
Abstract
This study shows that the supramolecular arrangement of proteins in nanoparticle structures predicts nanoparticle accumulation in neutrophils in acute lung inflammation (ALI). We observed homing to inflamed lungs for a variety of nanoparticles with agglutinated protein (NAPs), defined by arrangement of protein in or on the nanoparticles via hydrophobic interactions, crosslinking and electrostatic interactions. Nanoparticles with symmetric protein arrangement (for example, viral capsids) had no selectivity for inflamed lungs. Flow cytometry and immunohistochemistry showed NAPs have tropism for pulmonary neutrophils. Protein-conjugated liposomes were engineered to recapitulate NAP tropism for pulmonary neutrophils. NAP uptake in neutrophils was shown to depend on complement opsonization. We demonstrate diagnostic imaging of ALI with NAPs; show NAP tropism for inflamed human donor lungs; and show that NAPs can remediate pulmonary oedema in ALI. This work demonstrates that structure-dependent tropism for neutrophils drives NAPs to inflamed lungs and shows NAPs can detect and treat ALI.
Similar content being viewed by others


Main
Neutrophils are ‘first-responder’ cells in acute inflammation, rapidly ‘marginating’ in and adhering to inflamed vessels1,2,3,4,5,6,7. Neutrophils can be activated by a variety of pathogen- and damage-associated factors, such as bacterial lipopolysaccharides (LPS)8,9. In acute inflammation, neutrophils marginate in most organs, but by far most avidly in the lung capillaries. There, abundant narrow vessels provide an ideal setting for neutrophil accumulation by adhesion and by mechanical retention aided by changes in neutrophil stiffness during inflammation5,6,10,11,12,13,14,15. Neutrophils are therefore key cell types in acute lung inflammation (ALI). In ALI, marginated neutrophils can secrete tissue-damaging substances (proteases, reactive oxygen species) and extravasate into alveoli, leading to a disrupted endothelial barrier and accumulation of neutrophils and oedema in the alveoli (Fig. 1a)6,7,10,11,12,16. Patients suffering the worst outcomes in COVID-19 have elevated neutrophil counts and neutrophilic infiltration in the alveoli17,18,19.

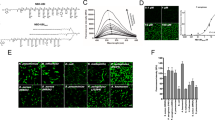
a, Schematic of neutrophil margination and extravasation in inflamed lungs (created with BioRender.com). ROS, reactive oxygen species. b, Biodistributions of lysozyme–dextran NGs in naive (n = 4 animals) and i.v.-LPS-affected (n = 8 animals) male C57BL/6 mice (red box, P < 1 × 10−10; *P = 0.00008). Inset: ratio of nanoparticle uptake in the lungs to nanoparticle uptake in the liver. c, Biodistributions of PEG–N-hydroxysuccinimide crosslinked human albumin NPs in naive (n = 3 animals) and i.v.-LPS-injured (n = 3 animals) mice (red box, P < 1 × 10−10; *P = 0.004). Inset: ratio of nanoparticle uptake in the lungs to nanoparticle uptake in the liver. d–k, Flow cytometry characterization of single-cell suspensions prepared from naive and i.v.-LPS-affected mouse lungs. Vertical axis in d and e indicates Ly6G staining for neutrophils and horizontal axis indicates signal from fluorescent NGs (d) or fluorescent albumin NPs (e). NG (f) and albumin NP (i) fluorescent signal from neutrophils in i.v.-LPS-injured mouse lungs (red/pink), compared to naive lungs (blue). Insets in f and i: flow cytometry data verifying increased neutrophil concentration in i.v.-LPS-injured mouse lungs (red/pink). Fraction of neutrophils positive for NGs (g) or albumin NPs (j) in naive or i.v.-LPS-injured lungs and fraction of NG-positive (h) or albumin NP-positive (k) cells that are neutrophils. For g and h, NGs/naive: n = 4 animals, NGs/LPS: n = 4 animals. For j and k, albumin NPs/naive: n = 3 animals, albumin NPs/LPS: n = 3 animals. *P = 2.6 × 10−7 (g), *P = 1.7 × 10−5 (h), *P = 0.0006 (j), *P = 0.007 (k). For l and m, fluorescence micrographs indicating association of NGs (red) with neutrophils (green, Ly6G stain) in the lungs of an i.v.-LPS-affected mouse (blue, tissue autofluorescence). Data are from histology for two naive mice and two i.v.-LPS-affected mice. l, Broad field of view indicating neutrophils and NGs alongside lung anatomy. m, Narrow field of view showing two neutrophils containing NGs. n, Single frame from real-time intravital imaging of NG (red) uptake in leukocytes (green) in the lungs of one i.v.-LPS-affected mouse (blue, Alexa Fluor 647-dextran). Statistical significance in b and c is derived from two-way analysis of variance (ANOVA) with Sidak’s multiple-comparisons test. Statistical significance in g,h,j,k is derived from paired two-tailed t tests. All error bars indicate mean ± s.e.m.
One of the challenges of treating ALI with drugs is poor tolerance of side effects in patients with the severe and heterogeneous system-wide maladies that can lead to ALI20,21,22,23. Targeted nanoparticle delivery to marginated neutrophils could provide treatment with lessened side effects, but delivery to marginated neutrophils remains an open challenge. Antibodies against Ly6G have achieved targeting to neutrophils in mice, but also deplete circulating neutrophils24,25,26,27. Additionally, while Ly6G marks neutrophils in mice, there is no analogous specific and ubiquitous marker on human neutrophils24. Therefore, antibody targeting has not been widely adopted for targeted drug delivery to neutrophils26. As another route to neutrophil targeting, previous studies observed activated neutrophils take up denatured and crosslinked albumin, concluding that denatured protein is critical in neutrophil–nanoparticle interactions28,29.
Nanoparticle structural properties such as shape, size and surface charge can define targeting behaviours30,31,32,33,34. In this article we screened a diverse panel of nanoparticles to identify structural properties that predict nanoparticle uptake in pulmonary marginated neutrophils in ALI. We identified more than ten nanoparticles with high selectivity for inflamed over naive lungs. Flow cytometry and immunohistochemistry showed these nanoparticles homing to marginated neutrophils. In many of the nanoparticles we tested, proteins were arranged at the supramolecular scale by different engineered intermolecular interactions or by evolved highly symmetrical arrangements of proteins (for example, viral capsids). Each nanoparticle with selectivity for inflamed lungs had structures with agglutinated protein in or on the nanoparticle. Nanoparticles with agglutinated protein (NAPs) are defined here as nanoparticles with supramolecular arrangement of protein in or on the nanoparticles determined by hydrophobic interactions, non-site-specific crosslinking or electrostatic interactions. In contrast to NAPs, we found that viral capsids and ferritin nanocages, defined by a regularly spaced and symmetric protein arrangement, have no selectivity for inflamed lungs. This study therefore shows that supramolecular arrangement of protein in nanoparticle structure predicts nanoparticle uptake in neutrophils in ALI. Our findings show that NAPs may improve diagnosis and treatment of acute lung inflammation.
Injury-selective uptake of nanoparticles in marginated neutrophils
Tracing of radiolabelled anti-Ly6G antibody, flow cytometry data and histology showed that intravenous (i.v.) LPS challenge in mice induces neutrophil margination in pulmonary vasculature 5 h after LPS administration (Supplementary Figs. 1 and 2). Following i.v. LPS, tracing of radiolabelled, fixed and inactivated Escherichia coli indicated marginated neutrophils in the lungs taking up the bacteria (Supplementary Fig. 3). To identify parameters in nanoparticle structure that correlate with nanoparticle uptake in marginated neutrophils in inflamed lungs, we therefore conducted an in vivo screen of biodistributions for a diverse array of radiolabelled nanoparticles in naive and i.v.-LPS-challenged mice. To show the radiotracer screen measures nanoparticle uptake in pulmonary marginated neutrophils, we more fully characterized the in vivo behaviour of two early hits in the screen.
Lysozyme–dextran nanogels (NGs) and poly(ethylene)glycol (PEG)-crosslinked albumin nanoparticles (albumin NPs) have been characterized as targeted drug-delivery agents35,36,37,38. 125I-Labelled NGs (diameter, 136.4 ± 3.6 nm; polydispersity index (PDI), 0.10 ± 0.02; zeta potential, −0.3 ± 0.1 mV; Supplementary Fig. 4a) and human albumin NPs (diameter, 317.8 ± 3.6 nm; PDI, 0.14 ± 0.05; zeta potential, −13.6 ± 0.6 mV; Supplementary Fig. 4b) were traced 30 min after i.v. injection in naive and i.v.-LPS-injured mice. Absolute NG lung uptake and lung:liver ratio increased 25-fold between naive and LPS-injured animals (Fig. 1b and Supplementary Table 2). Lung uptake of human albumin NPs increased 14-fold (Fig. 1c and Supplementary Table 2). Pharmacokinetics studies showed that NG uptake in inflamed lungs peaked 30 min after injection, followed by clearance over 24 h (Supplementary Fig. 5).
Flow cytometry identified cell types taking up fluorescent NGs or albumin NPs in the lungs. Flow data showed that the total number of lung cells containing NGs or albumin NPs increased between naive and LPS-injured lungs, corroborating radiotracer findings (Supplementary Figs. 5 and 6). Neutrophils accounted for the bulk of NG and albumin nanoparticle accumulation in i.v.-LPS-injured lungs (Fig. 1d,e). More than 70% of neutrophils contained nanoparticles in LPS-affected lungs, compared with <20% in naive lungs. Likewise, more than 70% of nanoparticle uptake in the lungs was accounted for by uptake in neutrophils (Fig. 1f–i and Supplementary Table 3). Leukocytes in general accounted for more than 90% of nanoparticle uptake (Supplementary Fig. 6c,d). Histology of LPS-injured lungs confirmed NG localization to neutrophils in lung vasculature (Fig. 1j). Slices in confocal images indicated that NGs were inside neutrophils (Fig. 1k). Intravital imaging of injured lungs showed real-time colocalizing of NGs with leukocytes in injured lungs (Fig. 1l and Supplementary Movie 1).
NGs accumulated in neutrophils in other models. Radiolabelled NGs also accumulated in the lungs following intratracheal (i.t.)39 and footpad LPS administration40. Lung:liver NG uptake ratio increased 45-fold following i.t.-LPS injury (Supplementary Fig. 8). Footpad LPS induced an 11-fold increase in NG lung uptake 6 h after insult and a 38-fold increase at 24 h (Supplementary Fig. 9). Footpad LPS also enhanced NG uptake in the legs. Intraplantar complete Freund’s adjuvant (CFA) injection was used to induce acute neutrophil accumulation local to the footpad, rather than the lungs41,42. CFA injury led to a threefold increase in NG uptake in the ipsilateral paw, but no change in lung uptake (Supplementary Fig. 10). Flow cytometry showed that ~90% of NG uptake in the feet was in leukocytes. CFA induced a fourfold increase in the proportion of NG uptake accounted for by neutrophils, demonstrating NG tropism for neutrophils outside of the lungs (Supplementary Fig. 11).
In vivo screen of diverse nanoparticle structures in acutely inflamed lungs
We screened biodistributions of nanoparticles with different sizes, shapes, compositions and surface charges in naive and i.v.-LPS-challenged mice. A subset of these nanoparticles had structures incorporating protein, including variant NGs (nanoparticles based on hydrophobic interactions between proteins), crosslinked protein nanoparticles, nanoparticles based on electrostatic interactions of charged proteins, viral capsids and ferritin nanocages.
Nanoparticles based on hydrophobic protein interactions
Atomic force microscopy and electron microscopy studies have shown that NGs form by lysozyme–lysozyme hydrophobic interactions, arranging a protein core stabilized by a dextran shell35,37,43. Noting previous work implicating denatured protein as a factor in nanoparticle uptake in neutrophils28,29, we obtained circular dichroism (CD) spectroscopy data indicating that the secondary structure of lysozyme in NGs is the same as lysozyme itself (Supplementary Fig. 12a). Lysozyme and NGs were exposed to the hydrophobic probe 8-anilino-1-naphthalenesulfonic acid (ANSA)44, demonstrating increased hydrophobicity on NGs versus lysozyme itself (Supplementary Fig. 12b). Our data are not consistent with lysozyme being denatured in NGs, but lysozyme arrangement in NGs alters the accessibility of hydrophobic domains.
NG structure was varied by modifying the lysozyme–dextran composition and the pH at which NGs were formed43. NGs of diameter ~75 nm (zeta potential, 7.1 ± 0.5 mV), ~200 nm (zeta potential, −0.2 ± 0.2 mV) and ~275 nm (zeta potential, −0.4 ± 0.1 mV) were traced, adding to data above for 130 nm NGs (Supplementary Figs. 4a and 13 and Supplementary Table 1). All the NG variants had consistent uptake in inflamed lungs, despite size-dependent variations in NG uptake in the liver and spleen (Fig. 2a and Supplementary Table 4).

a–c, NAPs accumulate in acutely inflamed lungs. a, Biodistributions of variant NGs indicating uptake of 75 nm NGs (n = 4 i.v.-LPS animals, n = 4 naive animals, red box: P < 1 × 10−10) and 200 nm NGs (n = 5 i.v.-LPS, n = 5 naive, red box: p < 1 × 10−10) in LPS-injured lungs, but not naive lungs. Data for 130 nm NGs are identical to that presented in Fig. 1b. b, Biodistributions of variant crosslinked albumin NPs indicating uptake of albumin nanorods (n = 3 i.v.-LPS animals, n = 3 naive animals; red box, P < 1 × 10−10) and bovine albumin NPs (n = 3 i.v.-LPS animals, n = 3 naive animals; red box, P < 1 × 10−10) in LPS-injured, but not naive lungs. Data for human albumin nanoparticles are identical to that presented in Fig. 1c. c, Biodistributions of charge-agglutinated protein NPs, indicating uptake of particles comprised of E-GFP and guanidine-tagged PONI or particles comprised of E-GFP and guanidine-tagged gold nanoparticles in LPS-injured (PONI: n = 5 animals; Au: n = 3 animals), but not naive (PONI: n = 4 animals; Au: n = 3 animals) lungs. PONI/E-GFP data reflect tracing of both 131I-labelled PONI and 125I-labelled E-GFP. For PONI tracer data: red box, P < 1 × 10−10. For E-GFP tracer data: red box, P = 0.0003. For Au/E−GFP data: red box, P = 1.6 × 10−9. d, NPs based on symmetric supramolecular arrangement of protein do not have tropism for inflamed lungs (schematics created with BioRender.com). Biodistributions of adenovirus (n = 5 i.v.-LPS animals, n = 5 naive animals; blue box, P = 0.88), adeno-associated virus (n = 3 i.v.-LPS animals, n = 3 naive animals; blue box, P = 0.56) and ferritin nanocages (n = 5 i.v.-LPS animals, n = 5 naive animals; blue box, P = 0.35) indicating no selectivity for LPS-injured versus naive lungs. e, Biodistributions of bare liposomes (schematic created with BioRender.com, n = 4 i.v.-LPS animals, n = 4 naive animals) indicating no selectivity for LPS-injured versus naive lungs (blue box, P = 0.31). Biodistributions of IgG-coated polystyrene NPs indicating low levels of uptake in both naive (n = 4 animals) and LPS-injured (n = 4 animals) lungs (blue box, P = 0.0004). Statistical significance in all panels is derived from two-way ANOVA with Sidak’s multiple comparisons test. All error bars indicate mean ± s.e.m.
Nanoparticles based on protein crosslinking
Electrohydrodynamic jetting has been used to form highly monodisperse PEG-crosslinked albumin micro- and nanoparticles, as characterized in electron microscopy studies38,45,46,47. CD spectroscopy indicated that albumin secondary structure was not altered in albumin NPs, while ANSA staining indicated that PEG–albumin NPs are more hydrophilic than albumin alone (Supplementary Fig. 14).
We varied the geometry and protein composition of crosslinked protein nanoparticles. Human albumin nanorods (aspect ratio, 3:1), bovine albumin nanoparticles (317.3 ± 38.5 nm; PDI, 0.17 ± 0.04; zeta potential, −11.2 ± 0.5 mV), human haemoglobin nanoparticles (328.1 ± 16.1 nm; PDI, 0.08 ± 0.01; zeta potential, −6.6 ± 0.8 mV), human transferrin nanoparticles (345.2 ± 10.2 nm; PDI, 0.12 ± 0.004; zeta potential, −5.6 ± 0.8 mV) and chicken lysozyme nanoparticles (298.6 ± 12.4 nm; PDI, 0.06 ± 0.01; zeta potential, −0.2 ± 0.7 mV) were traced in naive and IV LPS-treated mice (Fig. 2b, Supplementary Figs. 4b and Fig. 15 and Supplementary Table 1). Including human albumin NPs (Fig. 1c), five of the formulations had selectivity for inflamed lungs (Supplementary Table 5). Lysozyme nanoparticles were not selective for inflamed lungs, but accumulated in both naive and inflamed lungs in high concentrations.
Nanoparticles based on electrostatic protein interactions
Electron microscopy studies and functional assays have recently characterized nanoparticles based on interactions between charged proteins and oppositely charged polymers or gold nanoclusters48,49,50. We traced nanoparticles comprising poly(glutamate)-tagged green fluorescent protein (E-GFP) combined with arginine–gold nanoparticles (diameter, 89.0 ± 1.6 nm; PDI, 0.14 ± 0.04; zeta potential, −6.4 ± 0.5 mV) or with poly(oxanorborneneimide) (PONI) with guanidino and tyrosyl groups (diameter, 158.9 ± 6.2 nm; PDI, 0.17 ± 0.03; zeta potential, 18.3 ± 0.5 mV) (Supplementary Fig. 4c and Supplementary Table 1). For PONI/E-GFP nanoparticles, PONI was labelled with 131I and E-GFP was labelled with 125I to trace both components of the nanoparticles. PONI/E-GFP and arginine–gold/E-GFP nanoparticles were both selective for i.v.-LPS-affected lungs (Fig. 2c and Supplementary Fig. 16). PONI and E-GFP components of PONI/E-GFP nanoparticles accumulated in inflamed lungs at similar concentrations.
Nanoparticles based on symmetric protein arrangement
Adeno-associated virus, adenovirus and horse spleen ferritin are nanoparticles based on arrangement of protein in symmetric structures (sizes in Supplementary Fig. 4c, zeta potentials in Supplementary Table 1)51,52,53,54,55,56. Adeno-associated virus serotype 8, human adenovirus and horse spleen ferritin had no selectivity for i.v.-LPS-affected lungs (Fig. 2d, Supplementary Fig. 17 and Supplementary Table 6).
Non-protein nanoparticles
PEGylated liposomes (103.6 ± 8.7 nm; PDI, 0.09 ± 0.01; zeta potential, −2.2 ± 0.2 mV) were traced with 111In-DOTA-lipids and carboxylate polystyrene nanoparticles were conjugated to tracer 125I-immunoglobulin G (IgG) (230.5 ± 2.8 nm; PDI, 0.14 ± 0.0; zeta potential, −4.0 ± 0.8 mV; Supplementary Fig. 2c,d and Supplementary Table 1). As example nanoparticles with few or no proteins, these two nanoparticles did not home to i.v.-LPS-affected lungs (Fig. 2e and Supplementary Fig. 18).
Free protein controls
As compared to nanoparticles based on bovine albumin, hen lysozyme and human transferrin, our results indicate that albumin, lysozyme and transferrin themselves do not home to or have selectivity for i.v.-LPS-affected lungs (Supplementary Fig. 19 and Supplementary Table 7).
Protein-conjugated liposomes
Beyond data with PEGylated liposomes above, we traced protein-conjugated liposomes in naive and i.v.-LPS-treated mice. Liposomes were functionalized with rat IgG conjugated via N-succinimidyl S-acetylthioglycolate–maleimide (SATA–maleimide) chemistry (178.8 ± 6.9 nm; PDI, 0.23 ± 0.03; zeta potential, −2.1 ± 0.5 mV) or via click chemistry methods (128.3 ± 4.3 nm; PDI, 0.17 ± 0.03; zeta potential, −4.1 ± 0.2 mV), in which IgG was conjugated to dibenzocyclooctyne (DBCO) and the modified IgG was conjugated to azide-liposomes (Fig. 3a, Supplementary Fig. 4d and Supplementary Table 1)57. Compared to bare and SATA–IgG liposomes, DBCO–IgG liposomes had enhanced tropism for i.v.-LPS-affected lungs (Fig. 3b, Supplementary Fig. 20 and Supplementary Table 8). i.t.-LPS injury also enhanced lung uptake of DBCO–IgG liposomes (Supplementary Fig. 21). Flow cytometry with fluorescent DBCO–IgG liposomes confirmed liposome tropism for neutrophils in inflamed lungs (Fig. 3c,d and Supplementary Fig. 22). Approximately 90% of liposome signal in inflamed lungs was in neutrophils and >98% was in leukocytes.

a, Schematic of antibody-coated liposomes prepared via copper-free click reaction of azide-functionalized liposomes with DBCO-functionalized IgG (liposome schematic created with BioRender.com). b, Biodistributions in i.v.-LPS-injured mice for bare liposomes (n = 3 animals), liposomes conjugated to IgG via SATA-maleimide chemistry (n = 3 animals) and liposomes conjugated to IgG via DBCO–azide chemistry (n = 3 animals) (red box, P < 1 × 10−10 for DBCO–IgG liposomes versus bare liposomes and DBCO–IgG liposomes versus SATA–IgG liposomes). c, Mouse lung flow cytometry data indicating Ly6G anti-neutrophil staining density versus levels of DBCO–IgG liposome uptake as indicated by the green fluorescent TopFluor PC lipid signal. d, Flow cytometry data verifying increased DBCO–IgG liposome uptake in and selectivity for neutrophils following LPS insult. Inset: verification of increased concentration of neutrophils in the lungs following LPS. e, Fraction of neutrophils positive for DBCO–IgG liposomes in naive (n = 3 animals) or i.v.-LPS-injured (n = 3 animals) lungs (*P = 0.0003) and fraction of DBCO–IgG liposome-positive cells that are neutrophils (*P = 1.7 × 10−6). f, Biodistributions in i.v.-LPS-injured mice for azide-functionalized liposomes conjugated to IgG loaded with 2.5 (n = 3 animals), 5 (n = 3 animals), 10 (n = 4 animals) and 20 DBCO molecules per IgG (n = 3 animals; red box, P < 1 × 10−10 for DBCO(20×)–IgG liposomes compared to each of the other DBCO density groups). Insets in b and f: ratio of NP uptake in the lungs to NP uptake in the liver. Statistical significance in b and f is derived from two-way ANOVA with Tukey’s multiple-comparisons test. Statistical significance in e is derived from paired two-tailed t tests. All error bars indicate mean ± s.e.m.
We confirmed that DBCO modification of IgG confers neutrophil tropism on IgG liposomes by titrating the DBCO:IgG ratio (Supplementary Fig. 4d for liposome sizes, Supplementary Fig. 23, Supplementary Table 1 for liposome zeta potentials). The results in Fig. 3b used 20 DBCO per IgG. Reducing DBCO:IgG reduced DBCO–IgG liposome uptake in i.v.-LPS-affected lungs (Fig. 3e and Supplementary Table 9). DBCO–IgG itself did not accumulate in the lungs (Supplementary Fig. 24), showing that incorporation of the modified protein in nanoparticle structure was necessary for inflamed lung tropism. CD spectroscopy indicated DBCO did not change the IgG secondary structure (Supplementary Fig. 25).
In the above data, NAPs based on hydrophobic interactions, crosslinking and charge interactions all exhibited selectivity for inflamed lungs (Fig. 2a–c), with additional data indicating that this selectivity is based on tropism for marginated neutrophils (Fig. 1). Adding hydrophobic DBCO to protein on liposomes led to liposome uptake in neutrophils in inflamed lungs (Fig. 3), indicating that hydrophobic interactions between proteins on liposome surfaces also predict tropism for marginated neutrophils. Nanoparticles based on highly symmetric protein arrangements had no tropism for inflamed lungs (Fig. 2d). NAP selectivity for inflamed lungs did not linearly correlate with size or zeta potential (Supplementary Fig. 26), accommodated both spheres and rods, and was observed with eight different types of protein. Agglutinated protein was unique as a structural motif common to all nanoparticles with high selectivity for inflamed lungs. Principal component and linear discriminant analyses of our compiled biodistributions confirmed that (1) NAPs stood out from the other tested nanoparticles in their tropism for inflamed lungs; and (2) DBCO–IgG liposomes aligned with other NAPs (Supplementary Figs. 27 and 28).
Complement opsonization as a mechanism in NAP tropism for neutrophils
We conducted in vitro and in vivo studies isolating biological mechanisms by which neutrophils recognize NAPs. Exposure of NGs to serum before in vitro incubation with neutrophils led to a >30-fold increase in NG uptake (Fig. 4a and Supplementary Fig. 29a,b). We hypothesized that serum opsonins are a determinant in NG interactions with neutrophils. Mass spectrometry quantified the abundance of different proteins adsorbed on NGs after incubation in serum, identifying large quantities of complement proteins C3 and C5 (Fig. 4b and Supplementary Fig. 30a). C3 and C5 were significantly more abundant than complement C2 and C4 (C3 versus C2: P < 1 × 10−10, C3 versus C4: P < 1 × 10−10, C5 versus C2: P < 1 × 10−10, C5 versus C4: P = 0.011), signifying predominance of alternative pathway complement activation58.

a, Flow cytometry data indicating neutrophils take up lysozyme–dextran NGs after NG incubation in mouse serum (n = 18 biological replicates) but not after NG incubation in buffer (n = 18 biological replicates; *P = 6.9 × 10−9). b, Mass spectrometry characterization of proteins adsorbed on NGs after incubation with mouse serum as in a. Plotted data indicate the concentration of detected peptides associated with the five most abundant proteins on serum-incubated NGs (n = 3 serum/NG preparations). Complement proteins are highlighted in red text. c,d, Flow cytometric assessment of NG uptake in mouse neutrophils after NG incubation with buffer, mouse serum, heat-treated mouse serum and mouse serum treated with CVF: example histograms of NG fluorescence in neutrophils for different serum conditions (c); data reflecting NG mean fluorescence in neutrophils for different serum conditions (d). In d, bars with blue stripes indicate naive neutrophils (n = 18 biological replicates for naive serum, n = 10 for heat-treated serum, n = 11 for CVF-treated serum) and bars with red stripes indicate LPS-stimulated neutrophils (n = 12 biological replicates for naive serum, n = 5 for heat-treated serum, n = 7 for CVF-treated serum). For comparison to normal serum/naive, *P < 1 × 10−10 and **P < 1 × 10−10. For comparison to normal serum/LPS, ***P = 7.3 × 10−10 and ****P = 3.7 × 10−9. e, Biodistributions of NGs in naive mice (n = 4 animals), mice treated with CVF (n = 4 animals), mice treated with i.v. LPS (n = 4 animals) and mice treated with i.v. LPS and CVF (n = 4 animals). Naïve and i.v.-LPS data are identical to that presented in Supplementary Fig. 13, upper right panel. For comparison between LPS and CVF + LPS groups, *P = 1.6 × 10−10. f, Mass spectrometry characterization of complement opsonization of NGs (n = 3 serum/NG preparations) and human adenovirus (n = 3 serum/adenovirus preparations). Peptide count data as in b is normalized to peptide counts on NGs incubated with complement-depleted CVF-treated serum. Relative complement quantities below zero indicate complement opsonization at lower levels than on NGs after treatment with complement-depleted serum. Statistical significance in a is derived from paired two-tailed t tests. Statistical significance in d is derived from one-way ANOVA with Tukey’s multiple-comparisons test. Statistical significance in e is derived from two-way ANOVA with Tukey’s multiple-comparisons test. All error bars indicate mean ± s.e.m.
Serum was heat treated to inactivate complement59. Heat treatment eliminated the effect of serum on NG uptake in neutrophils (Fig. 4c and Supplementary Fig. 29a, b). Cobra venom factor (CVF) treatment to deplete serum complement by C3 activation60 also eliminated serum effects on NG–neutrophil interactions (Fig. 4c and Supplementary Fig. 29a,b). Serum from a mouse treated with CVF yielded identical results (Supplementary Fig. 29a,b). In vitro, LPS stimulation of neutrophils did not modify NG uptake (Fig. 4d and Supplementary Fig. 29c,d). In vitro, complement interactions with NAPs, rather than LPS stimulation of neutrophils, determined NAP uptake in neutrophils.
We treated mice with intraperitoneal CVF to deplete complement in vivo before NAP administration. CVF treatment reduced NG tropism for i.v.-LPS-inflamed lungs by >50%, with a >5-fold reduction in the lungs:blood ratio (Fig. 4e and Supplementary Fig. 31).
We also performed mass spectrometry analysis of serum proteins on adenovirus capsids, a protein nanoparticle with no tropism for neutrophils in inflamed lungs. All tested complement proteins were reduced on adenovirus versus NGs, with C5 and factor B absent on adenovirus (Fig. 4f and Supplementary Fig. 30a–c). CVF treatment of serum reduced the quantities of C3 and factor B on NGs, and the levels of these proteins on adenovirus capsid with naive serum were less than those on NGs with complement-depleted serum (Supplementary Fig. 30c).
Imaging lung inflammation with NAPs
Computerized tomography (CT) imaging can diagnose ALI by detecting pulmonary oedema, but cannot distinguish ALI from cardiogenic pulmonary oedema (CPO)61. CPO was induced in mice via propranolol infusion62. CT imaging confirmed pulmonary oedema after propranolol infusion (Fig. 5a,b, Supplementary Fig. 32 and Supplementary Movies 2 and 3). NGs homed to i.v.-LPS-affected lungs, but not CPO-affected lungs (Fig. 5c). We tested NGs as single-photon emission computerized tomography (SPECT)-CT contrast agents for ALI by imaging 111In-labelled NGs (Supplementary Fig. 33) in naive and i.v.-LPS-affected lungs. 111In SPECT signal was detectable in LPS-affected lungs, but at background level in naive lungs (Fig. 5d and Supplementary Movies 4–7). Previous studies have noted the potential for diagnostic imaging of lung inflammation based on neutrophils, including clinical data with autologous neutrophils in ALI and chronic obstructive pulmonary disease12,63. Our studies used NAP tropism for neutrophils to non-invasively image lung inflammation.

a, Three-dimensional reconstructions of chest CT data for naive mouse lungs and lungs with CPO. White/yellow indicates lower attenuation, corresponding to airspace in healthy lungs. Red/dark background indicates higher attenuation, corresponding to fluid in the lungs. b, Quantification of CT attenuation in naive (blue) and oedematous (yellow) lungs, averaged across axial slices (individual slice values in Supplementary Fig. 32). c, Biodistributions of 200 nm lysozyme–dextran NGs in naive mice, mice treated with i.v.-LPS and mice subject to CPO (n = 4 animals). Naïve and i.v.-LPS data are identical to that presented in Supplementary Fig. 13, upper right panel. Inset: ratio of NP uptake in the lungs to NP uptake in the liver. For comparison of CPO and i.v.-LPS values, *P < 1 × 10−10 for main panel and P = 0.005 for inset. d, Co-registered CT (greyscale) and SPECT (red/yellow) images indicating 111In-labelled lysozyme–dextran NG uptake in a naive and an i.v.-LPS-affected mouse. White/yellow indicates more nanogel uptake. Red/dark background indicates less lysozyme–dextran NG uptake. Statistical significance in c is derived from two-way ANOVA with Tukey’s multiple-comparisons test. All error bars indicate mean ± s.e.m.
NAP accumulation in inflamed human lungs ex vivo
We performed experiments to test NAP tropism for inflamed human lungs. NGs were added to single-cell suspensions prepared from human lungs. NGs accumulated in leukocytes in the suspensions in a dose-dependent manner (Supplementary Fig. 34a,b). Fluorescent or 125I-labelled NGs were infused via arterial catheter into ex vivo human lungs excluded from transplant due to lung damage64. Tissue dye indicated areas of the lungs perfused by NG suspensions (Supplementary Fig. 34c). For fluorescent NG experiments, high- and low-perfusion regions were selected for histology. NGs were detectable only in well-perfused tissue (Supplementary Fig. 34d). In experiments with 125I-NGs, 131I-ferritin was concurrently infused as a control particle without tropism for injured mouse lungs. NGs retained in human lungs at >5-fold higher concentration than ferritin (Supplementary Fig. 34e). NGs were concentrated in highly perfused lung regions, while ferritin was uniformly distributed (Supplementary Fig. 35). Our data indicate that NAP tropism for neutrophils in inflamed mouse lungs may be recapitulated in human lungs.
Effects of NAPs in acute lung inflammation
Mice were treated with nebulized LPS as a high-throughput model for severe acute respiratory distress syndrome (ARDS)65. Histology showed that nebulized LPS caused haemorrhage and airway infiltration by neutrophils 24 h after insult (Supplementary Fig. 36). In bronchoalveolar lavage fluid (BALF), nebulized LPS elevated neutrophil/leukocyte concentrations 50-fold and protein concentration 3-fold (Fig. 6a,b and Supplementary Fig. 37), indicating vascular disruption and oedema.

Timeline: nanoparticles or vehicle were administered as an i.v. bolus 2 h after nebulized LPS administration (liposome schematic created with BioRender.com). a,b, BALF was harvested 22 h after nanoparticle (30 mg kg−1) or vehicle administration. a, Protein concentration in BALF, reflecting quantity of oedema in naive mice (n = 5 animals), sham-treated mice with model ARDS (n = 15 animals) and mice with model ARDS treated with DBCO–IgG liposomes (n = 14 animals), NGs (n = 5 animals) or bare PEGylated liposomes (n = 5 animals). *P = 6.6 × 10−7, 0.0001 and 0.002 for comparison of DBCO–IgG liposome treatment with sham treatment, NG treatment and bare liposome treatment, respectively. b, Concentration of leukocytes in BALF for same groups as in a. *P = 1.1 × 10−6, 0.0001 and 0.002 for comparison of DBCO–IgG liposome treatment with sham treatment, NG treatment and bare liposome treatment, respectively. Quantities in a and b are represented as degree of protection against infiltration into alveoli, extrapolated from levels in naive mice (100% protection) and untreated mice with model ARDS (0% protection). c,d, Dose–response for oedema (c) and leukocyte infiltration (d) in alveoli of ARDS mice treated with DBCO–IgG liposomes. Data were obtained as in a and b, but with different liposome doses (n = 3 animals for 2.5, 5 and 10 mg kg−1 liposome doses). e, Chemokine CXCL2 levels in alveoli of LPS-injured mice with and without DBCO–IgG liposome treatment (n = 3 animals for all groups). Dashed line indicates CXCL2 levels in alveoli of naive mice. **P = 0.024, 0.079 and 0.034 for comparison of sham treatment with 2.5, 5 and 10 mg kg−1 DBCO–IgG liposome treatment, respectively. f, Concentration of neutrophils in BALF of naive mice (n = 5 animals), mice with model ARDS (n = 9 animals) and mice with model ARDS dosed with 30 mg kg−1 DBCO–IgG liposomes (n = 9 animals). For comparison of DBCO–IgG liposome treatment to sham treatment, *P = 0.009. g, Biodistributions of anti-Ly6G antibody in naive mice (n = 3 animals), LPS-injured mice (n = 3 animals) and mice treated with 10 mg kg−1 DBCO–IgG liposomes, with organs sampled at 1 h after treatment (n = 3 animals) or 22 h after treatment (n = 3 animals). Naive and untreated LPS-affected data are identical to data in Supplementary Fig. 1a. *P < 1 × 10−10 for all comparisons of anti-Ly6G uptake in lungs or spleen of liposome-treated mice versus sham-treated mice. h, Complete blood count analysis of circulating leukocyte concentrations in naive mice (n = 3 animals), LPS-injured mice (n = 3 animals) and mice treated with 10 mg kg−1 DBCO–IgG liposomes, with blood sampled 22 h after treatment (n = 3 animals). *P = 0.019, 0.025 and 0.047 for comparison of DBCO–IgG liposome-treated to sham-treated values for total white blood cell (WBC), lymphocyte (Lym.) and neutrophil (Neu.) counts, respectively. i, Schematic for the fate of neutrophils in mice with model ARDS, with and without DBCO–IgG liposome treatment, based on data in f–h (created with BioRender.com). Statistical significance in a, b, e and f is derived from one-way ANOVA with Tukey’s multiple-comparisons test. Statistical significance in g and h is derived from two-way ANOVA with Tukey’s multiple-comparisons test. All error bars indicate mean ± s.e.m.
DBCO–IgG liposomes, NGs and bare liposomes were administered (dose, 30 mg kg−1) 2 h after nebulized LPS. BALF leukocytes and protein were reduced by >50% by DBCO–IgG liposomes, but were unaffected by bare liposomes and NGs (Fig. 6a,b and Supplementary Fig. 37). DBCO–IgG liposome effects on BALF leukocytes and protein were dose dependent, with IC50s of 5.7 and 1.5 mg kg−1, respectively (Fig. 6c,d and Supplementary Fig. 38). CXCL2, a chemoattractant in neutrophil diapedesis66, was elevated in BALF and liver by nebulized LPS. BALF and liver CXCL2 were reduced by DBCO–IgG liposomes (Fig. 6e and Supplementary Fig. 39). Cytokine IL-6 is elevated in plasma in severe ARDS67. In our model, DBCO–IgG liposomes suppressed plasma IL-6 in a dose-dependent manner (Supplementary Fig. 40).
DBCO–IgG liposomes suppressed neutrophil accumulation in BALF in a manner consistent with suppression of BALF leukocytes (Fig. 6f and Supplementary Fig. 41). Quantifying intravascular neutrophils via radiotracer Ly6G antibody, we found DBCO–IgG liposomes reduced pulmonary marginated neutrophils, increased circulating neutrophils, and transiently increased spleen neutrophils, relative to untreated values (Fig. 6g and Supplementary Fig. 42). Complete blood count (CBC) confirmed DBCO–IgG liposome effects on circulating neutrophils (Fig. 6h and Supplementary Fig. 43). CBC also indicated that DBCO–IgG liposomes mitigate LPS suppression of lymphocytes and platelets and have insignificant effects on red blood cells (Supplementary Figs. 43 and 44).
These findings indicate that DBCO–IgG liposomes cause neutrophils to leave the lungs, getting sequestered in the spleen or returning to circulation. Accordingly, we observe lower neutrophil extravasation into alveoli and less oedema after DBCO–IgG liposome treatment (Fig. 6i). DBCO–IgG liposomes had no significant effects on body weight (Supplementary Fig. 45), but changes to signalling molecules CXCL2 and IL-6 and effects on platelets and lymphocytes indicate systemic effects for these NAPs.
Conclusions
This study identifies nanoparticle properties leading to tropism for neutrophils in inflamed lungs. These neutrophils take up NAPs, but not nanoparticles whose surface proteins are arranged with regular and symmetric spacing (for example, viral capsids and protein nanocages51,53,54,55,56). ‘Agglutination’ is defined here by the three types of protein–protein interactions enumerated in Fig. 2, which are designed without specific supramolecular arrangement. Electron microscopy data confirm there is no symmetry or regularity in protein–protein spacing in NAPs35,37,43,46,47,49,50. Thus, the structures of nanoparticles tested in our studies are differentiated by arrangement of protein at the supramolecular scale: nanoparticles with or without regular spacing and symmetry in protein arrangement. Differences in nanoparticle tropism for neutrophils in inflamed lungs aligned with differences in supramolecular-scale protein arrangement, but not differences in size, shape or zeta potential (Supplementary Figs. 26–28).
Supporting our conclusions, we re-engineered liposomes to behave like NAPs. We hypothesized that addition of the hydrophobic molecule DBCO to IgG on liposomes would recapitulate the protein ‘agglutination’ seen in NAPs based on hydrophobic interactions. Three lines of data confirm this: DBCO–IgG liposomes and NGs have similar tropism for neutrophils in inflamed lungs; replacing DBCO with less hydrophobic SATAmaleimide conjugation68 abrogates neutrophil tropism; and titrating down the amount of DBCO on IgG ratchets down neutrophil tropism of DBCO–IgG liposomes.
We showed that the mechanism by which neutrophils recognize NAPs is dependent on complement opsonization. While prior literature shows the importance of complement opsonization in nanoparticle uptake69, our observations suggest that the alternative pathway of complement responds to differences in supramolecular arrangement of protein.
Finally, we showed that some NAPs have anti-inflammatory properties. DBCO–IgG liposomes, without any cargo drug, provide dose-dependent reduction in protein and leukocyte extravasation into pulmonary alveoli in a model of the human lung disease ARDS. Neutrophil tracing showed that DBCO–IgG liposomes effect these benefits by decreasing neutrophil retention in inflamed pulmonary vasculature and reducing the neutrophils’ ability to cross the pulmonary vascular barrier. DBCO–IgG liposomes decreased systemic and local inflammatory cytokines and chemokines, which may indicate effects on other cell types (for example, CXCL2 is secreted primarily by monocytes and macrophages)66,70. These anti-inflammatory effects may be viewed as side effects, since they imply effects on multiple cell types and signalling molecules (a full discussion of immunological implications is included in the Supplementary Information). Not all NAPs have anti-inflammatory effects (for example, NGs), so NAPs may be loaded with drugs, such as antibiotics, that have benefits in ALI.
Supramolecular arrangement of protein in nanoparticle structure predicts nanoparticle uptake in pulmonary marginated neutrophils during acute inflammation. Agglutinated protein is a motif in nanoparticle structure correlated with nanoparticle tropism for neutrophils. The NAPs identified in this study, and future NAPs based on parameters outlined here, could be untapped resources for diagnosis and treatment of devastating inflammatory disorders such as ALI.
Methods
Nanoparticle synthesis
NGs, crosslinked protein nanoparticles, charged protein nanoparticles, liposomes,and polystyrene nanoparticles were prepared as previously described36,43. Details of the synthesis for each type of nanoparticle are included in the Supplementary Information. Nanoparticles, proteins and bacteria were labelled with 125I, 131I or 111In according to described methods57. Details of labelling techniques are provided in the Supplementary Information.
E. coli preparation
TOP10 E. coli (ThermoFisher #C404002) were grown overnight in Terrific broth with ampicillin. Bacteria were heat-inactivated by a 20 min incubation at 60 °C, then fixed by overnight incubation in 4% paraformaldehyde. After fixation, bacteria were pelleted by centrifugation at 1,000g for 10 min. Pelleted bacteria were washed three times in PBS, before resuspension by pipetting. Bacterial concentration was verified by optical density at 600 nm, before radiolabelling as described above. Bacteria were administered i.v. in mice (7.5 × 107 colony-forming units in a 100 µl suspension per mouse).
Nanoparticle and protein tracing in mice
Nanoparticle or protein biodistributions were tested by i.v. injecting nanoparticles or protein (suspended to 100 µl in PBS or 0.9% saline at a dose of 2.5 mg kg−1 with tracer quantities of radiolabelled material) in C57BL/6 male mice from Jackson Laboratories. For experiments tracing anti-Ly6G biodistributions to locate intravascular neutrophils, radiolabelled anti-Ly6G was administered i.v. at 0.1 mg kg−1. The quantity of injected radioactivity was measured by gamma counter (Perkin-Elmer) immediately before injection. To determine the nanoparticle masses for dosing, NGs and charged protein nanoparticles were prepared from reactants at known concentrations in synthetic methods involving no material loss; crosslinked protein nanoparticles were resuspended from weighed powder; polystyrene nanoparticles, ferritin and viral capsids were purchased at known concentrations. Liposomes were prepared from reactants at known concentrations and previous work with the same synthetic methods assessed low material losses during filtration and purification of liposomes57,71.
Biodistributions in naive mice were compared to biodistributions in injury models. Biodistribution data were collected at 30 min after nanoparticle or protein injection, unless otherwise stated. Blood was collected by vena cava draw and mice were killed by exsanguination and cervical dislocation. Organs were harvested and rinsed in saline, and blood and organs were examined for nanoparticle or protein retention by gamma counter. To calculate concentration in organs, quantity of retained radioactivity was normalized to organ weights.
For i.v. LPS, mice were anaesthetized with 3% isoflurane before retro-orbital injection of LPS from E. coli strain B4 at 2 mg kg−1 in 100 µl PBS. After 5 h, mice were anaesthetized with ketamine/xylazine (10 mg kg−1 ketamine, 100 mg kg−1 xylazine, intramuscular administration) and before jugular vein nanoparticle or protein injection. For i.t. LPS, B4 LPS was administered to mice (anaesthetized with ketamine/xylazine) at 1 mg kg−1 in 50 µl of PBS via tracheal catheter, followed by 100 µl of air71. NGs were injected 16 h after i.t. LPS and DBCO–IgG liposomes were injected 1, 2 or 6 h after i.t. LPS. For footpad LPS, B4 LPS was administered at 1 mg kg−1 in 50 µl PBS via footpad injection. NGs were injected i.v. 6 or 24 h after footpad LPS. For cardiogenic pulmonary oedema, mice were anaesthetized with ketamine/xylazine and administered propranolol in saline (3 µg ml−1) via jugular vein catheter at 83 µL min−1 over 120 min62, before i.v. NG injection. For localized footpad inflammation, mice were anaesthetized with 3% isoflurane and 20 µl of CFA or 20 µl of sham saline was injected subcutaneously in the central plantar region of the left hind paw42, 6 h before i.v. NG injection.
Single-cell suspension flow cytometry
Single-cell suspensions were prepared from male C57BL/6 mouse lungs for flow cytometry. Fluorescent nanoparticles were administered at 2.5 mg kg−1 30 min before the animals were killed and their lungs extracted. Mice were anaesthetized with ketamine/xylazine (10 mg kg−1 ketamine, 100 mg kg−1 xylazine, intramuscular administration) before installation of a tracheal catheter secured by suture. After the animals had been killed by vena cava exsanguination, lungs were perfused via the right ventricle with ~10 ml of cold PBS. Lungs were then infused via a tracheal catheter with 1 ml of cold PBS solution with 5 U ml−1 dispase, 2.5 mg ml−1 collagenase I and 1 mg ml−1 of DNAse I. Immediately after infusion, the trachea was sutured shut while removing the tracheal catheter. Lungs with intact trachea were removed via thoracotomy and kept on ice before manual disaggregation.
Single-cell suspensions were also prepared from mouse feet. Feet were removed immediately following after the animals had been killed by cervical dislocation and 100 µl of dispase/collagenase/DNAse was injected subcutaneously in the feet. Tissue was separated from the bones while the feet were held in 1 ml of dispase/collagenase/DNAse.
Disaggregated lung or foot tissue was aspirated in an additional 2 ml of dispase/collagenase/DNAse and incubated at 37 °C for 45 min, vortexing every 10 min. After addition of 1 ml of fetal calf serum, tissue suspensions were strained through 100 µm filters and centrifuged at 400g for 5 min. Pelleted material was resuspended in 10 ml of cold ACK lysing buffer. The resulting suspensions were strained through a 40 µm filter, incubated for 10 min on ice, and centrifuged at 400g for 5 min. The pelleted material was rinsed in 10 ml of FACS buffer (2% fetal calf serum and 1 mM EDTA in PBS). After 400g/5 min centrifugation, pellets were resuspended in 2% PFA in 1 ml FACS buffer for a 10 min room temperature incubation. Fixed cell suspensions were centrifuged at 400g for 5 min and resuspended in 1 ml of FACS buffer.
To stain fixed cells, 100 µl aliquots of cell suspensions were pelleted at 400g for 5 min, then resuspended in labelled antibody diluted in FACS buffer (1:150 dilution for anti-Ly6G, APC-anti-CD31 or PE-anti-F480 antibodies and 1:500 dilution for anti-CD45 andtibodies). Samples were incubated with staining antibodies for 20 min at room temperature in the dark, diluted with 1 ml of FACS buffer and pelleted at 400g for 5 min. Stained pellets were resuspended in 200 µl of FACS buffer immediately before flow cytometry (BD Accuri). Data were gated to exclude debris and doublets. Controls with no stain, obtained from naive and i.v.-LPS-injured mice, established gates for negative/positive staining with FITC/AlexFluor 488, PE, PerCP/Cy5.5 and APC/Alexa Fluor 647. Single-stain controls allowed automatic generation of compensation matrices in FCS Express software.
To analyse intravascular leukocyte populations in lungs, mice received i.v. FITC-anti-CD45 5 min before they were killed. Populations of intravascular versus extravascular leukocytes were assessed by also staining fixed cell suspensions with PerCP-conjugated anti-CD45 and/or Alexa Fluor 647-conjugated anti-Ly6G. Comparing PerCP anti-CD45 with FITC anti-CD45 signal indicated intravascular versus extravascular leukocytes. Comparison of Alexa Fluor 647 anti-Ly6G, PerCP anti-CD45 and FITC anti-CD45 signal indicated intravascular versus extravascular neutrophils.
To characterize nanoparticle distribution among different cells in lungs or feet, fluorescent nanoparticles were administered at 2.5 mg kg−1 via jugular vein injection and circulated for 30 min. Fixed single-cell suspensions were stained and coincidence of nanoparticle fluorescence with anti-CD45, anti-Ly6G, PE anti-CD31 or PE anti-F480 fluorescence was assessed.
In vitro neutrophil uptake of nanoparticles
Bone marrow was collected from pooled femurs of C57BL/6 mice. Neutrophils were isolated with the StemCell Technologies RoboSep Mouse Neutrophil Enrichment Kit by magnetic-bead-mediated depletion of non-neutrophils. To serum-treat NGs before incubation with neutrophils, 5 × 109 FITC-labelled NGs in 10 µl PBS were incubated with 10 µl serum for 1 h at 37 °C; then 1 × 106 neutrophils were rotated with 5 × 109 NGs in 20 µl PBS for 15 min at 37 °C. For flow cytometry (BD Accuri C6), neutrophils were washed and stained with PerCP/Cy5.5 Ly6G antibodies (BD Biosciences, 1:100 dilution) and non-neutrophils were excluded from analysis via Ly6G staining (see Supplementary Fig. 27 for gating). NG fluorescence in neutrophils was quantified.
To probe the role of complement in neutrophil–nanoparticle interactions, complement was depleted from serum via two methods: heat treatment and CVF treatment. For heat treatment, serum was incubated at 56 °C for 1 h and denatured proteins were removed by centrifugation at 10,000g for 15 min. For CVF treatment, 10 units CVF per ml serum were incubated for 1 h at 37 °C, then centrifuged at 10,000g for 15 min. Nanoparticle incubation with heat- and CVF-treated serum followed the same protocol described above for naive serum.
In vivo effects of complement on nanoparticle tropism for neutrophils
Mice were dosed with 100 µg CVF per kg via intraperitoneal injection 24 h before NG administration or blood draw to test the in vivo effects of complement depletion on nanoparticle–neutrophil interactions. For experiments with both LPS and CVF, B4 LPS from E. coli was administered i.v. at 2 mg kg−1 19 h after CVF and 5 h before i.v. NGs (2.5 mg kg−1), as described in Nanoparticle and protein tracing in mice.
Mass spectrometry profiles of protein coronae on nanoparticles
A 25 µl volume of NGs or adenovirus capsids in a 5 mg ml−1 suspension were incubated with and equivalent volume of wild-type or CVF-treated (as above) mouse serum or saline sham for 1 h at 37 °C. Nanoparticles were pelleted by centrifugation and washed with 1 ml PBS three times to separate from unbound serum proteins.
Opsonized and sham-opsonized nanoparticles were prepared for mass spectrometry analysis as follows. Samples were solubilized and digested with the iST kit (PreOmics GmbH) per the manufacturer’s protocol. Nanoparticle pellets were resuspended, reduced and alykylated by addition of sodium deoxycholate buffer containing tris(2-carboxyethyl)phosphine and 2-chloroacetamide. The resulting suspensions were heated at 95 °C for 10 min. Proteins were enzymatically hydrolysed for 1.5 h at 37 °C by endoproteinase Lys-C and trypsin. The resulting peptides were desalted, dried by vacuum centrifugation and reconstituted in 0.1% trifluoroacetic acid containing indexed retention time peptides (Biognosys Schlieren). Ultraperformance liquid chromatography–mass spectrometry data were obtained and analysed by published methods72,73, as detailed in the Supplementary Information.
SPECT/CT imaging
Imaging techniques, as described previously57, are detailed in the Supplementary Information. SPECT and CT data, in NIFTI format, were opened with ImageJ software (FIJI package) and processed for background removal, pseudocolour assignment and three-dimensional reconstruction, as detailed in the Supplementary Information.
Nanoparticle administration in human lungs
Human lungs were obtained after organ harvest from transplant donors whose lungs were in advance deemed unsuitable for transplantation. Lungs were kept at 4 °C and used within 24 h of organ harvest. Lungs were inflated with low-pressure oxygen and oxygen flow was maintained at 0.8 l min−1 for gentle inflation. Pulmonary artery subsegmental branches were endovascularly cannulated, then tested for retrograde flow by perfusing for 5 min with Steen solution containing a small amount of green tissue dye at 25 cm H2O pressure. Pulmonary veins through which efflux of perfusate emerged were noted, allowing collection of solutions after passage through the lungs. A 2 ml mixture of 125I-labelled NGs and 131I-labelled ferritin was injected through the arterial catheter. Approximately 100 ml of 3% BSA in PBS was passed through the same catheter to rinse unbound nanoparticles. A solution of green tissue dye was subsequently injected through the same catheter. The cannulated lung lobe was dissected into ~1 g segments, which were evaluated for density of tissue dye staining. Segments were weighed, divided into ‘high’, ‘medium’, ‘low’ and ‘null’ levels of dye staining, and measured for 131I and 125I signal in a gamma counter.
For experiments with cell suspensions derived from human lungs (chosen for research use as above), single-cell suspensions were generously provided by Edward Morrisey at the University of Pennsylvania. Aliquots of 600,000 cells were pelleted at 400g for 5 min and resuspended in 100 µl FACs buffer containing different quantities of FITC-dextran NGs. Cells and NGs were incubated at room temperature for 60 min before twofold pelleting at 400g with 1 ml PBS washes. Cells were resuspended in 200 µl FACS buffer for staining with APC anti-human CD45 (1:500 dilution, 20 min room temperature incubation). Cells were pelleted at 400g for 5 min and resuspended in 200 µl PBS for immediate analysis with flow cytometry (BD Accuri). Negative/positive NG or anti-CD45 signal was established by comparison to unstained cells. Single-stained controls indicated no spectral overlap between FITC-NG fluorescence and anti-CD45 APC fluorescence.
Effects of nanoparticles in nebulized LPS model
Mice were exposed to nebulized B4 LPS in a whole-body exposure chamber, with separate compartments for each mouse (MPC-3 AERO; Braintree Scientific). To maintain adequate hydration, mice were injected with 1 ml sterile saline warmed to 37 °C, intraperitoneally, immediately before LPS exposure. LPS was reconstituted in PBS to 10 mg ml−1 and stored at −80 °C until use. Immediately before nebulization, LPS was thawed and diluted to 5 mg ml−1 with PBS. Then, 5 ml of diluted LPS was aerosolized via a jet nebulizer connected to the exposure chamber (NEB-MED H, Braintree Scientific). Nebulization was performed until all liquid was nebulized (~20 min).
DBCO–IgG liposomes (20:1 DBCO:IgG, 2.5, 5,10 or 30 mg kg−1), bare liposomes (30 mg kg−1), NGs (30 mg kg−1) or saline sham were administered via retro-orbital injections of 100 µl of suspension 2 h after LPS exposure. Mice were anaesthetized with 3% isoflurane to facilitate injections. Blood draws and BALF were collected 24 h after LPS exposure, as previously described and detailed in the Supplementary Information71. Mice were weighed before administration of nebulized LPS and before BALF and blood draws.
To stain for flow cytometry, BALF samples were centrifuged at 300g for 4 min, the supernatant was aspirated and 100 µl of staining buffer (1:1,000 APC-anti-CD45 or 1:150 Alexa Fluor 488-anti-Ly6G in FACS buffer) was added. Samples were stained for 30 min at room temperature in the dark, then 1 ml of FACS buffer was added, samples were centrifuged at 300g for 4 min and supernatant was aspirated. Cells were resuspended in 900 µl of FACS buffer for flow cytometry analysis (BD Accuri). Forward scatter (area) versus side scatter (area) plots gated-out non-cellular debris and forward scatter (area) versus forward scatter (height) plots gated-out doublets. Unstained controls set gates for APC and Alexa Fluor 488 signal. Single-stained controls showed no spectral overlap between APC-anti-CD45 and Alexa Fluor 488-anti-Ly6G. CD45- and Ly6G-positive cells determined leukocyte and neutrophil concentrations, respectively.
To trace intravascular neutrophils after nebulized LPS treatment and 10 mg kg−1 DBCO–IgG liposome dosing, 125I-anti-Ly6G (0.1 mg kg−1) was administered 1 or 22 h after liposomes, and biodistributions were determined as described above. In mice treated with DBCO–IgG liposomes, blood was drawn into EDTA via the vena cava before exsanguination. Lungs and liver were removed after obtaining BALF and stored at −80 °C. Blood was immediately evaluated with CBC measurements (Abaxis VetScan HM5). Blood remaining after CBC was centrifuged at 1,500g for 10 min at 4 °C and plasma was extracted and stored at −80 °C. Chemokine CXCL2 and cytokine IL-6 were measured in BALF, plasma, and lung and liver homogenates according to published methods detailed in the Supplementary Information74.
CD spectroscopy
Proteins were prepared in deionized and filtered water at concentrations of 0.155 mg ml−1 for human albumin, 0.2 mg ml−1 for hen lysozyme and 0.48 mg ml−1 for IgG. Albumin NPs, NGs and IgG-coated liposomes were diluted such that albumin, lysozyme and IgG concentrations in the suspensions matched concentrations of corresponding protein solutions. Protein and nanoparticle solutions were analysed in quartz cuvettes with 10 mm path length in an Aviv CD spectrometer. The instrument was equilibrated in nitrogen at 25 °C for 30 min before use and samples were analysed with sweeps between 185 and 285 nm in 1 nm increments. Each data point was obtained after a 0.333 s settling time, with a 2 s averaging time. CDNN75 software deconvoluted CD data (expressed in millidegrees) via a neural network algorithm assessing alignment of spectra with library-determined spectra for helices, antiparallel sheets, parallel sheets, β turns and random coils75.
8-Anilino-1-naphthalenesulfonic acid nanoparticle staining
8-Anilino-1-naphthalenesulfonic acid (ANSA) at 0.06 mg ml−1 was mixed with lysozyme, human albumin or IgG at 1.5 mg ml−1 in PBS. For nanoparticle analysis, nanoparticle suspensions were prepared such that albumin, lysozyme and IgG concentrations in the suspensions matched the 1.5 mg ml−1 concentration of protein solutions. Protein or nanoparticles and ANSA were reacted at room temperature for 30 min. Excess ANSA was removed from solutions by three centrifugations against 3 kDa cut-off centrifugal filters (Amicon). After resuspension to original volume, ANSA-stained protein/nanoparticle solutions/suspensions were examined for fluorescence (excitation, 375 nm) and absorbance maxima corresponding to ANSA.
Histology
For imaging neutrophils in naive and i.v.-LPS-affected lungs, mice were given i.v. anti-Ly6G and killed 30 min later. Lungs were embedded in M1 medium, flash frozen and sectioned in 10 µm slices. Sections were stained with Alexa Fluor 594-goat anti-rat secondary antibody (1:200 dilution) and imaged with epifluorescence microscopy. Similarly, rhodamine–dextran NGs were administered i.v. in i.v.-LPS mice 30 min before the mice were killed. Lungs were sectioned as above and stained with clone 1A8 anti-Ly6G antibody, followed by Alexa Fluor 350-goat anti-rat secondary antibody (1:150 dilution), before epifluorescence and confocal imaging of NG and neutrophil fluorescence.
For histological verification of injury following nebulized LPS treatment, one set of injured mouse lungs and one set of naive lungs was infused intratracheally with 4% paraformaldehyde. The trachea was tied off and lungs and trachea were removed via thoracotomy. The lungs were suspended in 4% paraformaldehyde for overnight fixation before embedding in paraffin, sectioning and haematoxylin/eosin staining.
Sections of human lungs were obtained after administration of rhodamine–dextran NGs. NG-perfused and non-perfused tissue regions were harvested, embedded in M1 medium, flash frozen and sectioned in 10 µm slices. NG fluorescence and tissue autofluorescence were detected with epifluorescence imaging.
Live lung imaging
A mouse was anaesthetized with ketamine/xylazine 5 h after i.v. LPS. A jugular vein catheter was placed for injection of NGs, anti-CD45 and fluorescent dextran. A patch of skin on the back of the mouse, around the juncture between the ribcage and the diaphragm, was denuded. The mouse was maintained on mechanical ventilation and the lungs were exposed via incision at the juncture between the ribs and the diaphragm. A coverslip affixed to a rubber O-ring was sealed to the incision by vacuum. The exposed lungs were focused under the objective using autofluorescence. With 100 ms exposure, channels corresponding to violet, green, near-red and far-red fluorescence were sequentially imaged. Rhodamine–dextran NGs (2.5 mg kg−1), Brilliant Violet-anti-CD45 (0.8 mg kg−1) and Alexa Fluor 647–70 kDa dextran (40 mg kg−1) were injected via the jugular vein. Images were recorded for 30 min in SlideBook software and opened in ImageJ (FIJI distribution) for composition in movies with co-registration of the four fluorescent channels.
Animal and human study protocols
All animal studies were carried out in strict accordance with Guide for the Care and Use of Laboratory Animals as adopted by National Institutes of Health and approved by University of Pennsylvania Institutional Animal Care and Use Committee. All animal experiments used male C57BL/6 J mice, 6–8 weeks old, purchased from Jackson Laboratories. Mice were maintained at 20–25 °C, 50% ± 20% humidity, and on a 12/12 h dark/light cycle with food and water ad libitum.
Human lungs were obtained by the University of Pennsylvania Lung Biology Institute’s Human Lung Tissue Bank (HLTB) from Gift of Life Donor Program (Philadelphia, PA, USA). Lungs provided to the HLTB were determined unsuitable for transplantation into a recipient, and would have been discarded if not used for our studies. Lungs provided by the HLTB for these studies are deidentified and cannot be linked to individual donors. Clinical staff procuring and distributing the tissue through the HLTB are not involved in the research after distribution of the deidentified tissue. Studies employing deidentified tissue from the HLTB were therefore determined by the University of Pennsylvania Institutional Review Board (IRB) to be IRB-exempt and were not considered human research subjects as defined by the Office of Human Research Protection of the National Institutes of Health. Deidentified patient data for human lungs (age, sex and cause of death) are tabulated in Supplementary Table 11.
Statistical analysis
Error bars indicate the standard error of the mean throughout. Significance tests are described in captions. Statistical power was determined for statements of statistical significance and tabulated in the supplementary materials.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are presented graphically or in tables in the paper and Supplementary Information. Raw numerical imaging, flow cytometry, gamma counter, mass spectrometry and spectroscopy data provided graphically in this study are available as tabulated values from the corresponding author upon reasonable request.
Code availability
Linear discriminant and principal component analysis were completed with Gnu Octave 6.1.0 scripts adapted from https://www.bytefish.de/blog/pca_lda_with_gnu_octave/ and are available in full in the Supplementary Information.
References
Wright, H. L., Moots, R. J., Bucknall, R. C. & Edwards, S. W. Neutrophil function in inflammation and inflammatory diseases. Rheumatology 49, 1618–1631 (2010).
Sônego, F. et al. Paradoxical roles of the neutrophil in sepsis: protective and deleterious. Front. Immunol. 7, 155 (2016).
Jickling, G. C. et al. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J. Cereb. Blood Flow. Metab. 35, 888–901 (2015).
Mehta, J. et al. Neutrophil function in ischemic heart disease. Circulation 79, 549–556 (1989).
Grommes, J. & Soehnlein, O. Contribution of neutrophils to acute lung injury. Mol. Med. 17, 293–307 (2011).
Summers, C. et al. Neutrophil kinetics in health and disease. Trends Immunol. 31, 318–324 (2010).
Gee, M. H. & Albertine, K. H. Neutrophil–endothelial cell interactions in the lung. Annu. Rev. Physiol. 55, 227–248 (1993).
Mayadas, T. N., Cullere, X. & Lowell, C. A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 9, 181–218 (2014).
Mantovani, A., Cassatella, M. A., Costantini, C. & Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 11, 519–531 (2011).
Williams, A. E. & Chambers, R. C. The mercurial nature of neutrophils: still an enigma in ARDS? Am. J. Physiol. Lung Cell. Mol. Physiol. 306, L217–L230 (2014).
Zemans, R. L. & Matthay, M. A. What drives neutrophils to the alveoli in ARDS? Thorax 72, 1–3 (2017).
Summers, C. et al. Pulmonary retention of primed neutrophils: a novel protective host response, which is impaired in the acute respiratory distress syndrome. Thorax 69, 623–629 (2014).
Doerschuk, C. M. Mechanisms of leukocyte sequestration in inflamed lungs. Microcirculation 8, 71–88 (2001).
Worthen, G. S., Schwab, B., Elson, E. L. & Downey, G. P. Mechanics of stimulated neutrophils: cell stiffening induces retention in capillaries. Science 245, 183–186 (1989).
Kuebler, W. M. & Goetz, A. E. The marginated pool. Eur. Surg. Res 34, 92–100 (2002).
Doyle, N. A. et al. Neutrophil margination, sequestration, and emigration in the lungs of L-selectin-deficient mice. J. Clin. Invest. 99, 526–533 (1997).
Zhou, Z. et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe 27, 883–890.e2 (2020).
Zeng, F. et al. Can we predict the severity of coronavirus disease 2019 with a routine blood test? Pol. Arch. Intern. Med. 130, 400–406 (2020).
Wang, D. et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 323, 1061–1069 (2020).
Brenner, J. S. Nanomedicine for the treatment of acute respiratory distress syndrome. The 2016 ATS Bear Cage Award-winning proposal. Ann. Am. Thorac. Soc. 14, 561–564 (2017).
Brenner, J. S., Greineder, C., Shuvaev, V. & Muzykantov, V. Endothelial nanomedicine for the treatment of pulmonary disease. Expert Opin. Drug Deliv. 12, 239–261 (2015).
Gao Smith, F. et al. Effect of intravenous β-2 agonist treatment on clinical outcomes in acute respiratory distress syndrome (BALTI-2): a multicentre, randomised controlled trial. Lancet 379, 229–235 (2012).
National, Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network, et al. Randomized, placebo-controlled clinical trial of an aerosolized β2-agonist for treatment of acute lung injury. Am. J. Respir. Crit. Care Med. 184, 561–568 2011).
Lee, P. Y., Wang, J.-X., Parisini, E., Dascher, C. C. & Nigrovic, P. A. Ly6 family proteins in neutrophil biology. J. Leukoc. Biol. 94, 585–594 (2013).
Daley, J. M., Thomay, A. A., Connolly, M. D., Reichner, J. S. & Albina, J. E. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J. Leukoc. Biol. 83, 64–70 (2008).
Vij, N., Min, T., Bodas, M., Gorde, A. & Roy, I. Neutrophil targeted nano-drug delivery system for chronic obstructive lung diseases. Nanomedicine 12, 2415–2427 (2016).
Bartneck, M. & Wang, J. Therapeutic targeting of neutrophil granulocytes in inflammatory liver disease. Front. Immunol. 10, 2257 (2019).
Wang, Z., Li, J., Cho, J. & Malik, A. B. Prevention of vascular inflammation by nanoparticle targeting of adherent neutrophils. Nat. Nanotechnol. 9, 204–210 (2014).
Chu, D., Gao, J. & Wang, Z. Neutrophil-mediated delivery of therapeutic nanoparticles across blood vessel barrier for treatment of inflammation and infection. ACS Nano 9, 11800–11811 (2015).
Myerson, J. W. et al. Non-affinity factors modulating vascular targeting of nano- and microcarriers. Adv. Drug Deliv. Rev. 99, 97–112 (2016).
Mitragotri, S. & Lahann, J. Physical approaches to biomaterial design. Nat. Mater. 8, 15–23 (2009).
Anselmo, A. C. & Mitragotri, S. Impact of particle elasticity on particle-based drug delivery systems. Adv. Drug Deliv. Rev. 108, 51–67 (2017).
Ernsting, M. J., Murakami, M., Roy, A. & Li, S.-D. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J. Control. Release 172, 782–794 (2013).
Anselmo, A. C. & Mitragotri, S. Cell-mediated delivery of nanoparticles: taking advantage of circulatory cells to target nanoparticles. J. Control. Release 190, 531–541 (2014).
Ferrer, M. C. C. et al. Icam-1 targeted nanogels loaded with dexamethasone alleviate pulmonary inflammation. PLoS ONE 9, e102329 (2014).
Myerson, J. W. et al. Flexible nanoparticles reach sterically obscured endothelial targets inaccessible to rigid nanoparticles. Adv. Mater. 30, e1802373 (2018).
Myerson, J. W. et al. Cross-linker-modulated nanogel flexibility correlates with tunable targeting to a sterically impeded endothelial marker. ACS Nano 13, 11409–11421 (2019).
Rahmani, S. et al. Long-circulating Janus nanoparticles made by electrohydrodynamic co-jetting for systemic drug delivery applications. J. Drug Target 23, 750–758 (2015).
Brenner, J. S. et al. Mechanisms that determine nanocarrier targeting to healthy versus inflamed lung regions. Nanomedicine 13, 1495–1506 (2017).
Lu, M. & Munford, R. S. The transport and inactivation kinetics of bacterial lipopolysaccharide influence its immunological potency in vivo. J. Immunol. 187, 3314–3320 (2011).
Gregory, N. S. et al. An overview of animal models of pain: disease models and outcome measures. J. Pain 14, 1255–1269 (2013).
Ghasemlou, N., Chiu, I. M., Julien, J.-P. & Woolf, C. J. CD11b+Ly6G− myeloid cells mediate mechanical inflammatory pain hypersensitivity. Proc. Natl Acad. Sci. USA 112, E6808–E6817 (2015).
Li, J., Yu, S., Yao, P. & Jiang, M. Lysozyme–dextran core–shell nanogels prepared via a green process. Langmuir 24, 3486–3492 (2008).
Bertsch, M. & Kassner, R. J. Selective staining of proteins with hydrophobic surface sites on a native electrophoretic gel. J. Proteome Res. 2, 469–475 (2003).
Roh, K., Martin, D. C. & Lahann, J. Biphasic Janus particles with nanoscale anisotropy. Nat. Mater. 4, 759–763 (2005).
Gregory, J. V. et al. Systemic brain tumor delivery of synthetic protein nanoparticles for glioblastoma therapy. Nat. Commun. 11, 5687 (2020).
Hwang, S. et al. Anisotropic hybrid particles based on electrohydrodynamic co-jetting of nanoparticle suspensions. Phys. Chem. Chem. Phys. 12, 11894–11899 (2010).
Lee, Y. et al. In vivo editing of macrophages through systemic delivery of CRISPR-Cas9-ribonucleoprotein-nanoparticle nanoassemblies. Adv. Ther. 2, 1900041 (2019).
Mout, R. et al. Direct cytosolic delivery of CRISPR/Cas9-ribonucleoprotein for efficient gene editing. ACS Nano 11, 2452–2458 (2017).
Lee, Y.-W. et al. Direct cytosolic delivery of proteins through coengineering of proteins and polymeric delivery vehicles. J. Am. Chem. Soc. 142, 4349–4355 (2020).
Russo, C. J. & Passmore, L. A. Electron microscopy: ultrastable gold substrates for electron cryomicroscopy. Science 346, 1377–1380 (2014).
Mietzsch, M. et al. Comparative analysis of the capsid structures of aavrh.10, aavrh.39, and AAV8. J. Virol. 94, e01769-19 (2020).
Yu, X. et al. Cryo-EM structure of human adenovirus D26 reveals the conservation of structural organization among human adenoviruses. Sci. Adv. 3, e1602670 (2017).
Drouin, L. M. & Agbandje-McKenna, M. Adeno-associated virus structural biology as a tool in vector development. Future Virol. 8, 1183–1199 (2013).
Nemerow, G. R., Stewart, P. L. & Reddy, V. S. Structure of human adenovirus. Curr. Opin. Virol. 2, 115–121 (2012).
Pontillo, N., Pane, F., Messori, L., Amoresano, A. & Merlino, A. Cisplatin encapsulation within a ferritin nanocage: a high-resolution crystallographic study. Chem. Commun. 52, 4136–4139 (2016).
Hood, E. D. et al. Vascular targeting of radiolabeled liposomes with bio-orthogonally conjugated ligands: single chain fragments provide higher specificity than antibodies. Bioconjug. Chem. 29, 3626–3637 (2018).
Dunkelberger, J. R. & Song, W.-C. Complement and its role in innate and adaptive immune responses. Cell Res. 20, 34–50 (2010).
Soltis, R. D., Hasz, D., Morris, M. J. & Wilson, I. D. The effect of heat inactivation of serum on aggregation of immunoglobulins. Immunology 36, 37–45 (1979).
Haihua, C., Wei, W., Kun, H., Yuanli, L. & Fei, L. Cobra venom factor-induced complement depletion protects against lung ischemia reperfusion injury through alleviating blood-air barrier damage. Sci. Rep. 8, 10346 (2018).
Saguil, A. & Fargo, M. Acute respiratory distress syndrome: diagnosis and management. Am. Fam. Physician 85, 352–358 (2012).
Fang, X. et al. Novel role for CFTR in fluid absorption from the distal airspaces of the lung. J. Gen. Physiol. 119, 199–207 (2002).
Tregay, N. et al. Use of autologous 99mtechnetium-labelled neutrophils to quantify lung neutrophil clearance in COPD. Thorax 74, 659–666 (2019).
Brenner, J. S. et al. Red blood cell-hitchhiking boosts delivery of nanocarriers to chosen organs by orders of magnitude. Nat. Commun. 9, 2684 (2018).
Matute-Bello, G. et al. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am. J. Respir. Cell Mol. Biol. 44, 725–738 (2011).
De Filippo, K. et al. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 121, 4930–4937 (2013).
Meduri, G. U. et al. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest 107, 1062–1073 (1995).
Duncan, R. J., Weston, P. D. & Wrigglesworth, R. A new reagent which may be used to introduce sulfhydryl groups into proteins, and its use in the preparation of conjugates for immunoassay. Anal. Biochem. 132, 68–73 (1983).
Szebeni, J., Muggia, F., Gabizon, A. & Barenholz, Y. Activation of complement by therapeutic liposomes and other lipid excipient-based therapeutic products: prediction and prevention. Adv. Drug Deliv. Rev. 63, 1020–1030 (2011).
Qin, C.-C., Liu, Y.-N., Hu, Y., Yang, Y. & Chen, Z. Macrophage inflammatory protein-2 as mediator of inflammation in acute liver injury. World J. Gastroenterol. 23, 3043–3052 (2017).
Hood, E. D. et al. Antioxidant protection by PECAM-targeted delivery of a novel NADPH-oxidase inhibitor to the endothelium in vitro and in vivo. J. Control. Release 163, 161–169 (2012).
Nesvizhskii, A. I., Keller, A., Kolker, E. & Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 (2003).
Bielow, C., Mastrobuoni, G. & Kempa, S. Proteomics quality control: quality control software for maxquant results. J. Proteome Res. 15, 777–787 (2016).
Parhiz, H. et al. PECAM-1 directed re-targeting of exogenous mRNA providing two orders of magnitude enhancement of vascular delivery and expression in lungs independent of apolipoprotein E-mediated uptake. J. Control. Release 291, 106–115 (2018).
Böhm, G., Muhr, R. & Jaenicke, R. Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 5, 191–195 (1992).
Acknowledgements
This research was supported by NIH R01HL125462 (V.R.M.). We acknowledge funding from the Defense Threat Reduction Agency (HDTRA-1-15-0045, J.L., V.R.M.). V.M.R. acknowledges NIH EB022641. J.W.M. was supported by NIH T32HL07915 while conducting this research. O.A.M.-C. received funding from the American Heart Association (19CDA345900001). L.L.M. was supported in part by NIH UL1TR001878. CD spectroscopy data were obtained at the University of Pennsylvania Biological Chemistry Resource Center. SPECT-CT images were obtained by Eric Blankenmyer in collaboration with the University of Pennsylvania Perelman School of Medicine Small Animal Imaging Facility. Single-cell suspensions prepared from human lungs were generously provided by Edward E. Morrisey’s group at the University of Pennsylvania.
Author information
Authors and Affiliations
Contributions
J.W.M, P.N.P, N.H., L.R.W., D.C.L, Y.-W.L., O.A.M.-C., E.D.H., T.S., J.V.G. and J.N. designed and prepared nanoparticles used in the study. J.W.M., P.M.G. and R.Y.K. prepared E. coli for tracing in mice. J.W.M., P.N.P., L.R.W., O.A.M.-C., L.L.M., P.M.G., I.J., J.N. and C.F.G. performed studies tracing nanoparticles in mice. J.W.M., P.N.P., M.H.Z., M.E.Z., J.W. and H.-Y.Z. performed studies of therapeutic efficacy. J.W.M., P.N.P. and L.T.F. performed studies tracing nanoparticles in human lungs. J.W.M., L.T.F. and K.M.R. performed studies tracing nanoparticle uptake in isolated cells. J.W.M., P.N.P., V.R.M. and J.S.B. analysed all data. The manuscript was written through contributions from all authors. All authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
Findings in this study contributed to US provisional patent application number 62/943469, with J.W.M., P.N.P., L.R.W., V.R.M. and J.S.B. as inventors. The remaining authors declare no competing interests.
Additional information
Peer review information Nature Nanotechnology thanks Edwin Chilvers, Bengt Fadeel, Gaurav Sahay and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–45, Discussion, Methods, Tables 1–11, description of Videos 1–7, data analysis code and tabulated analyses of statistical power.
Supplementary Video 1
Intravital imaging of lysozyme-dextran nanogel uptake in IV-LPS-injured mouse lungs.
Supplementary Video 2
Three-dimensional reconstruction of chest CT images of naive mouse lungs.
Supplementary Video 3
Three-dimensional reconstruction of chest CT images of oedematous mouse lungs.
Supplementary Video 4
Three-dimensional reconstruction of SPECT-CT imaging of lysozyme-dextran nanogel uptake in IV-LPS-injured mouse lungs.
Supplementary Video 5
Three-dimensional reconstruction of SPECT-CT imaging of lysozyme-dextran nanogel uptake in naive mouse lungs.
Supplementary Video 6
Three-dimensional reconstruction of SPECT-CT imaging of lysozyme-dextran nanogel biodistribution in an IV-LPS-injured mouse.
Supplementary Video 7
Three-dimensional reconstruction of SPECT-CT imaging of lysozyme-dextran nanogel biodistribution in a naive mouse.
Rights and permissions
About this article

Cite this article
Myerson, J.W., Patel, P.N., Rubey, K.M. et al. Supramolecular arrangement of protein in nanoparticle structures predicts nanoparticle tropism for neutrophils in acute lung inflammation. Nat. Nanotechnol. 17, 86–97 (2022). https://doi.org/10.1038/s41565-021-00997-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41565-021-00997-y
This article is cited by
-
Cholesterol-modified sphingomyelin chimeric lipid bilayer for improved therapeutic delivery
Nature Communications (2024)
-
Cell-mediated nanoparticle delivery systems: towards precision nanomedicine
Drug Delivery and Translational Research (2024)
-
Engineering nanomaterial physical characteristics for cancer immunotherapy
Nature Reviews Bioengineering (2023)
-
Targeted modulation of immune cells and tissues using engineered biomaterials
Nature Reviews Bioengineering (2023)
-
Bioimaging guided pharmaceutical evaluations of nanomedicines for clinical translations
Journal of Nanobiotechnology (2022)
