
Abstract
Ticks have a profound impact on public health. Haemaphysalis is one of the most widespread genera in Asia, including Japan. The taxonomy and genetic differentiation of Haemaphysalis spp. is challenging. For instance, previous studies struggled to distinguish Haemaphysalis japonica and Haemaphysalis megaspinosa due to the dearth of nucleotide sequence polymorphisms in widely used barcoding genes. The classification of H. japonica japonica and its related sub-species Haemaphysalis japonica douglasi or Haemaphysalis jezoensis is also confused due to their high morphological similarity and a lack of molecular data that support the current classification. We used mitogenomes and microbiomes of H. japonica and H. megaspinosa to gain deeper insights into the phylogenetic relationships and genetic divergence between two species. Phylogenetic analyses of concatenated nucleotide sequences of protein-coding genes and ribosomal DNA genes distinguished H. japonica and H. megaspinosa as monophyletic clades, with further subdivision within the H. japonica clade. The 16S rRNA and NAD5 genes were valuable markers for distinguishing H. japonica and H. megaspinosa. Population genetic structure analyses indicated that genetic variation within populations accounted for a large proportion of the total variation compared to variation between populations. Microbiome analyses revealed differences in alpha and beta diversity between H. japonica and H. megaspinosa: H. japonica had the higher diversity. Coxiella sp., a likely endosymbiont, was found in both Haemaphysalis species. The abundance profiles of likely endosymbionts, pathogens, and commensals differed between H. japonica and H. megaspinosa: H. megaspinosa was more diverse.
Similar content being viewed by others


Introduction
Ticks are arachnids that feed on blood of humans, domestic animals, and wildlife. During the process of blood feeding, ticks can transmit a variety of pathogens including the causative agents of anaplasmosis, ehrlichiosis, borreliosis, piroplasmosis and tick-borne encephalitis1. In addition, ticks can cause irritation, allergy and toxicosis2. Approximately, nine hundreds of tick species are distributed around the world3. However, the taxonomy of ticks remains uncertain due to the limited phylogenetic information on mitochondrial and nuclear genomes4. In the field of tick taxonomy and speciation, the classification of closely related tick species often presents a complex challenge. This challenge is not limited to subspecies alone but extends to distinct species with remarkably similar morphological features. Instances of such ambiguity are prevalent among various tick species, where differentiation based solely on visual attributes becomes complex. Notably, the classification of tick species like Ixodes scapularis and Ixodes pacificus5,6, both black-legged ticks inhabiting different regions, underscores this difficulty. Similarly, Dermacentor variabilis and Dermacentor andersoni7, distinguished by their distinct habitats in North America, exhibit analogous morphologies that warrant molecular scrutiny for accurate differentiation. Likewise, species like Amblyomma maculatum and Amblyomma triste8, as well as Rhipicephalus appendiculatus and Rhipicephalus zambeziensis9,10, exemplify the need for genetic techniques to unravel taxonomic uncertainties. Even in Europe, ticks such as Ixodes hexagonus and Ixodes canisuga11 demand molecular methods for accurate identification due to their close resemblance. These cases highlight the importance of molecular, genetic, and morphological approaches in untangling the complexities of tick taxonomy, leading to enhanced comprehension of their ecological roles and disease transmission potential.
A good example for the uncertain taxonomy is the phylogenetic classification of Haemaphysalis species, which are predominantly distributed in Asia including Japan. The genus Haemaphysalis (Koch, 1844), is considered the second largest genus of ticks (166 species) after the genus Ixodes (243 species)12. Albeit Haemaphysalis japonica and Haemaphysalis megaspinosa are morphologically distinct from each other, the previous studies could not differentiate between them genetically due to the lack of sequence polymorphisms in the partial sequences of two mitochondrial genes 16S rRNA and NADH dehydrogenase subunit 2 (ND2) genes13,14. Moreover, a confusing classification of H. japonica and its related species Haemaphysalis japonica douglasi and Haemaphysalis jezoensis has been debated. H. japonica was firstly described by Warburton using male specimens which were collected in Kyushu Island, Japan15. The monograph written by Nuttall and Warburton supplemented the information on this species and introduced the description of H. japonica var. douglasi using a male specimen collected in northern China16 Thereafter, Pomerantzev elevated this variety as a subspecies H. japonica douglasi17. Meanwhile, H. jezoensis was described using specimens collected in Hokkaido Island, Japan by Ogura and Takada18. However, H. jezoensis was suggested to be morphologically identical to H. japonica douglasi from China and Russia and collectively named as Haemaphysalis douglasi without taxonomic descriptions19. Most recently, H. douglasi from Hokkaido and H. japonica from Honshu Islands were suggested to be the same species based on comparing the partial sequences of the large subunit ribosomal RNA gene and internal transcribed spacer 220.
Recently, the whole mitochondrial genomes (mitogenomes) are being used to describe the deep phylogenetic structure of ticks21,22 and to investigate the demographic patterns and population structure of sympatric tick species23. For instance, this approach has yielded a robust phylogenetic hypothesis to resolve Amblyomma cajennense species complex through utilizing the mitogenomes for A. cajennense s. s., Amblyomma mixtum, Amblyomma tonelliae, Amblyomma patinoi, and Amblyomma sculptum24. In addition, mitogenome sequencing has become instrumental in uncovering cryptic tick species. For example, in the case of Rhipicephalus ticks, sequencing mitogenomes revealed two Rhipicephalus microplus clades and ticks presumed to be R. microplus from Southern China and Northern India were identified as a cryptic species closely related to Rhipicephalus annulatus25. Another example is the detection of a potentially novel tick species closely related to Amblyomma testudinarium from Myanmar23. Simultaneously, the tick microbiome plays a crucial role in tick physiology, host–pathogen interactions, and vector-borne disease transmission26. Recent advancements in the field of tick microbiome research have revealed the complex microbial communities harbored by ticks and their potential impact on tick biology and vector competence27,28. Tick microbiomes exhibit considerable variation across different tick species29. For example, the microbiome of Ixodes granulatus is characterized by the presence of Rickettsiales and Borrelia30, whereas Haemaphysalis hystricis predominantly harbors Acinetobacter and Rickettsiales31. Haemaphysalis shimoga, on the other hand, is marked by a significant abundance of Coxiella and varying levels of Rickettsiales, Candidatus Rhabdochlamydia, and Stenotrophomonas31. Meanwhile, Dermacentor steini displays a unique microbial profile, with prominent Acinetobacter and Francisella, along with Rickettsiales and Burkholderiales32. These examples underscore the distinct microbial communities existing in different tick species, even those closely related, and emphasize the importance of studying tick microbiomes for a comprehensive understanding of tick physiology and their potential roles in disease transmission31. Investigating the microbiome of several tick species could provide valuable insights into the development of targeted tick control strategies, potentially disrupting the microbial balance and reducing tick-borne disease burden33.
To gain deeper insights into the phylogenetic relationships and genetic divergence between H. japonica and H. megaspinosa, we employed whole mitogenome sequencing and comprehensive microbiome analysis. These cutting-edge approaches allowed us to elucidate the mitogenomic and microbial characteristics that differentiate these closely related tick species, providing valuable information on their evolutionary history, potential ecological niches.
Results
Characteristics of the analyzed mitogenomes
The mitogenomes of 29 H. japonica and 18 H. megaspinosa specimens from Japan were sequenced: these ranged from 14,678 to 14,683 bp in length. A total of 37 genes were identified, including 13 protein-coding genes (PCGs), 22 transfer RNA (tRNA) genes, and two ribosomal DNA (rDNA). Additionally, two control regions were observed in the mitogenome. Notably, no gene rearrangements were detected among the 47 newly sequenced Haemaphysalis mitogenomes.
The nucleotide sequences of 47 ticks were aligned with the reference mitogenome sequences of H. japonica from China (accession number: NC_037246), as well as Haemaphysalis flava (accession number: NC_005292). The 29 mitogenomes of H. japonica had an average nucleotide identity of 99.4% (99.0–99.9) whereas the 18 mitogenomes of H. megaspinosa had an average nucleotide identity of 99.8% (99.4–99.9). On the other hand, the 29 mitogenomes of H. japonica had an average nucleotide identity of 99.0% (98.9–99.1) with the 18 mitogenomes of H. megaspinosa.
Phylogenetic relationships inferred from the concatenated sequences of 13 protein-coding genes and two ribosomal DNA genes
The phylogeographic analysis included 29 H. japonica and 18 H. megaspinosa specimens collected from 10 prefectures in Japan, as well as two reference sequences retrieved from GenBank: H. japonica from China (accession number: NC_037246) and H. flava (accession number: NC_005292). The maximum clade credibility (MCC) tree topology revealed that the H. japonica sequences formed a clade separate from the H. megaspinosa sequences (Fig. 1). Furthermore, the H. japonica sequences exhibited further subdivision into two additional clades (hereinafter: Hj1 and Hj2). The MCC analysis of the concatenated and translated 13 PCGs further supported the findings of the phylogeographic tree based on the concatenated 15 mitochondrial genes (Fig. 2). To identify suitable markers for molecular differentiation between H. japonica and H. megaspinosa, we examined the parsimony information sites in 15 mitochondrial genes. Among these genes, the 16S rDNA exhibited the highest number of parsimony information sites, with a total of 30 sites. Additionally, the NAD5 gene showed a substantial number of parsimony information sites, with a total of 37 sites. These markers provide valuable molecular information that can aid in distinguishing between H. japonica and H. megaspinosa (Supplementary table 1).

Genetic diversity and phylogenetic relationships of H. japonica and H. megaspinosa specimens from Japan revealed by Bayesian MCC tree of concatenated of 15 mitochondrial gene sequences. The tree was rooted to H. flava which is the putative sister-species to H. japonica and H. megaspinosa (Barker unpublished data). In the tree, the clade representing specimens from H. megaspinosa are highlighted in blue (Hm) while H. japonica specimens were subdivided into two clades that are highlighted in light cyan (Hj1) and green (Hj2). The nucleotide differences among the 47 newly sequenced mitogenomes of Haemaphysalis collected in Japan were examined. The positions of single nucleotide variations are indicated by vertical lines in the mitogenome sequences.

Genetic diversity and phylogenetic relationships among H. japonica and H. megaspinosa specimens from Japan using a Bayesian MCC tree based on the translated concatenated sequences of 13 protein coding genes. The tree was rooted to H. flava which is the putative sister-species to H. japonica and H. megaspinosa (Barker unpublished data). In the tree, the clade representing specimens from H. megaspinosa is highlighted in blue, while H. japonica specimens are further subdivided into two clades highlighted in light cyan and green.
Genetic structure of H. japonica populations
Our acquired H. japonica sequences were grouped into two subclades, Hj1 and Hj2. Hj1 emerged as the prevailing subspecies of H. japonica on Honshu Island, while Hj2 dominated Hokkaido Island. Statistical analysis of population genetic structure was performed to assess the influence of geographic factors on the genetic variation within and between populations of H. japonica. The results of the Analysis of Molecular Variance (AMOVA) indicated that the genetic structure of H. japonica is not influenced by geographic factors, with a lower proportion of genetic variation observed among populations (38.05%) compared to within populations (61.95%). The global FST values (p < 0.01) indicated significant genetic differentiation within the Fukushima and Yamagata population (p = 0.0000) (Table 1).
Comparing microbiome diversity between H. megaspinosa and H. japonica (Hj1 and Hj2)
After demultiplexing and quality filtering, 1,466,794 high-quality reads were obtained from the Illumina MiSeq sequencer for the microbial analysis, corresponding to 770 features identified through DADA2 quality control analysis. Among the 33 ticks representing two Haemaphysalis species, one sample was excluded from the diversity analysis due to the significantly low number of obtained sequences (n = 4123). For the microbial analysis, H. megaspinosa was represented by one group (Hm) and H. japonica was categorized into two subgroups (Hj1 and Hj2) representing the phylogenetic subclades as illustrated in Fig. 1.
The Kruskal–Wallis test was employed to determine the significance of alpha diversity between H. japonica and H. megaspinosa. The results indicated that H. japonica exhibited higher alpha diversity compared to H. megaspinosa. This was evident through the analysis of various metrics, including Shannon diversity (p < 0.09), Faith’s PD (p-value < 0.01), observed features (p-value < 0.05), and Pielou’s evenness (p < 0.12) (Fig. 3a).

Diversity analyses of microbial populations of H. japonica Hj1 (n = 7), Hj2 (n = 12), and H. megaspinosa (n = 13) samples. Each dot shows the microbial population from an individual ixodid tick and color represents sample species. (a) A box and whisker plot was used to compare the alpha diversity of microbiome communities in H. japonica and H. megaspinosa. The results showed a significant difference between the two species in terms of Faith PD (p < 0.01) and Observed OTUs (p < 0.05), but no significant difference was observed for Shannon and Evenness indices (p < 0.09 and 0.12, respectively), as determined by the Kruskal–Wallis test. (b) The beta diversity of the microbiome in H. japonica and H. megaspinosa was examined. Pairwise PERMANOVA analysis revealed significant differences in community dissimilarity among the different tick species. Specifically, only Unweighted UniFrac showed a significant difference (p < 0.001, pseudo-F). (c) The PCoA plots, based on Unweighted UniFrac, Weighted UniFrac, Jaccard, and Bray–Curtis dissimilarity metrics, displayed overlapping clusters of samples from each Haemaphysalis species.
The Pairwise Permutational multivariate analysis of variance (PERMANOVA) analysis revealed that beta diversity metrics, including unweighted UniFrac, weighted UniFrac, Jaccard, and Bray–Curtis, differed between H. japonica and H. megaspinosa (pseudo-F, p < 0.001, p < 0.4, p < 0.08, and p < 0.2, respectively) (Fig. 3b). The PCoA plots supported the findings from the pairwise PERMANOVA analysis, showing overlapping clusters of samples from each Haemaphysalis species (Fig. 3c). Moreover, there was some separation in the clustering of samples based on unweighted UniFrac (Fig. 3c).
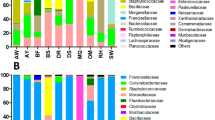
The composition of the most abundant bacterial families varied significantly across the different Haemaphysalis tick subgroups, as shown in Table 2 and Fig. 4. We provided a comprehensive overview of the total number of reads for each identified bacterial feature in this study. Our findings indicate that every analyzed Haemaphysalis species exhibited the presence of Coxiella as a predominant potential endosymbiont in their microbiome (Fig. 4a).

(a) The relative abundance (%) of bacterial taxa present in the microbiome of H. japonica and H. megaspinosa. The top 30 most abundant taxa are shown individually, while the remaining taxa are grouped together. Each bar represents the bacterial taxa identified in a single sample. (b) The LEfSe analysis revealed the taxa that were significantly differentially abundant (p < 0.05) within H. megaspinosa.
By analyzing the relative abundance and conducting linear discriminant analysis effect size (LEfSe) analysis on bacterial genera, we found that the two examined Haemaphysalis species displayed similar profiles of potential endosymbionts, specifically Coxiella and potential pathogens, specifically Ehrlichia. However, they exhibited distinct profiles of commensals, with H. megaspinosa samples displaying higher diversity compared to H. japonica (Fig. 4b).
Discussion
The investigation of the phylogeographic structure of tick populations may play a crucial role in enhancing our understanding of tick distribution and the prevalence of tick-borne pathogens23. In the present study, we present the first assessment of the genetic diversity and microbiome structure of H. japonica and H. megaspinosa, which are widely distributed in East and Northeast Asia34. Our research stands out as we employed whole mitogenomes of multiple individuals from each tick species to investigate the genetic diversity on a regional scale in Japan. In contrast to earlier studies that struggled to molecularly differentiate H. japonica and H. megaspinosa or classify H. japonica subspecies in the country13,20, we successfully analyzed the nucleotide variations in the complete mitogenomes between H. japonica and H. megaspinosa. Additionally, we examined the intraspecies genetic relationships and population differentiation among H. japonica subspecies. Our study benefited from a wide spatial scale of sample collection, enabling us to construct a comprehensive phylogenetic tree using 29 new H. japonica and 18 H. megaspinosa mitogenome sequences. Intriguingly, the 29 mitogenomes of H. japonica were on average only 1% (1.1–0.9%) different to the 18 mitogenomes of H. megaspinosa (genetic identity of 99.0% (98.9–99.1). Since Mans et al. (2021) found that pairwise nucleotide differences greater than 5% indicated different species of ticks35, the morphology of the voucher specimens of the 29 H. japonica and 18 H. megaspinosa ticks we studied will be studied in detail, in the future, to test the idea that H. japonica and H. megaspinosa may be conspecific. Furthermore, we provided valuable insights into the microbiome structure of both tick species based on amplicon sequences obtained from 32 tick samples from Japan.
In our study, tick specimens collected from various biogeographic regions in Japan were examined. Despite the different ecoclimatic habitats, phylogenetic analysis revealed that all sequences from the same Haemaphysalis sp. formed a monophyletic group with a high percentile identity of 99.0–99.9%. We found that the 16S rDNA had the highest number of parsimony information sites, totaling 30 sites. Additionally, the NAD5 gene exhibited a significant number of parsimony information sites, with a total of 37 sites. These markers, as presented in Supplementary Table 1, offer valuable molecular information that can aid in the molecular differentiation of H. japonica and H. megaspinosa. By utilizing these markers, researchers and practitioners can enhance their ability to accurately distinguish between these two tick species, contributing to a better understanding of their distribution and epidemiological significance. Moreover, our analysis revealed that the mitogenomes of H. japonica ticks exhibited a biphyletic grouping, with a sequence identity range of 99.0–99.9%. This suggests that H. japonica in Japan can be further classified into two subspecies where H. japonica found in Honshu Island (Hj1) formed a single clade and H. japonica found predominantly in Hokkaido (Hj2) formed the other cluster with the same species from China. This finding supports the previous observation that H. japonica (H. jezoensis at the time) was morphologically identical to H. japonica douglasi from China and Russia19.
The statistical analysis of population genetic structure aimed to investigate the genetic differentiation among the Hj1 and Hj2 populations of H. japonica. From the MCC tree analysis, it was observed that the Hj2 population consisted of some specimens from Honshu Island. However, no sequences from Hokkaido were found within the Hj1 population. The results of the AMOVA indicated that the genetic structure of H. japonica is not significantly influenced by geographic factors, as the proportion of genetic variation among populations (38.05%) was lower compared to within populations (61.95%). This suggests that there is more genetic diversity within each population, and the populations are not highly structured. Despite the clustering of some Hokkaido specimens with Fukushima individuals in the MCC tree, the absence of Hokkaido sequences within the Hj1 population suggests a lack of gene flow or limited genetic exchange between these populations. This may indicate some level of genetic differentiation between the Hj1 and Hj2 populations. The significant genetic differentiation within the Hj1 populations (p = 0.0000) further supports the presence of distinct genetic clusters within these regions. It is possible that factors other than geographic proximity, such as local adaptation or genetic drift, are contributing to the observed genetic differentiation.
The comparison of polymorphic sites revealed that relying solely on a single mitochondrial gene for phylogenetic analysis of Haemaphysalis spp. may lead to incorrect or incomplete conclusions. Current studies focusing on population structure and phylogenetic analysis of ticks commonly employ multiple mitochondrial genes to draw comprehensive conclusions36,37. Moreover, our findings indicate that the H. megaspinosa population in Japan exhibits lesser genetic diversity compared to the closely related H. japonica population. It is important to note that the evolution of mitochondrial genes can be influenced by various ecological factors, including endosymbionts and host diversity38. The disparity in diversity between H. japonica and H. megaspinosa in Japan may be attributed to variations in endosymbiont genetics that can lead to mitogenome divergence in tick species39.
Advancements in sequencing technology and bioinformatic tools have greatly expanded research on vector-associated microbiomes. This progress has significantly contributed to our understanding of tick systematics and the identification of novel tick-borne pathogens26,40. Acknowledging the influence of diverse ecological factors and endosymbionts is crucial in understanding the evolutionary dynamics of mitochondrial genes41,42. In our study, we conducted a comprehensive investigation of the microbiomes of 32 Haemaphysalis ticks, providing valuable insights into the relationship between microbiome composition and species variations. Notably, we observed a predominant presence of a Coxiella-like endosymbiont (CLE) within the microbiomes of both H. japonica and H. megaspinosa ticks. This finding aligns with previous studies that have detected CLE in other Haemaphysalis tick species such as H. longicornis43, H. shimoga31 and Haemaphysalis punctata44. The differences in microbiome diversity that we observed between H. japonica and H. megaspinosa can likely be attributed to various external factors, including the sources and histories of tick blood meals, the presence or absence of pathogenic bacteria and protozoa, and the surrounding environmental conditions45. Our study elucidated that the variability among individuals of H. japonica and H. megaspinosa is primarily driven by the presence of environmental bacteria, as depicted in Fig. 4b. Furthermore, we revealed that both tick species share similar potential pathogens, such as Ehrlichia species. This suggests that H. japonica and H. megaspinosa occupy similar ecological niches and possess comparable physiological characteristics, facilitating the presence of shared potential pathogens.
Our study provides valuable insights into the phylogenetic classification, genetic diversity, and microbiome structure of genetically closely related Haemaphysalis species. Through the utilization of complete tick mitogenomes, we successfully differentiated between H. japonica and H. megaspinosa and examined the intraspecies genetic relationships within H. japonica. The identified markers, particularly the 16S rDNA and NAD5 gene, offer valuable tools for accurate molecular differentiation between these tick species. Our findings support the presence of distinct genetic clusters within H. japonica populations, with limited gene flow between different regions. The investigation of tick microbiomes revealed the predominance of a CLE and highlighted differences in microbiome diversity between H. japonica and H. megaspinosa. These findings enhance our understanding of the genetic and ecological characteristics of Haemaphysalis ticks and contribute to the knowledge of tick-borne disease epidemiology and control efforts. Further studies exploring the relationship between endosymbiont genetics, mitogenome divergence, and tick microbiomes are warranted to gain a comprehensive understanding of these complex interactions.
Materials and methods
Tick samples and DNA extraction
Ticks were collected from Japan during 2013 to 2018 by flagging a flannel cloth on the plants. The collected ticks were kept in sterile plastic tubes till identification in the laboratory. The morphological identification was conducted by using a stereomicroscope as previously described46. A total of 18 H. megaspinosa and 29 H. japonica (Fig. 5) were morphologically identified and kept separately in Eppendorf tubes at – 20 °C for the molecular analysis (Fig. 5 and Table 3). In the current study, we used the name H. japonica regardless of their geographic origin. For DNA extraction, each tick was washed twice with ethanol (70%) and molecular grade phosphate-buffered saline (PBS) and homogenized at 3,000 rpm for 30 s in 100 μL of Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Life Technologies) by using a Micro Smash MS-100R (TOMY, Tokyo, Japan). A blackPREP Tick DNA/RNA Kit (Analytikjena, Germany) was used to extract DNA from a 50 μL of the tick homogenate47.

Geographic distribution of Haemaphysalis samples used in the present study in Japan. Sample collection sites are illustrated in circles (F: females), triangles (M: males) and squares (N: nymphs). Samples were collected from ten prefectures in Japan.
Whole mitogenome sequencing and assembly
Amplification of the complete mitogenomes of H. megaspinosa and H. japonica was achieved through long- and short-range PCRs, based on a modified protocol from a previous study22. The long-range PCR primers, mtG_K23 (5′-TCCTACATGATCTGAGTTYAGACCG-3′) and K26 (5′-ACGGGCGATATGTRCATATTTTAGAGC-3′), along with the short-range PCR primers, H_gap_F1 (5′-YAAYTCCAAAAATTGATGCAAA-3′) and H_gap_R1 (5′-AAGTCAAGRTGCARCAWAAR-3′), were designed by aligning the complete mitogenomes of Haemaphysalis genus available in the database. For the long-range PCR, a 50-μl reaction mixture was prepared, consisting of 10 μl of 5 × PrimeSTAR GXL Buffer (Mg2+ Plus) (TaKaRa Bio Inc., Shiga, Japan), 4.0 μl of dNTP Mixture (2.5 mM each), 200 nM of each primer, 1.0 μl of PrimeSTAR® GXL DNA Polymerase (TaKaRa Bio Inc.), and 2.0 μl of template DNA. The reaction conditions were set as follows: 45 cycles of denaturation at 98 °C for 10 s, annealing at 60 °C for 15 s, and extension at 68 °C for 10 min. As for the short-range PCR, a 25-μl reaction mixture was prepared, containing 12.5 μl of 2 × Gflex PCR Buffer (Mg2+, dNTP plus) (TaKaRa Bio Inc.), 0.5 μl of Tks Gflex DNA Polymerase (1.25 units/μl) (TaKaRa Bio Inc.), 200 nM of each primer, and 1.0 μl of template DNA. The reaction conditions consisted of an initial denaturation step at 94 °C for 60 s, followed by 45 cycles of denaturation at 98 °C for 10 s, annealing at 55 °C for 15 s, extension at 68 °C for 60 s, and a final extension at 68 °C for 5 min. To analyze the amplified PCR products, electrophoresis was performed using a 1.5% agarose gel stained with Gel-Red™ (Biotium, Hayward, CA). The PCR products were then purified using the NucleoSpin Gel and PCR Clean-Up Kit (TaKaRa Bio Inc.).
The long-range and short-range PCR products were combined at equal concentrations, with a ratio of 7:1, respectively. The final DNA concentration of the mixed PCR products from each sample was normalized to 0.2 ng/μl. Subsequently, Illumina sequencing libraries were generated from the purified PCR amplicons using the Nextera DNA Library Prep Kit (Illumina, Hayward, CA). Sequencing was performed on the Illumina MiSeq platform, utilizing the MiSeq reagent kit v3 for 600 cycles. To obtain the complete mitogenome sequences for each H. megaspinosa and H. japonica sample, the reads were assembled using CLC Genomics Workbench v20.0.4 (Qiagen, Hilden, Germany).
Comparative analysis of mitogenomes and phylogenetic inference
The complete mitogenome sequences were imported into Geneious version 10.2.6 (Biomatters Ltd., Auckland, New Zealand) and aligned with the mitogenome sequences of H. flava (accession number: NC_005292) and H. japonica (accession number: NC_037246). Subsequently, 13 PCG sequences and two rDNA sequences were extracted from each mitogenome sequence and concatenated to create a combined alignment. The concatenated sequences of the 15 mitochondrial genes from H. megaspinosa and H. japonica samples collected in Japan were aligned using the MAFFT software48. To determine the appropriate substitution model, PHYML 3.0 software was utilized, employing the Akaike Information Criterion49. Furthermore, a Bayesian phylogenetic tree was constructed using BEAST version 1.4, a cross-platform program for Bayesian analysis of molecular sequences through Markov chain Monte Carlo (MCMC) simulations. The GTR nucleotide substitution model with discrete gamma-distributed rate variation was employed to model sequence evolution. Additionally, a strict clock model was selected to assume a constant evolution rate across the entire tree. The Bayesian skyline coalescent model, a demographic model within a Bayesian framework, was used. The MCMC sampling was performed for 50 million generations, with samples collected every 50,000 steps after a burn-in period of 500,000 steps. The MCC tree was determined using TreeAnnotator50. The resulting MCC tree was visualized using FigTree version 1.4.4 (http://beast.bio.ed.ac.uk/figtree), with branch lengths proportional to posterior values. Additionally, another tree was constructed using the same Bayesian phylogenetic analysis technique, but this time utilizing the translated sequences of the concatenated 13 PCG sequences. The evolutionary models, MCMC sampling, burn-in period, and visualization methods remained consistent with the previous tree construction.
Analysis of the genetic structure within H. japonica populations
Population genetic structure analyses were conducted using the AMOVA implemented in Arlequin software version 3.5.2.251. The genetic variance among and within populations of H. japonica collected from Hokkaido Island (n = 13) was compared to Fukushima and Yamagata prefectures (n = 16). The number of permutations was set to 1000, and significance was assessed at a p-value threshold of < 0.05 based on the calculated fixation indices (F-statistics). FST, which indicates the degree of differentiation within populations, was used to measure the extent of allelic fixation or identity within populations52. FSC, on the other hand, estimated the differentiation among populations within the assigned group. A higher value of FSC suggests greater heterogeneity among populations. In cases where a strong population genetic structure is present at the analyzed population scale, FSC is expected to be higher relative to FST.
Data deposition and accession numbers
The entire mitogenome sequences of 29 H. japonica and 18 H. megaspinosa have been submitted to the DNA Data Bank of Japan (http://www.ddbj.nig.ac.jp). The accession numbers for H. japonica samples are LC567908-LC567936, and for H. megaspinosa samples are LC567937-LC567954.
MiSeq 16S rDNA amplicon sequencing
A total of 33 genomic DNA samples from H. japonica (n = 20) and H. megaspinosa (n = 13) (Table 3), along with one DNA extraction blank controls and one negative controls with distilled water, were subjected to PCR amplification targeting the V3-V4 regions of the bacterial 16S rDNA. The Illumina barcoded primers, Illumina_16S_341F and Illumina_16S_805R, were used for the PCR amplification, following the protocols described in references53,54. Kapa HiFi HotStart Ready Mix (KAPA Biosystems, Wilmington, MA, USA) was utilized for the PCR reactions. Each PCR reaction consisted of 12.5 μL of 2 × KAPA HiFi HotStart ReadyMix, 5.0 μL of each primer, and 2.5 μL of the tick genomic DNA samples or negative controls. The PCR products were confirmed by agarose gel electrophoresis using a 1.5% agarose gel stained with Gel-RedTM (Biotium, Hayward, CA, USA) and visualized under UV light. The amplicons were purified using AMPure XP beads (Beckman Coulter Life Sciences, IN, USA). Subsequently, libraries were prepared using the Nextera Index Kit (Illumina, San Diego, CA, USA), and sequencing was performed with a MiSeq Reagent Kit v3 (600 cycles) on an Illumina MiSeq instrument following the manufacturer's instructions. The raw sequence data have been deposited in the DNA Data Bank of the Japan Sequence Read Archive under the accession number DRA017340.
Bioinformatics analysis
The tick microbiome analysis was processed using the obtained sequencing data and the quantitative insights into microbial ecology 2 software (QIIME2) version 2020.255. Initially, the raw sequencing data, obtained from BaseSpace (Illumina), were demultiplexed, quality-checked, and filtered using the q2-demux plugin. The quality-filtered reads were then subjected to denoising using the DADA2 pipeline version 2019.1056. The resulting amplicon sequence variants (ASVs) were aligned using the q2-alignment plugin with mafft48, and a phylogenetic tree was constructed using the q2-phylogeny plugin with fasttree257. For diversity analysis, a sampling depth of 21,773 reads was selected for comparing the diversity among the examined Haemaphysalis tick species. Alpha diversity measures such as Shannon diversity58, Faith’s Phylogenetic Diversity (Faith's PD)59, observed features60, and Pielou's evenness61 were calculated. The results were exported and visualized in R using the qiime2R, ggplot2, and phyloseq packages62. Beta diversity measures, including unweighted UniFrac distance63, weighted UniFrac distance64, Jaccard similarity index65, and Bray–Curtis dissimilarity66, were calculated using QIIME2. The clustering of ASVs according to species was visualized through Principal Coordinates Analysis (PCoA) using the EMPeror plugin in QIIME267 and R. Taxonomic assignment was performed using the q2-feature-classifier plugin68 with the classify-sklearn naïve Bayes taxonomy classifier and SILVA classifier reference sequences (release 132). The Decontam package69 in R was used to identify likely contaminants introduced during processing. Archaea, eukaryota, potential contaminants, and sequences not assigned to the kingdom level were manually removed for further analysis in QIIME2. A heatmap phylogenetic tree was constructed using the heatmap method in QIIME270. Differential abundance of the 30 most abundant taxonomic groups was visualized using the taxa_heatmap function in the qiime2R package in R. To identify the bacteria contributing to the dissimilarity of the microbiome among tick groups, the LEfSe in the Huttenhower lab Galaxy pipeline71 was implemented. Statistical analyses were performed to assess differences in alpha diversities among both Haemaphysalis species using a Kruskal–Wallis test. Alpha diversity measures, including Shannon diversity, Faith's PD, observed features, and Pielou's evenness, were considered as response variables, with tick species as fixed effect variables. Subsequently, we examined the impact of species on beta diversity in both Haemaphysalis ticks using Adonis PERMANOVA with 999 permutations72.
Data availability
The unprocessed sequence data have been submitted to the DNA Data Bank of the Japan Sequence Read Archive and can be accessed via the DRA accession number DRA017340.
References
de la Fuente, J., Estrada-Pena, A., Venzal, J. M., Kocan, K. M. & Sonenshine, D. E. Overview: Ticks as vectors of pathogens that cause disease in humans and animals. Front. Biosci. 13, 6938–6946 (2008).
Guo, D. H. et al. Complete mitochondrial genomes of Dermacentor silvarum and comparative analyses with another hard tick Dermacentor nitens. Exp. Parasitol. 169, 22–27. https://doi.org/10.1016/j.exppara.2016.07.004 (2016).
Barker, S. C. & Murrell, A. Systematics and evolution of ticks with a list of valid genus and species names. Parasitology 129(Suppl), S15-36 (2004).
Liu, Z. Q. et al. Sequencing of complete mitochondrial genomes confirms synonymization of Hyalomma asiaticum asiaticum and kozlovi, and advances phylogenetic hypotheses for the Ixodidae. PLoS One 13, e0197524. https://doi.org/10.1371/journal.pone.0197524 (2018).
Eisen, R. J., Eisen, L. & Beard, C. B. County-scale distribution of Ixodes scapularis and Ixodes pacificus (Acari: Ixodidae) in the continental United States. J. Med. Entomol. 53, 349–386. https://doi.org/10.1093/jme/tjv237 (2016).
Kain, D. E., Sperling, F. A. H., Daly, H. V. & Lane, R. S. Mitochondrial DNA sequence variation in Ixodes pacificus (Acari: Ixodidae). Heredity 83, 378–386. https://doi.org/10.1038/sj.hdy.6886110 (1999).
Dergousoff, S. J. & Chilton, N. B. Differentiation of three species of ixodid tick, Dermacentor andersoni, D. variabilis and D. albipictus, by PCR-based approaches using markers in ribosomal DNA. Mol. Cell. Probes 21, 343–348. https://doi.org/10.1016/j.mcp.2007.04.003 (2007).
Lado, P. et al. The Amblyomma maculatum Koch, 1844 (Acari: Ixodidae) group of ticks: Phenotypic plasticity or incipient speciation?. Parasit. Vectors 11, 610. https://doi.org/10.1186/s13071-018-3186-9 (2018).
Madder, M., Speybroeck, N., Bilounga, A., Helleputte, D. & Berkvens, D. Survival of unfed Rhipicephalus appendiculatus and Rhipicephalus zambeziensis adults. Med. Vet. Entomol. 19, 245–250. https://doi.org/10.1111/j.1365-2915.2005.00566.x (2005).
Speybroeck, N. et al. Variation in body size in the tick complex Rhipicephalus appendiculatus/Rhipicephalus zambeziensis. J. Vector Ecol. 29, 347–354 (2004).
Hornok, S. et al. Contributions to the phylogeny of Ixodes (Pholeoixodes) canisuga, I. (Ph.) kaiseri, I. (Ph.) hexagonus and a simple pictorial key for the identification of their females. Parasit. Vectors 10, 545. https://doi.org/10.1186/s13071-017-2424-x (2017).
Alberto, A. et al. The Hard Ticks of the World (Acari: Ixodida: Ixodidae) 1st edn. (Springer Dordrecht, 2014).
Takano, A. et al. Construction of a DNA database for ticks collected in Japan: Application of molecular identification based on the mitochondrial 16S rDNA gene. Med. Entomol. Zool. 65, 13–21. https://doi.org/10.7601/mez.65.13 (2014).
Fukunaga, M., Yabuki, M., Hamase, A., Oliver, J. H. Jr. & Nakao, M. Molecular phylogenetic analysis of ixodid ticks based on the ribosomal DNA spacer, internal transcribed spacer 2, sequences. J. Parasitol. 86, 38–43. https://doi.org/10.1645/0022-3395(2000)086[0038:Mpaoit]2.0.Co;2 (2000).
Warburton, C. On some new and obscure species of the genus Haemaphysalis of the ixodidae. P. Camb. Philos. Soc. 14, 508–519 (1908).
Nuttall, G., Warburton, C., Cooper, W. & Robinson, L. Ticks, a Monograph of the Ixodoidea. Part III. The Genus Haemaphysalis 349–550 (Cambridge University Press, 1915).
Pomerantsev, B. I. & Zoologicheskii, I. Fauna of the USSR. New series no. 41, Arachnida. Vol. 4, no. 2. Ixodid ticks (Ixodidae). 199 pages; 26 cm (American Institute of Biological Sciences Washington, D.C., 1959).
Ogura, K. & Takada, K. The ticks parasitic on cattle and horses in Hokkaido, Japan. J. Coll. Agric. Hokkaido Imperial Univ. 18, 199–206 (1927).
Kitaoka, S. Keys to the species in immature stages of the Japanese Haemaphysalis ticks (Ixodidae). Bull. Natl. Inst. Anim. Health (Jpn.) 88, 49–63 (1985).
Nakao, M. & Ito, T. Haemaphysalis japonica, Haemaphysalis jezoensis and Haemaphysalis douglasi (Acari: Ixodidae): Which tick is distributed in Hokkaido?. Med. Entomol. Zool. 65, 33–35. https://doi.org/10.7601/mez.65.33 (2014).
Burger, T. D., Shao, R. & Barker, S. C. Phylogenetic analysis of the mitochondrial genomes and nuclear rRNA genes of ticks reveals a deep phylogenetic structure within the genus Haemaphysalis and further elucidates the polyphyly of the genus Amblyomma with respect to Amblyomma sphenodonti and Amblyomma elaphense. Ticks Tick Borne Dis. 4, 265–274. https://doi.org/10.1016/j.ttbdis.2013.02.002 (2013).
Kelava, S. et al. Phylogenies from mitochondrial genomes of 120 species of ticks: Insights into the evolution of the families of ticks and of the genus Amblyomma. Ticks Tick Borne Dis. 12, 101577. https://doi.org/10.1016/j.ttbdis.2020.101577 (2021).
Mohamed, W. M. A. et al. Comparative mitogenomics elucidates the population genetic structure of Amblyomma testudinarium in Japan and a closely related Amblyomma species in Myanmar. Evol. Appl. 15, 1062–1078. https://doi.org/10.1111/eva.13426 (2022).
Cotes-Perdomo, A. P. et al. Phylogenetic relationships of the Amblyomma cajennense complex (Acari: Ixodidae) at mitogenomic resolution. Ticks Tick Borne Dis. 14, 102125. https://doi.org/10.1016/j.ttbdis.2023.102125 (2023).
Burger, T. D., Shao, R. & Barker, S. C. Phylogenetic analysis of mitochondrial genome sequences indicates that the cattle tick, Rhipicephalus (Boophilus) microplus, contains a cryptic species. Mol. Phylogenet. Evol. 76, 241–253. https://doi.org/10.1016/j.ympev.2014.03.017 (2014).
Wang, J., Gao, L. & Aksoy, S. Microbiota in disease-transmitting vectors. Nat. Rev. Microbiol. https://doi.org/10.1038/s41579-023-00901-6 (2023).
Paulson, A. R., Lougheed, S. C., Huang, D. & Colautti, R. I. Multiomics reveals symbionts, pathogens, and tissue-specific microbiome of blacklegged ticks (Ixodes scapularis) from a Lyme disease hot spot in southeastern Ontario, Canada. Microbiol. Spectr. https://doi.org/10.1128/spectrum.01404-23 (2023).
Fountain-Jones, N. M. et al. Positive associations matter: Microbial relationships drive tick microbiome composition. Mol. Ecol. https://doi.org/10.1111/mec.16985 (2023).
Narasimhan, S. et al. Grappling with the tick microbiome. Trends Parasitol. 37, 722–733. https://doi.org/10.1016/j.pt.2021.04.004 (2021).
Che Lah, E. F., Ahamad, M., Dmitry, A., Md-Zain, B. M. & Yaakop, S. Metagenomic profile of the bacterial communities associated with Ixodes granulatus (Acari: Ixodidae): A potential vector of tick-borne diseases. J. Med. Entomol. 60, 753–768. https://doi.org/10.1093/jme/tjad044 (2023).
Lau, A. C. C. et al. The dynamics of the microbiome in ixodidae are shaped by tick ontogeny and pathogens in Sarawak, Malaysian Borneo. Microb. Genom. https://doi.org/10.1099/mgen.0.000954 (2023).
Lim, F. S. et al. Bacterial communities in Haemaphysalis, Dermacentor and Amblyomma ticks collected from wild boar of an Orang Asli Community in Malaysia. Ticks Tick-borne Dis. 11, 101352. https://doi.org/10.1016/j.ttbdis.2019.101352 (2020).
Wu-Chuang, A., Obregon, D., Mateos-Hernández, L. & Cabezas-Cruz, A. Anti-tick microbiota vaccines: How can this actually work?. Biologia 77, 1555–1562. https://doi.org/10.1007/s11756-021-00818-6 (2022).
Kelava, S. et al. Seventy-eight entire mitochondrial genomes and nuclear rRNA genes provide insight into the phylogeny of the hard ticks, particularly the Haemaphysalis species, Africaniella transversale and Robertsicus elaphensis. Ticks Tick-borne Dis. 14, 102070. https://doi.org/10.1016/j.ttbdis.2022.102070 (2023).
Mans, B. J. et al. Nuclear (18S–28S rRNA) and mitochondrial genome markers of Carios (Carios) vespertilionis (Argasidae) support Carios Latreille, 1796 as a lineage embedded in the Ornithodorinae: re-classification of the Carios sensu Klompen and Oliver (1993) clade into its respective subgenera. Ticks Tick-borne Dis. 12, 101688. https://doi.org/10.1016/j.ttbdis.2021.101688 (2021).
Gui, Z. et al. Genetic diversity analysis of Dermacentor nuttalli within Inner Mongolia, China. Parasit. Vectors 14, 1–12 (2021).
Regilme, M. A. F. et al. Comparative population genetic structure of two ixodid tick species (Acari: Ixodidae) (Ixodes ovatus and Haemaphysalis flava) in Niigata prefecture, Japan. Infect. Genet. Evolut. 94, 104999 (2021).
Kaufman, E. L. et al. Range-wide genetic analysis of Dermacentor variabilis and its Francisella-like endosymbionts demonstrates phylogeographic concordance between both taxa. Parasit. Vectors 11, 1–11 (2018).
Sassera, D. et al. ‘Candidatus Midichloria mitochondrii’, an endosymbiont of the tick Ixodes ricinus with a unique intramitochondrial lifestyle. Int. J. Syst. Evolut. Microbiol. 56, 2535–2540 (2006).
Maitre, A. et al. Rickettsial pathogens drive microbiota assembly in Hyalomma marginatum and Rhipicephalus bursa ticks. Mol. Ecol. https://doi.org/10.1111/mec.17058 (2023).
Coimbra-Dores, M. J. et al. Mitochondrial sequences of Rhipicephalus and Coxiella endosymbiont reveal evidence of lineages co-cladogenesis. FEMS Microbiol. Ecol. https://doi.org/10.1093/femsec/fiaa072 (2020).
Al-Khafaji, A. M. et al. Multi-locus sequence typing of Ixodes ricinus and its symbiont Candidatus Midichloria mitochondrii across Europe reveals evidence of local co-cladogenesis in Scotland. Ticks Tick Borne Dis. 10, 52–62. https://doi.org/10.1016/j.ttbdis.2018.08.016 (2019).
Zhang, X.-Y. et al. Growth dynamics and tissue localization of a Coxiella-like endosymbiont in the tick Haemaphysalis longicornis. Ticks Tick-borne Dis. 13, 102005. https://doi.org/10.1016/j.ttbdis.2022.102005 (2022).
Portillo, A. et al. Exploring the bacteriome in anthropophilic ticks: To investigate the vectors for diagnosis. PLoS One 14, e0213384. https://doi.org/10.1371/journal.pone.0213384 (2019).
Moustafa, M. A. M. et al. Novel symbionts and potential human pathogens excavated from argasid tick microbiomes that are shaped by dual or single symbiosis. Comput. Struct. Biotechnol. J. 20, 1979–1992. https://doi.org/10.1016/j.csbj.2022.04.020 (2022).
Thu, M. J. et al. Diversity of spotted fever group rickettsiae and their association with host ticks in Japan. Sci. Rep. 9, 1500. https://doi.org/10.1038/s41598-018-37836-5 (2019).
Nakao, R., Magona, J. W., Zhou, L., Jongejan, F. & Sugimoto, C. Multi-locus sequence typing of Ehrlichia ruminantium strains from geographically diverse origins and collected in Amblyomma variegatum from Uganda. Parasit. Vectors 4, 137. https://doi.org/10.1186/1756-3305-4-137 (2011).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evolut. 30, 772–780 (2013).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. https://doi.org/10.1093/sysbio/syq010 (2010).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolut. Biol. 7, 214. https://doi.org/10.1186/1471-2148-7-214 (2007).
Excoffier, L., Laval, G. & Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online 1, 47–50 (2007).
Holsinger, K. E. & Weir, B. S. Genetics in geographically structured populations: Defining, estimating and interpreting F(ST). Nat. Rev. Genet. 10, 639–650. https://doi.org/10.1038/nrg2611 (2009).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1–e1 (2013).
Herlemann, D. P. et al. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 5, 1571–1579 (2011).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490 (2010).
Shannon, C. E. A mathematical theory of communication. ACM SIGMOBILE Mobile Comput. Commun. Rev. 5, 3–55 (2001).
Faith, D. P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61, 1–10 (1992).
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072 (2006).
Pielou, E. C. The measurement of diversity in different types of biological collections. J. Theor. Biol. 13, 131–144 (1966).
McMurdie, P. J. & Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217 (2013).
Lozupone, C. & Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235 (2005).
Lozupone, C. A., Hamady, M., Kelley, S. T. & Knight, R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–1585 (2007).
Jaccard, P. Nouvelles recherches sur la distribution florale. Bull. Soc. Vaud. Sci. Nat. 44, 223–270 (1908).
Sorensen, T. A. A method of establishing groups of equal amplitude in plant sociology based on similarity of species content and its application to analyses of the vegetation on Danish commons. Biol. Skar. 5, 1–34 (1948).
Vázquez-Baeza, Y., Pirrung, M., Gonzalez, A. & Knight, R. EMPeror: A tool for visualizing high-throughput microbial community data. Gigascience https://doi.org/10.1186/2047-217X-2-16 (2013).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 1–17 (2018).
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A. & Callahan, B. J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 1–14 (2018).
Hunter, J. D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 9, 90–95 (2007).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, 1–18 (2011).
Anderson, M. J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26, 32–46 (2001).
Acknowledgements
The authors would like to express their deep gratitude to all collaborators who supported in collection of ticks in each prefecture.
Funding
This study was supported in part by the Japan Society for the Promotion of Science (JSPS) KAKENHI, Japan (19F19097, 20K21358, and 20KK0151, 22H02505, and 23H02373); the Japan Agency for Medical Research and Development (AMED), Japan (JP22fk0108614j0002, JP23fk0108644, and JP 23fk0108625); AMED SCARDA World-leading institutes for vaccine research and development Hokkaido Synergy Campus (223fa627005h0001); the Ministry of Health, Labour and Welfare (MHLW) (23HA2010).
Author information
Authors and Affiliations
Contributions
Conceptualization, R.N.; Data curation, M.A.M.M., W.M.A.M., E.C., and D.N.; Formal analysis, M.A.M.M., W.M.A.M., and R.N.; Investigation, M.A.M.M., W.M.A.M., E.C., and D.N.; Methodology, M.A.M.M., W.M.A.M., and R.N.; Project administration, R.N.; Funding acquisition, K.M. and R.N.; Resources, K.M., N.N., and R.N.; Software, W.M.A.M.; Supervision, S.C.B. and R.N.; Visualization, M.A.M.M.; Writing-original draft, M.A.M.M.; Writing-review and editing, E.C., A.W.G., S.C.B, and R.N. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Moustafa, M.A.M., Mohamed, W.M.A., Chatanga, E. et al. Unraveling the phylogenetics of genetically closely related species, Haemaphysalis japonica and Haemaphysalis megaspinosa, using entire tick mitogenomes and microbiomes. Sci Rep 14, 9961 (2024). https://doi.org/10.1038/s41598-024-60163-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-60163-x
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
