acs.estlett.0c00357 (2020).
Mlejnková, H. et al. Preliminary study of SARS-COV-2 occurrence in wastewater in the Czech Republic. Int. J. Environ. Res. Public. Health. 17, 5508. https://doi.org/10.3390/ijerph17155508 (2020).
Nemudryi, A. et al. Temporal detection and phylogenetic assessment of SARS-COV-2 in municipal wastewater. Cell. Rep. Med.1, 100098. https://doi.org/10.1016/j.xcrm.2020.100098 (2020).
Randazzo, W. et al. SARS-CoV-2 RNA in wastewater anticipated COVID-19 occurrence in a low prevalence area. Water Res.181, 115942. https://doi.org/10.1016/j.watres.2020.115942 (2020).
Sherchan, S. P. et al. First detection of SARS-CoV-2 RNA in wastewater in North America: a study in Louisiana, USA. Sci. Total Environ.743, 140621. https://doi.org/10.1016/j.scitotenv.2020.140621 (2020).
Maida, C. M. et al. Wastewater-based epidemiology for early warning of SARS-COV-2 circulation: a pilot study conducted in Sicily, Italy. Int. J. Hyg. Environ. Health. 242, 113948. https://doi.org/10.1016/j.ijheh.2022.113948 (2022).
Sharif, S. et al. Detection of SARS-CoV-2 in wastewater using the existing environmental surveillance network: a potential supplementary system for monitoring COVID-19 transmission. PloS One. 16, e0249568. https://doi.org/10.1371/journal.pone.0249568 (2021).
Schumann, V. F. et al. SARS-CoV-2 infection dynamics revealed by wastewater sequencing analysis and deconvolution. Sci. Total Environ.853, 158931. https://doi.org/10.1016/j.scitotenv.2022.158931 (2022).
Carcereny, A. et al. Monitoring emergence of the SARS-CoV-2 B.1.1.7 variant through the Spanish National SARS-CoV-2 Wastewater Surveillance System (VATar COVID-19). Environ. Sci. Technol.55, 11756–11766. https://doi.org/10.1016/j.scitotenv.2022.158931 (2021).
Trigo-Tasende, N. et al. Wastewater early warning system for SARS-CoV-2 outbreaks and variants in a Coruña, Spain. Environ. Sci. Pollut Res. Int.30, 79315–79334. https://doi.org/10.1007/s11356-023-27877-3 (2023).
Deng, Y. et al. Use of sewage surveillance for COVID-19 to guide public health response: a case study in Hong Kong. Sci. Total Environ.821https://doi.org/10.1016/j.scitotenv.2022.153250 (2022).
Karthikeyan, S. et al. Wastewater sequencing reveals early cryptic SARS-CoV-2 variant transmission. Nature. 609, 101–108. https://doi.org/10.1038/s41586-022-05049-6 (2022).
Sapoval, N. et al. Enabling accurate and early detection of recently emerged SARS-CoV-2 variants of concern in wastewater. Nat. Commun.14https://doi.org/10.1038/s41467-023-38184-3 (2023).
Jahn, K. et al. Early detection and surveillance of SARS-CoV-2 genomic variants in wastewater using COJAC. Nat. Microbiol.7, 1151–1160. https://doi.org/10.1038/s41564-022-01185-x (2022).
Deák, G. et al. Evaluating recovery control concentrations of bovine coronavirus (EVAG 015V-02282) used for SARS-COV-2 wastewater surveillance. E3S Web Conf.437, 02011. https://doi.org/10.1051/e3sconf/202343702011 (2023).
Gerrity, D., Papp, K., Stoker, M., Sims, A. & Frehner, W. Early-pandemic wastewater surveillance of SARS-CoV-2 in Southern Nevada: methodology, occurrence, and incidence/prevalence considerations. Water Res. X10, 100086 (2021).
Suo, T. et al. ddPCR: a more accurate tool for SARS-CoV-2 detection in low viral load specimens. Emerg. Microbes Infect.9, 1259–1268. https://doi.org/10.1080/22221751.2020.1772678 (2020).
Boogaerts, T. et al. An alternative approach for bioanalytical assay optimization for wastewater-based epidemiology of SARS-CoV-2. Sci. Total Environ.789, 148043. https://doi.org/10.1016/j.scitotenv.2021.148043 (2021).
Champredon, D. et al. Emergence and spread of SARS-CoV-2 variants of concern in Canada: a retrospective analysis from clinical and wastewater data. BMC Infect. Dis.24https://doi.org/10.1186/s12879-024-08997-8 (2024).
Lawal, O. U. et al. Metagenomics of Wastewater Influent from Wastewater Treatment Facilities across Ontario in the era of emerging SARS-CoV-2 variants of concern. Microbiol. Resour. Announc. 11https://doi.org/10.1128/mra.00362-22 (2022).
Eurostat. Regions in Europe –interactive edition. Eurostat (2022). https://ec.europa.eu/eurostat/cache/digpub/regions/#total-population
Deák, G. et al. Preconcentration system using qualitative filtration of water/wastewater samples for identification especially SARS-CoV-2, or other types of pathogens (Patent 137598 A0). OSIM (2023). https://www.osim.ro/images/Publicatii/Inventii/2023/inv_08_2023.pdf
Fonseca, M. S. et al. Evaluation of SARS-CoV-2 concentrations in wastewater and river water samples. Case Stud. Chem. Environ. Eng.6, 100214. https://doi.org/10.1016/j.cscee.2022.100214 (2022).
Gonzales-Gustavson, E. et al. Characterization of the efficiency and uncertainty of skimmed milk flocculation for the simultaneous concentration and quantification of water-borne viruses, bacteria and protozoa. J. Microbiol. Methods. 134, 46–53. https://doi.org/10.1016/j.mimet.2017.01.006 (2017).
AllPrep PowerViral DNA/RNA Kit Handbook. QIAGEN (2018). https://www.qiagen.com/us/resources/resourcedetail?id=41a1323f-581c-4e65-8d66-2491be4c625e&lang=en
National Wastewater Surveillance System. Centers for Disease Control and Prevention (2023). https://www.cdc.gov/nwss/wastewater-surveillance.html
Data Analysis Statistical & Process Improvement Tools. MiniTAB (2024). https://www.minitab.com/en-us/
Illumina, D. N. A. & Prep Reference Guide. Illumina (2023). https://support.illumina.com/downloads/illumina-dna-prep-reference-guide-1000000025416.html
O’Toole, Á. et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol.https://doi.org/10.1093/ve/veab064 (2021).
Aksamentov, I., Roemer, C., Hodcroft, E. B. & Neher, R. A. Nextclade: clade assignment, mutation calling and quality control for viral genomes. J. Open. Source Softw.6, 3773. https://doi.org/10.21105/joss.03773 (2021).
Letunić, I. & Bork, P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res.49, W293–W296. https://doi.org/10.1093/nar/gkab301 (2021).
Ministry of Health (Romania) (2024). https://www.ms.ro/en/
Li, L. et al. Detecting SARS-CoV-2 variants in wastewater and their correlation with circulating variants in the communities. Sci. Rep.12https://doi.org/10.1038/s41598-022-20219-2 (2022).
Nyaruaba, R. et al. Digital PCR applications in the SARS-CoV-2/COVID-19 era: a Roadmap for Future outbreaks. Clin. Microbiol. Rev.35https://doi.org/10.1128/cmr.00168-21 (2022).
Lambisia, A. W. et al. Optimization of the SARS-COV-2 ARTIC Network v4 primers and whole genome sequencing protocol. Front. Med.9https://doi.org/10.3389/fmed.2022.836728 (2022).
Amman, F. et al. Viral variant-resolved wastewater surveillance of SARS-CoV-2 at national scale. Nat. Biotechnol.40, 1814–1822. https://doi.org/10.1038/s41587-022-01387-y (2022).
Iwamoto, R. et al. Identification of SARS-CoV-2 variants in wastewater using targeted amplicon sequencing during a low COVID-19 prevalence period in Japan. Sci. Total Environ.887, 163706. https://doi.org/10.1016/j.scitotenv.2023.163706 (2023).
Kuroiwa, M. et al. Targeted amplicon sequencing of wastewater samples for detecting SARS-CoV-2 variants with high sensitivity and resolution. Sci. Total Environ.893, 164766. https://doi.org/10.1016/j.scitotenv.2023.164766 (2023).
Tamáš, M., Potocarova, A., Konečná, B., Klučár, Ľ. & Mackulak, T. Wastewater Sequencing—An innovative method for variant monitoring of SARS-COV-2 in populations. Int. J. Environ. Res. Public. Health. 19, 9749. https://doi.org/10.3390/ijerph19159749 (2022).
Rambaut, A. et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol.5, 1403–1407. https://doi.org/10.1038/s41564-020-0770-5 (2020).
Cov-Lineages Lineage List. Cov-Lineages (2024). https://cov-lineages.org/lineage_list.html
Kitamura, K., Sadamasu, K., Muramatsu, M. & Yoshida, H. Efficient detection of SARS-CoV-2 RNA in the solid fraction of wastewater. Sci. Total Environ.763, 144587. https://doi.org/10.1016/j.scitotenv.2020.144587 (2021).
Isanovic, M., Velez, K. E. C. & Norman, R. S. Dispersion of SARS-CoV‐2 RNA across a wastewater treatment plant and its workers. Water Environ. J.36, 713–722. https://doi.org/10.1111/wej.12812 (2022).
Peccia, J. et al. Measurement of SARS-CoV-2 RNA in wastewater tracks community infection dynamics. Nat. Biotechnol.38, 1164–1167. https://doi.org/10.1038/s41587-020-0684-z (2020).
Anantharajah, A. et al. How to choose the right real-time RT-PCR primer sets for the SARS-CoV-2 genome detection? J. Virol. Methods. 295, 114197. https://doi.org/10.1016/j.jviromet.2021.114197 (2021).
D’Aoust, P. M. et al. Quantitative analysis of SARS-CoV-2 RNA from wastewater solids in communities with low COVID-19 incidence and prevalence. Water Res.188, 116560. https://doi.org/10.1016/j.watres.2020.116560 (2021).
Muenchhoff, M. et al. Multicentre comparison of quantitative PCR-based assays to detect SARS-CoV-2, Germany, March 2020. Euro. Surveill. 25https://doi.org/10.2807/1560-7917.ES.2020.25.24.2001057 (2020).
Navarro, A. et al. SARS-CoV-2 detection in wastewater using multiplex quantitative PCR. Sci. Total Environ.797, 148890. https://doi.org/10.1016/j.scitotenv.2021.148890 (2021).
Hong, P. et al. Estimating the minimum number of SARS-CoV-2 infected cases needed to detect viral RNA in wastewater: to what extent of the outbreak can surveillance of wastewater tell us? Environ. Res.195, 110748. https://doi.org/10.1016/j.envres.2021.110748 (2021).
Wong, Y. H. M. et al. Positive association of SARS-CoV-2 RNA concentrations in wastewater and reported COVID-19 cases in Singapore – A study across three populations. Sci. Total Environ.902, 166446. https://doi.org/10.1016/j.scitotenv.2023.166446 (2023).
Ahmed, W. et al. Comparison of virus concentration methods for the RT-qPCR-based recovery of murine Hepatitis virus, a surrogate for SARS-CoV-2 from untreated wastewater. Sci. Total Environ.739, 139960 (2020).
La Rosa, G. et al. SARS-CoV-2 has been circulating in northern Italy since December 2019: evidence from environmental monitoring. Sci. Total Environ.750, 141711. https://doi.org/10.1016/j.scitotenv.2020.141711 (2021).
González, R. et al. COVID-19 surveillance in Southeastern Virginia using wastewater-based epidemiology. Water Res.186, 116296. https://doi.org/10.1016/j.watres.2020.116296 (2020).
Salvo, M. et al. Evaluation of low-cost viral concentration methods in wastewaters: implications for SARS-CoV-2 pandemic surveillances. J. Virol. Methods. 297, 114249. https://doi.org/10.1016/j.jviromet.2021.114249 (2021).
Decaro, N. et al. Detection of bovine coronavirus using a TaqMan-based real-time RT-PCR assay. J. Virol. Methods. 151, 167–171. https://doi.org/10.1016/j.jviromet.2008.05.016 (2008).
Pasha, A. B. T. et al. Monitoring SARS-CoV-2 RNA in wastewater from a shared septic system and sub-sewershed sites to expand COVID-19 disease surveillance. J. Water Health. 22 (6), 978–992. https://doi.org/10.2166/wh.2024.303 (2024).
Al-Duroobi, H. et al. Wastewater-based surveillance of COVID-19 and removal of SARS-CoV-2 RNA across a major wastewater treatment plant in San Antonio, Texas. Environ. Sci. Adv.2 (5), 709–720. https://doi.org/10.1039/d3va00015j (2023).
Qiu, Y. et al. Validating and optimizing the method for molecular detection and quantification of SARS-CoV-2 in wastewater. Sci. Total Environ.812, 151434. https://doi.org/10.1016/j.scitotenv.2021.151434 (2022).
Jafferali, M. H. et al. Benchmarking virus concentration methods for quantification of SARS-CoV-2 in raw wastewater. Sci. Total Environ.755, 142939. https://doi.org/10.1016/j.scitotenv.2020.142939 (2021).
Christensen, E. & Myrmel, M. Coagulant residues’ influence on virus enumeration as shown in a study on virus removal using aluminium, zirconium and chitosan. J. Water Health. 16, 600–613. https://doi.org/10.2166/wh.2018.028 (2018).
Tracking, S. A. R. S. CoV-2 variants. World Health Organisation (2024). https://www.who.int/activities/tracking-SARS-CoV-2-variants
The European Respiratory Virus Surveillance Summary (ERVISS). European Centre for Disease Prevention and Control (2023). https://www.ecdc.europa.eu/en/covid-19/country-overviews
Analysis of confirmed cases of COVID-19. National Institute of Public Health Romania (2024). https://insp.gov.ro/centrul-national-de-supraveghere-si-control-al-bolilor-transmisibile-cnscbt/infectia-cu-noul-coronavirus-sars-cov-2/analiza-cazuri-confirmate-covid19/
D’Aoust, P. M. et al. Wastewater to clinical case (WC) ratio of COVID-19 identifies insufficient clinical testing, onset of new variants of concern and population immunity in urban communities. Sci. Total Environ.853, 158547. https://doi.org/10.1016/j.scitotenv.2022.158547 (2022).
Zhu, Y. et al. COVID-19 case prediction via wastewater surveillance in a low-prevalence urban community: a modeling approach. J. Water Health. 20 (2), 459–470. https://doi.org/10.2166/wh.2022.183 (2022).
Galani, A. et al. SARS-CoV-2 wastewater surveillance data can predict hospitalizations and ICU admissions. Sci. Total Environ.804, 150151. https://doi.org/10.1016/j.scitotenv.2021.150151 (2022).
Bagutti, C. et al. Wastewater monitoring of SARS-CoV-2 shows high correlation with COVID-19 case numbers and allowed early detection of the first confirmed B.1.1.529 infection in Switzerland: results of an observational surveillance study. Schweiz. Med. Wochenschr. 152 (2526), w30202. https://doi.org/10.4414/smw.2022.w30202 (2022).
Rezaeitavabe, F. et al. Beyond linear regression: modeling COVID-19 clinical cases with wastewater surveillance of SARS-CoV-2 for the city of Athens and Ohio University campus. Sci. Total Environ.912, 169028. https://doi.org/10.1016/j.scitotenv.2023.169028 (2024).
Acknowledgements
This work was supported by the European Commission DG Environment [060701/2021/864662/SUB/ENV]. C2] Emergency Support under Council Regulation (EU) 2016/369 as amended by Council Regulation (EU) 2020/521 Support to Member States for the establishment of national systems, local collection points and digital infrastructure for the monitoring of Covid 19 and its variants in wastewater – Romania programme coordinated by the Ministry of Environment, Water and Forests.
Funding
This work was supported by the European Commission DG Environment [060701/2021/864662/SUB/ENV]. C2] Emergency Support under Council Regulation (EU) 2016/369 as amended by Council Regulation (EU) 2020/521 Support to Member States for the establishment of national systems, local collection points and digital infrastructure for the monitoring of Covid 19 and its variants in wastewater – Romania.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design as follows: Deák György: Writing – review & editing, Supervision. Prangate Raluca: Investigation, Methodology, Writing – original draft. Matei Monica: Conceptualization, Writing – review & editing. Croitoru Cristina: Formal analysis, Writing – original draft. Boboc Mădălina: Writing – review & editing, Conceptualization.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
“Not applicable”, research does not report on or involve the use of any animal or human data or tissue.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Deák, G., Prangate, R., Croitoru, C. et al. The first detection of SARS-CoV-2 RNA in the wastewater of Bucharest, Romania. Sci Rep 14, 21730 (2024). https://doi.org/10.1038/s41598-024-72854-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-72854-6
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
