
Abstract
The increasing number of approved nucleic acid therapeutics demonstrates the potential to treat diseases by targeting their genetic blueprints in vivo. Conventional treatments generally induce therapeutic effects that are transient because they target proteins rather than underlying causes. In contrast, nucleic acid therapeutics can achieve long-lasting or even curative effects via gene inhibition, addition, replacement or editing. Their clinical translation, however, depends on delivery technologies that improve stability, facilitate internalization and increase target affinity. Here, we review four platform technologies that have enabled the clinical translation of nucleic acid therapeutics: antisense oligonucleotides, ligand-modified small interfering RNA conjugates, lipid nanoparticles and adeno-associated virus vectors. For each platform, we discuss the current state-of-the-art clinical approaches, explain the rationale behind its development, highlight technological aspects that facilitated clinical translation and provide an example of a clinically relevant genetic drug. In addition, we discuss how these technologies enable the development of cutting-edge genetic drugs, such as tissue-specific nucleic acid bioconjugates, messenger RNA and gene-editing therapeutics.
Similar content being viewed by others
Main
Nearly half a century ago, Friedmann and Roblin conceptualized that inherited genetic disorders resulting in dysfunctional gene products could be treated by introducing a functional gene copy1. Today, technologies enabling nucleic acid delivery are at the forefront of global efforts to fight the COVID-19 pandemic2,3,4.
Targeting the genetic bases of many other diseases is rapidly becoming a reality, as demonstrated by the recent approval of various nucleic-acid-based therapeutics by the United States Food and Drug Administration (FDA) and the European Medicines Agency (EMA). In contrast to conventional drugs, which generally target proteins, genetic drugs modulate gene expression to induce therapeutic effects. Introducing exogenous nucleic acids into cells to counteract defective genes is an attractive way to achieve highly specific, durable and possibly curative therapeutic effects in inherited and acquired disorders. However, employing nucleic acids as therapeutics is challenging because they are susceptible to degradation by nucleases, contribute to immune activation and have unfavourable physicochemical characteristics that prevent facile transmission into cells. Safe and effective nucleic acid therapeutics therefore require sophisticated delivery platform technologies. In this Review we use the term ‘nucleic acid therapeutics’ to include all nucleic-acid-based approaches that modulate gene expression by inhibiting, adding, replacing, or editing at the DNA or RNA level.
Groundbreaking research over the past 30 years has resulted in safe, effective delivery platforms enabling nucleic acid therapeutics. Several ex vivo and in vivo genetic drugs have recently been approved (or are in late-stage development) for treating infections, cancer, muscular and retinal dystrophies, and other inherited disorders. Typical ex vivo gene therapies involve retrovirally transduced patient-derived hematopoietic stem cells or T cells, which are transplanted back into patients. Currently approved therapies include Strimvelis—patient-derived CD34+ cells transduced with a γ-retroviral vector to express DNA encoding the human adenosine deaminase (ADA) enzyme, treating severe combined immunodeficiency due to ADA deficiency (ADA-SCID)5—and the chimeric antigen receptor T cell products Kymriah (tisagenlecleucel), Yescarta (axicabtagene ciloleucel) and Tecartus (brexucabtagene autoleucel), for treating various types of leukaemia or lymphoma6. These latter approaches involve engineering autologous T cells using lentiviral or γ-retroviral vectors to express chimeric antigen receptors specific for the CD19 protein common on B cells6.
In parallel, several in vivo nucleic acid therapeutics have been approved (Table 1). The effectiveness of these treatments critically depends on chemical modifications and/or technologies designed to protect the nucleic acids from degradation and to ensure their stability in the circulation, to enable localization to the target tissue, and to ensure effective intracellular delivery. Here, we provide an overview of viral and non-viral technologies that have facilitated clinical translation of in vivo nucleic acid therapeutics. The vast majority of nucleic acid therapeutics that are either already approved or currently in late-stage clinical evaluation rely on four platform technologies: chemically modified antisense oligonucleotides (ASOs), N-acetylgalactosamine (GalNAc) ligand-modified short interfering RNA (siRNA) conjugates, lipid nanoparticles (LNPs), and adeno-associated virus (AAV) vectors (Fig. 1). For each platform technology, we explain the mode of action (Fig. 2), provide the rationale behind its development, highlight key technological aspects that have facilitated its clinical translation, and discuss its therapeutic effectiveness and adverse reactions using a clinically relevant drug product as an example. In addition, we address how these platform technologies are now enabling the next generation of nucleic acid therapeutics, such as targeted nucleic acid conjugates, messenger RNA (mRNA) and gene-editing therapeutics.

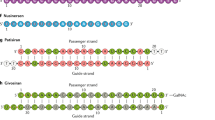
Four platform technologies that have helped herald a new age of medicine in which diseases can be treated at a genetic level. For ASOs (top left), backbone, sugar or nucleobase modifications of approved ASO therapeutics enhance affinity to target RNA, improve nuclease resistance, alter circulation characteristics and modulate immunological properties. Advances in chemical modifications have been reviewed elsewhere132,133. Schematic representation of an LNP (top right) containing siRNA or mRNA including key lipid components. A trivalent ligand with terminal GalNAc moieties (bottom left) is covalently linked to siRNA at the 3′-end of the sense strand. Multivalency and optimal spatial arrangement of the (GalNAc)3 ligand enable hepatocyte-specific targeting of siRNA via the asialoglycoprotein receptor. Notably, advanced chemical modifications, including 2′-OMe, 2′-fluoro, and phosphorothiorate linkages, are also utilized for approved GalNAc-siRNA therapeutics. Schematic representation (bottom right) of an AAV vector containing a 4.7-kb ssDNA with inverted terminal repeats (ITR). LNP schematic adapted with permission from ref. 76, American Chemical Society.

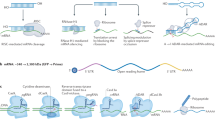
Subcutaneous, intravitreal and intrathecal administration of ASO modulates pre-mRNA splicing or induces RNAse H1- or interfering RNA (RNAi)-dependent transcript degradation. The cellular uptake mechanisms of ASOs and their endocytotic pathways have been reviewed elsewhere133. GalNAc−siRNA conjugates are delivered to hepatocytes via the asialoglycoprotein receptor (ASGPR) expressed on the sinusoidal surface. LNPs containing siRNA mediate hepatocyte gene silencing following internalization via the low-density lipoprotein receptor (LDLR) when administered intravenously. Loading siRNA into the RNA-induced silencing complex (RISC) results in target mRNA cleavage. LNP-mRNA vaccines administered intramuscularly elicit potent immune responses following expression of virus antigen. AAV vectors for treating retinal disorders are administered locally into the eye via subretinal injection for therapeutic transgene delivery. Other AAV vector drug products in development are administered intravenously. The capsid protein sequences dictate AAV tropism and uptake mechanism.
ASOs
ASOs are short synthetic nucleic acids that hybridize with cellular RNA using classic Watson−Crick base pairing to modulate gene expression. By binding pre-mRNA or mRNA, ASOs can post-transcriptionally regulate protein synthesis through mechanisms including modification of pre-mRNA processing and splicing, competitive inhibition, steric blockade of translational machinery, and degradation of bound target RNA. The latter mechanism leverages endogenous RNA degradation pathways mediated by RNase H1 (recruited by DNA:RNA duplexes) or Argonaute 2 (recruited by RNA:RNA duplexes through the RNA interference pathway)7. The ability to interact with pre-mRNA enables ASOs to target splicing processes and dramatically increases the amount of RNA sequence that can be selected for ASO binding, which can also limit off-target effects. For example, in the HTT gene, which encodes for the huntingtin protein, only 7% of 2,842 known single nucleotide polymorphisms could be targeted in the mature mRNA (using siRNAs) compared to 100% targeting of these single nucleotide polymorphisms by ASOs8.
Potential off-target binding toxicities are an important consideration for any nucleic-acid-based therapeutic strategy. Molecular size and precise sequence design give ASOs particularly robust therapeutic potential compared to other nucleic acid drugs. For example, artificial microRNA (miRNA)-enabled gene regulation relies on sequence complementarity between the target mRNA 3′ untranslated region and the first seven to eight 5′-nucleotides of the miRNA (the seed sequence)9. mRNA−miRNA binding outside the miRNA seed is variable, such that a single miRNA may interact with multiple mRNAs with different affinities10,11. ASO−RNA binding is rigorously regulated by complementarity between target RNA and the full ASO molecule, which is typically between 13 and 30 nucleotides in length. This stringent binding specificity directly correlates with ASO efficacy but can exacerbate toxicity if off-target binding does occur12,13,14.
The modest size of ASOs also means distribution to target tissues is achievable through multiple administration routes (Fig. 2). In vivo, unmodified phosphodiester ASOs are rapidly degraded by serum nucleases and cleared from circulation by renal filtration15. As a result, chemical modifications of ASO nucleosides, nucleobases, and the internucleoside backbone are critical for improving pharmacokinetics and pharmacodynamics while maintaining target affinity and efficacy (Fig. 1). Therapeutically effective ASOs are heavily modified, so they do not require a delivery carrier. This limited downstream processing during manufacturing decreases production-associated costs.
Platform technology design
ASOs were first indicated for translationally repressing Rous sarcoma virus (RSV) RNA to treat cytomegalovirus retinitis. Vitravene (fomivirsen), a deoxyribonucleotide-based ASO, binds terminal repetitive regions of RSV genomic RNA. Through competitive inhibition, Vitravene directly interferes with viral genome circularization and recruits RNase H1 to block viral protein translation13,14. Target gene expression can also be indirectly modified using ASOs complementary to endogenous miRNA or using miRNA mimetic ASOs. Although less specific, this approach enables simultaneous targeting of multiple genes regulated by shared miRNAs, usually to increase their expression11,16,17. ASOs can also upregulate gene expression through RNA cleavage-independent mechanisms, such as steric blockade. By selectively targeting upstream open reading frames and binding motifs for translational repressors within noncoding RNA, ASO binding can redirect translation to downstream open reading frames and increase mRNA translation by blocking the accessibility of negative regulators18,19. Similarly, ASOs can be used to alter pre-mRNA processing, by modifying 5′ capping and polyadenylation, and to influence mRNA splicing. Exondys 51 (eteplirsen) and Spinraza (nusinersen), both approved in 2016, respectively induce exon skipping and intron inclusion for the treatment of Duchenne muscular dystrophy and spinal muscular atrophy (SMA) (Table 1)20,21.
Early studies of phosphodiester ASOs achieved RNA target binding in vitro but were impeded by nuclease degradation and clearance in vivo18. Phosphorothiorate linkages within the ASO backbone (that is, first-generation ASO modifications) were introduced to increase nuclease resistance, decrease hydrophilicity and promote serum protein binding to improve circulation lifetime while maintaining RNase H1 activity and anionic charge22. First-generation ASOs, however, are immunostimulatory and have lower target binding affinities than their unmodified counterparts. Comparatively, phosphorodiamidate-modified morpholino oligomers have superior target RNA affinity and stability but are uncharged (which decreases serum protein binding and circulation lifetime) and rely exclusively on steric blockade-enabled regulatory mechanisms because they do not activate RNAse H123.
Extensive progress has been made in ASO nucleoside chemistry (Fig. 1). 2′ ribose or second-generation modifications include 2′-O-methyl (2′-OMe), 2′-O-methoxyethyl (2′-MOE), 2′-O-aminopropyl (2′-O-AP), and 2′-fluoro. These 2′ substituents influence ASO molecular conformation, resulting in improved RNA target binding affinity and, with the exception of 2′-fluoro, increased nuclease resistance24,25. Like phosphorodiamidate-modified morpholino oligomers, fully 2′-modified ASOs do not recruit RNase H1. Although less common, nucleobase modifications can also be incorporated into ASO design. Numerous modifications have been identified, but only replacing cytosine with 5-methylcytosine has proved beneficial: 5-methylcytosine substitution reduces ASO immunostimulatory effects without compromising Watson−Crick complementarity26. Third-generation modifications more extensively alter ASO chemistry to further enhance stability and potency post-administration and provide greater control over both target affinity and cellular tropism. These modifications may alter nucleoside connectivity and restrict ASO stereochemistry, as in locked nucleic acids, constrained ethyl nucleoside analogues and artificial amido-bridged nucleic acids27,28,29; change the backbone charge (phosphorodiamidate-modified morpholino oligomers are third-generation modified ASOs); or link ASOs to ligands, as in cholesterol- and GalNAc-conjugated ASOs30.
Single ASOs frequently incorporate multiple modifications to combine their advantageous properties and mitigate associated complications (Fig. 1). For example, adding second-generation modifications to a fully phosphorothiorate-modified ASO backbone increases binding affinity and maintains phosphorothiorate-conferred nuclease resistance24. Similarly, chimeric gapmer ASOs contain a central region of unmodified deoxynucleotides flanked by terminal 2′-modified nucleotides. The deoxynucleotide region preserves RNAse H1 activity, while peripheral 2′-modifications improve potency31. Tegsedi (inotersen), approved for treating polyneuropathy caused by hereditary transthyretin amyloidosis, is a gapmer ASO comprised of a 10-base deoxynucleotide core flanked by five 5′ and 3′ 2′-MOE-modified wings32.
Clinical example
Nusinersen, marketed as Spinraza, is a fully phosphorothioate- and 2′-MOE-modified ribonucleotide ASO approved for treating SMA33. SMA is an autosomal recessive disorder caused by loss-of-function mutations in the survival motor neuron 1 gene (SMN1). SMN1 mutations result in survival motor neuron protein (SMN) deficiency and cause motor neuron degeneration34. The DNA sequences of SMN1 and its paralogue SMN2 both encode SMN, but a base substitution within an intragenic SMN2 enhancer means that the major mRNA products of the two genes are different splice isoforms. SMN1 mRNA produces full-length SMN, while SMN2 mRNA lacks exon 7 and produces truncated, non-functional SMN35. A small proportion of SMN2 transcripts retain exon 7, and homozygous SMN1 mutation carriers with larger SMN2 copy numbers have milder SMA phenotypes36,37. The mechanism of Spinraza leverages this effect for SMA therapy. The ASO binds SMN2 pre-mRNA, modifies splicing to promote exon 7 inclusion and increases the proportion of full-length SMN translated from SMN2 mRNA38. Because Spinraza does not target the causative gene for SMA, ASO binding does not depend on SMN1 mutation type, meaning that Spinraza is a viable treatment option for all SMA patients.
The first phase 1 ascending-dose clinical trial of Spinraza, in which one intrathecal dose of 1 mg, 3 mg, 6 mg or 9 mg of the drug was administered, reported only mild and moderate adverse events in 89% of subjects. At nine to fourteen months post-treatment, cerebrospinal fluid SMN concentrations were unchanged in the 1 mg and 3 mg dose groups, and the 6 mg and 9 mg dose groups had respective mean increases in cerebrospinal fluid SMN of 118% and 161%. Compared to baseline, subjects in the 9 mg dose group showed a 17.6% improvement in the Hammersmith Functional Motor Scale Extended (HFMSE) score (used to assess motor function) 85 days post-treatment and a 32.8% improvement nine to fourteen months post-treatment. The 1 mg, 3 mg and 6 mg dose groups did not have better HFMSE results38.
Two phase 3 clinical trials assessed the efficacy of multiple Spinraza doses in infant and later-onset SMA patients. Spinraza-treated infants showed reduced risk of death or permanent ventilation (47%) and better motor-milestone response (51%) compared to untreated infants, and later-onset SMA patients had clinically meaningful improvements in HFMSE scores. In both trials, adverse event frequency was similar between treated and sham-injected groups. Adverse events included vomiting, pyrexia, constipation and lower respiratory infection21,39. Low platelet counts and elevated urine protein were measured in 16% and 56% of treated infants and later-onset patients, compared to 14% and 34% in untreated patients. Spinraza is approved for SMA treatment in both paediatric and adult patients33 and is administered by intrathecal injection. Patients receive four initial loading doses (at days 1, 15, 31 and 61) of 12 mg each and subsequent maintenance doses every four months33. Delivering Spinraza directly to the cerebrospinal fluid ensures the drug reaches its target within the central nervous system, as ASOs in peripheral circulation do not permeate the blood−brain barrier40.
GalNAc−siRNA conjugates
GalNAc conjugation represents an efficient way of increasing siRNA target organ accumulation and of facilitating their cellular uptake. In the absence of a protective delivery vector, siRNA has to be chemically modified to ensure stability in the circulation following parenteral administration41. To silence disease-causing genes in hepatocytes, these therapeutics are composed of siRNA conjugated to a triantennary GalNAc moiety targeting the asialoglycoprotein receptor (ASGPR) (Fig. 1)42. This receptor is predominantly expressed on hepatocytes and thus provides access to a defined cell type within the liver. ASGPR specifically binds carbohydrates with terminal galactose or GalNAc residues43. After ligand binding, the receptor−ligand complex is internalized by clathrin-dependent receptor-mediated endocytosis. Abundant (around 500,000 ASGPR per cell) and predominantly expressed (>95% of total expression) on the hepatocyte sinusoidal membrane, ASGPR is an ideal receptor for hepatic siRNA delivery. Further, its high internalization and recycling rate (within minutes) allow continuous uptake of siRNA molecules, thereby increasing target cell concentration. Different species exhibit the same carbohydrate recognition pattern, which is an important consideration in preclinical and translational study design.
Platform design
Selective and efficient ASGPR targeting ligands, optimized siRNA design and a favourable administration route have been key factors for clinically translating GalNAc−siRNA. ASGPR is a hetero-oligomeric receptor complex comprising numerous subunits with carbohydrate recognition domains44. Increasing the number of carbohydrates that simultaneously bind to several receptor subunits exponentially elevates avidity (the cluster glycoside effect)45. The carbohydrates’ spatial arrangement and the sugar moieties are crucial for specific and efficient ASGPR binding46. Of note, ASGPR has a higher (up to 100-fold) affinity for GalNAc as compared to galactose. To further improve affinity, triantennary GalNAc ligands were developed with dissociation constants in the low nanomolar range as compared to the millimolar range for monovalent ligands47. Additionally, triantennary GalNAc ligand compatibility with siRNA synthesis streamlined large-scale manufacturing processes41.
Advances in backbone chemistry initially designed for ASO therapeutics have also been applied to siRNA therapeutics (Fig. 1)41. Notable modifications used for siRNAs include 2′-OMe, 2′-fluoro and phosphorothiorate linkages to improve metabolic stability, reduce recognition by Toll-like receptor 3 and 7, and increase binding to target mRNA. Iterative siRNA design optimization resulted in Alnylam’s proprietary Enhanced Stabilization Chemistry (ESC)48,49. The augmented stability of ESC−GalNAc−siRNA conjugates substantially increased liver exposure and prolonged gene silencing duration50, resulting in efficacy ten times greater than standard template chemistry. Stability in acidic intracellular compartments of hepatocytes is therefore key to extended activity (long-term depot)50. Careful siRNA sequence selection, especially of seed regions, reduces the risk of hybridization-dependent off-target effects. Additional siRNA modifications, including glycol nucleic acid substitution in the seed region (ESC+) or altriol nucleic acid residues, might be implemented in future conjugates to further boost RNAi potency and curtail adverse effects51,52 Chirality-dependent properties could also offer additional functionalities53. Balancing further chemical modifications and compatibility with RNAi machinery will be vital to the next generation of siRNA therapeutics.
Finally, the administration route has affected the clinical translation of GalNAc−siRNA conjugates and influenced patient compliance. Owing to their low molecular weight, GalNAc−siRNA conjugates are amenable to subcutaneous administration54 (Fig. 2). This delays siRNA delivery to the liver, but efficient and steady knockdown is still achieved owing to the high-capacity recycling of ASGPR. Several GalNAc−siRNA conjugates are currently undergoing late-stage clinical evaluation for treating cardiometabolic or genetic disorders (Table 2).
Clinical example
Givosiran (marketed as Givlaari) is an FDA-approved RNAi therapeutic based on GalNAc−siRNA technology indicated for acute hepatic porphyria, which is caused by mutations in genes associated with the heme biosynthesis pathway55. Physical triggers that increase aminolevulinic acid synthase (ALAS1) expression cause a build-up of aminolevulinic acid (ALA) and porphobilinogen, which are toxic intermediates that harm nervous tissue and contribute to the onset of acute, life-threatening episodes of severe abdominal pain, neuropathy (central and peripheral) and neuropsychiatric symptoms. At the time of givosiran’s development, the only treatment for acute hepatic porphyria was Hematin (an exogenous heme source), which was approved for post-symptomatic patients but not as a prophylactic treatment. Givlaari leverages the RNAi machinery to reduce and prevent elevated ALAS1 expression.
One limitation of this technology’s clinical translation was the lack of an appropriate diagnostic biomarker other than liver biopsies. Co-developing a biomarker based on ALAS1−mRNA concentrations in serum and urine both improved clinical diagnosis and provided a pharmacodynamic biomarker that could be used to monitor therapeutic endpoints without invasive procedures56. A striking similarity across species (that is, rodents and non-human primates) was the presence of circulating RNA, which strongly correlated to hepatic expression. Moreover, patient samples also showed similarly elevated circulating ALAS1−mRNA levels.
In a three-part phase 1 trial, 33 subjects received givosiran and 10 received placebo. The data suggested that a single subcutaneous dose at 2.5 mg kg–1 resulted in >80% less circulating ALAS1−mRNA, and correspondingly reduced ALA and porphobilinogen levels in circulation57. This established a dosing regimen of 2.5 mg kg–1 monthly. In the phase 3 trial, the primary outcome measure was the annualized rate of composite porphyria attacks (hospitalization, urgent care, or Hematin administration) over six months58. Among the 48 patients receiving givosiran, the rate of attacks dropped by 74%. The secondary outcome measures were urinary ALA and porphobilinogen levels, which were markedly lower in subjects receiving givosiran. Adverse events more frequently seen in treated individuals were elevated liver enzymes, chronic kidney disease, injection site reactions and nausea.
LNPs
The permeability requirements of biological membranes severely limit the transfer of large, charge-dense macromolecules (that is, nucleic acids) across the plasma membrane. To overcome this, lipid polymorphic phase behaviours could temporarily compromise the permeability barrier and allow nucleic acids to enter the cell. Landmark work on cationic lipids by Felgner et al. paved the way for a new class of lipid-mediated delivery systems59.
LNPs provide a protected compartment, sequestered from serum nuclease activity and immune components, and a drug-biodistribution profile dictated by the carrier. In this Review, we focus on the approved LNP technology developed for hepatic siRNA delivery following intravenous administration (Fig. 2). The discovery of ionizable cationic lipids has been essential for the clinical translation of LNP-based RNA therapeutics. These lipids ensure efficient siRNA encapsulation (>85%), maintain neutral LNP surface charge at physiological pH, and play a major part in endosomal escape60. After systemic administration, apolipoprotein E (ApoE) adsorbs to the LNP surface and promotes internalization by hepatocytes via the low-density lipoprotein receptor (Fig. 1)61, resulting in >80% accumulated dose in the liver62.
Platform design
LNP systems represent 25 years of evolution from initial formulations composed merely of phospholipids and cholesterol. These developments drew from knowledge of lipid carriers for small-molecule therapeutics. Translating such systems to nucleic acids required additional functionalities such as enabling efficient entrapment, maintaining neutral surface charge and evading the immune system63.
LNPs are typically composed of four components: ionizable cationic lipids, phospholipids, cholesterol and polyethylene glycol (PEG)-lipids (Fig. 1). LNP formulations used for gene silencing in hepatocytes entrap siRNA at a ratio of 0.095 (w/w, siRNA/lipid) and generate ~50 nm particles with narrow size distributions64; this size is crucial to allowing these particles to pass through the fenestrated liver vasculature65. We discuss the following factors that have been critical for clinical translation: ionizable cationic lipid optimization, diffusible PEG-lipid design and a robust, scalable manufacturing process.
The ionizable lipid shape and acid-dissociation constant optimization dramatically improved LNP potency, thereby enabling clinical translation. Permanently cationic lipids such as DOTMA demonstrated toxicities that hindered rapid translation in vivo66. Comparatively, ionizable tertiary amine moieties in the lipid headgroup allowed for a net-positive charge at acidic pH and a neutral charge in circulation; the decreased toxicity and lower immune stimulation suggested improved clinical utility67,68. Rational design and iterative screening using the murine factor VII (FVII) model identified DLin−MC3−DMA60; its development has been reviewed elsewhere69. Optimizing previous-generation lipids, such as DLin−DAP, improved the therapeutic index 8,000-fold.
Another key component in determining LNP−siRNA transfection competency is the PEG-lipid, which was required to prevent aggregation during particle formation70. However, PEG-lipids inhibited uptake into target cells and were therefore counter-productive for transfection71. To find an optimal balance between stability and transfection competency, diffusible PEG-lipids containing C14 alkyl chains were developed72,73. In the presence of a lipid sink, these lipids rapidly dissociate from the LNP, generating transfection-competent systems. Preclinical studies determined that LNP with diffusible PEG-lipids rapidly accumulate in the liver, with circulation half-lives of less than 15 minutes62,73. This rapid accumulation and potency for hepatocyte transfection stemmed from ApoE adsorption.
LNP−siRNA manufacturing required a rapid, reproducible procedure. This was accomplished through an ethanol-loading technique that involved mixing preformed LNP (at pH 4) with nucleic acids in the presence of high ethanol concentrations (~40% v/v)74. A subsequently developed higher-throughput rapid mixing process combined lipids dissolved in ethanol with nucleic acids in an aqueous buffer (pH 4) through a T-junction, resulting in efficient nucleic acid loading into LNP systems75. Rapid-mixing techniques produced LNP−siRNA systems with high entrapment efficiencies (>85%) and narrow size distributions. Recent work suggests that such structures include a hydrophobic oil core primarily consisting of neutral ionizable lipid surrounded by siRNA complexed to lipids in a bilayer arrangement76 with heterogeneity in the siRNA copy number per particle77.
Clinical example
Patisiran (marketed as Onpattro) is an RNAi therapeutic that relies on LNP delivery technology to treat hereditary transthyretin amyloidosis (hATTR). This disease is caused by mutations in the gene for transthyretin (TTR) that make the TTR protein abnormally aggregate and accumulate. As a result, the disease manifests in a number of systemic pathologies affecting many organ systems. Transthyretin is expressed by hepatocytes in the liver and secreted into circulation (note similarity to the FVII screening model).
Onpattro is composed of D-Lin−MC3−DMA, phosphatidylcholine, cholesterol and PEG-lipid at a ratio of 50/10/38.5/1.5 (mol%)78. As with the ASO Tegsedi, at the time Onpattro was developed, liver transplantation was the only treatment option for patients with hATTR. Onpattro leverages the high LNP−siRNA accumulation in hepatocytes (source of TTR) and the potency for gene silencing in these cells to suppress mutated TTR expression. In doing so, Onpattro drastically reduces the amount of nucleic acid required to achieve efficient gene silencing79.
During clinical development, a key finding was that the doses required to achieve 80% gene silencing in non-human primate models were very similar to those required in humans (0.3 mg kg–1)80,81. In a phase 2 study, a total of 29 hATTR patients received two patisiran infusions at a dose of 0.01−0.3 mg kg–1 every 4 weeks or 0.3 mg kg–1 every 3 weeks (Q3W). A mean TTR knockdown level >85% was achieved after the second dose for the Q3W protocol79. In the phase 3 APOLLO study, 148 patients received patisiran at a dose of 0.3 mg kg–1 Q3W and 77 patients received placebo infusions. The primary endpoint measured the change in the modified Neuropathy Impairment Score (mNIS+7) from baseline to after 18 months. Results indicated that the patisiran group showed a median reduction in serum TTR levels >80% and mNIS+7 of −6 compared to the placebo group measuring +28. This suggested patisiran can halt and reverse disease progression82. More recently, post hoc analyses of data from the phase 2 open-label extension and the phase 3 trial suggested that patients benefit from patisiran even when simultaneously receiving oral small-molecule TTR stabilizers (tafamidis or diflunisal)83.
AAV vector
Since gene therapy was initially proposed several decades ago, the development of safe and efficient mammalian retroviruses, adenoviruses and adeno-associated viruses has been instrumental to clinical translation. Although early studies provided clear evidence that viral vectors could successfully deliver therapeutic transgenes, clinical benefits were observed only in a small number of studies, and significant immunotoxicities occurred (in some occasions leading to patient death)84. In addition, serious genotoxicity caused by the use of viral vectors capable of genome integration has also been observed. Although these outcomes raised considerable doubts about whether these viral strategies would be viable, they also initiated major efforts that improved our understanding of vector biology and its interactions with target cells and tissues. This ultimately resulted in the development and clinical translation of recombinant viral vectors, which are more effective and incur lower risk85,86.
Viral vectors are effective DNA delivery vehicles, as they have evolved to display tropism for specific cell types and, compared to the aforementioned approaches, are the only system able to actively deliver their payload to the nucleus. The importance of this latter feature cannot be overstated, as DNA whole-gene-encoding payloads are non-functional outside the nucleus. Typically, non-viral systems rely on a cell division stage when nuclear membrane collapse and reformation allows DNA to passively diffuse into the nucleus.
Several drug products based on recombinant viral vector-mediated transgene delivery have been approved by the FDA and the EMA. These treatments involve either ex vivo or in vivo approaches, whereby transgenes are directly delivered to cells, mainly mediated by AAV vectors, following parenteral administration87. This Review discusses AAV vector-mediated in vivo therapies, which in addition to being used for retinal disorders are also in late-stage clinical evaluation for treating diseases related to the liver and the central nervous system (Tables 1 and 2)87.
Platform design
Wild-type AAV is a small, non-enveloped parvovirus (~25 nm) with a ~4.7-kb single-stranded DNA (ssDNA) genome. Recombinant AAV vectors exploited as delivery vehicles generally contain the same capsid components and structure as wild-type AAV but all viral coding sequences are replaced by therapeutic gene expression cassettes, maximizing packing capacity and reducing immunogenicity (Fig. 1)87. Because AAV tropism has broad range, AAVs can be used for targeted transduction in a variety of tissues, potentially providing cellular specificity. Variable regions within capsid protein sequences define AAV serotypes and dictate tropism by modifying the binding interactions between capsids and receptors that facilitate their cellular uptake88,89. Receptor-bound AAVs enter cells by endocytosis, are released into the cytosol by acidification of the early endosome, and, following perinuclear accumulation, enter the nucleus90,91 (Fig. 2). Tissue specificity and transduction efficiency can be further modified by viral pseudotyping, chimeric capsid engineering and peptide library screening, among others92,93.
Additionally, AAV vectors have a number of other features that make them an attractive platform for therapeutic transgene delivery. First, AAVs require a helper virus for replication and are therefore considered non-pathogenic. Second, following AAV uncoating in the nucleus, inverted terminal repeats flanking the vector genome drive recombination to form episomal constructs. Genomic integration occurs at low frequency, reducing the potential for genotoxicity. Third, the 12 different AAV serotypes and hundreds of additional variants identified to date mean AAV-mediated nucleic acid therapy has extensive utility. Nevertheless, AAVs also have several limitations. Most prominently, AAV vectors have a packaging capacity for genes of less than about 5.0 kb, thereby restricting their application to treating disorders for which the transgene can be packaged in a single vector. To overcome this, strategies are being developed to accommodate larger transgenes, or multiple transgenes (for multigenetic disorder treatment), by co-administrating multiple vectors that carry separate (halves of) genes94. Following release in the nucleus, the ssDNA needs to be converted to a double-stranded DNA prior to transgene expression. Self-complementary AAV vectors have been designed to prevent expression delay, but their packaging capacity is halved. AAVs are most suitable for DNA delivery to long-lived, non-dividing or slowly dividing cells such as hepatocytes because episomal genomes are not replicated and are lost during mitosis95. Further, AAVs occur naturally in multiple species; in humans, neutralizing antibodies are widely prevalent, which dramatically reduces AAV-mediated gene delivery following intravenous administration (especially critical for repeated injections) and limits their broad applicability96.
Clinical example
Voretigene neparvovec-rzyl (marketed as Luxturna) is an AAV serotype 2 (AAV2) vector containing cDNA encoding the human RPE65 gene for treating inherited retinal dystrophy LCA type 2. The recombinant vector genome contains a cytomegalovirus enhancer and a hybrid chicken β-actin promoter that facilitate RPE65 expression while the vector remains episomal in the nucleus97. Prior to Luxturna’s development, there was no pharmacological treatment for LCA type 2, which is an autosomal recessive disease caused by mutations in the RPE65 gene. Although rare, this disorder was an appealing target for gene therapy, in part because it can be treated by introducing a single copy of the wild-type gene. Additionally, because it is a relatively small, easily accessible organ, the eye requires a lower number of vector particles for a therapeutic dose, thereby reducing the risk of adverse effects. This simplified access also facilitates multiple administration routes (mostly intravitreal or subretinal, as is the case for Luxturna) (Fig. 2). Moreover, the eye is immune privileged by the blood−ocular barrier, which prevents vector particles from spreading to other parts of the body and promoting an immune response. In addition, because the AAV targets post-mitotic retinal pigment epithelium cells, the treatment is expected to be long-lasting without episomal DNA dilution.
Following studies in small animals, the development of Luxturna was advanced by canine studies, in which vision could be restored following treatment with AAV-RPE65 in naturally occurring RPE65−/− dogs98. Importantly, improved visual function and protection against photoreceptor degradation persisted even five to eleven years later99. Phase 1/2 trials were conducted by multiple groups which concomitantly demonstrated that AAV2−RPE65 could be safely delivered to the retina, resulting in improved visual function for varied lengths of time100,101,102. In a randomized phase 3 trial, 29 patients with confirmed RPE65 mutations received a bilateral, subretinal injection of 1.5 × 1011 vector genomes of Luxturna (n = 20) or underwent the same efficacy outcome testing without intervention (n = 9)97. The primary endpoint was a change in multi-luminance mobility testing (MLMT) one year after intervention. The change in MLMT performance was determined based on the light intensity level by which participants could successfully complete an obstacle course after the intervention compared to baseline97. This test and other secondary outcomes, such as visual tests, demonstrated that treatment with Luxturna improved light sensitivity, visual fields and navigational ability in RPE65-mediated LCA patients who were previously untreatable. In patients who were treated during a phase 1/2 trial, clinical benefits are still present—lasting at least three years100. These results led to Luxturna’s approval in 2017.
Outlook and discussion
Since using gene therapy to treat diseases was first proposed half a century ago, major biological and technological breakthroughs have led to several safe, effective platform technologies (Fig. 3). Importantly, currently approved nucleic acid therapeutics and those now in development are often valuable therapeutic options for patients who previously had limited or no treatment options. The successful clinical application of these technologies has sparked great interest in improving tissue specificity (for example, for ASO and siRNA conjugates) and broadening potential applications, particularly for LNP and AAV vector systems. This Review focuses on approved nucleic acid therapeutics, but the extremely rapid development of mRNA and gene-editing therapeutics, which are already in clinical trials or have even been approved for treating infectious diseases and cancer, is worth mentioning103. Although newly developed treatments also include a number of ex vivo drug products, such as modified dendritic cells, we briefly highlight several in vivo approaches and conclude by discussing the socioeconomic impact of gene therapy as a whole.

This timeline depicts key discoveries and events during the development of nucleic acid therapeutics, with a specific focus on the delivery platform technologies reviewed herein. Each timeline point has a colour-coded circle: major developments (black), ASO (green), GalNAc−siRNA (blue), lipid (purple) and viral (yellow). Box 1 provides a detailed historical overview of the same process. AHP, acute hepatic porphyria; CMV, cytomegalovirus; H/N, head and neck cancer; FHChol, familial hypercholesterolemia; LCA, Leber congenital amaurosis.
Various bioconjugation strategies have been developed to overcome some limitations of approved ASO and siRNA therapeutics, specifically by modulating their pharmacokinetics and facilitating receptor-specific interactions. Modified ASOs conjugated to GalNAc can increase target gene silencing up to 60-fold in hepatocytes by increasing accumulation in these cells relative to non-parenchymal cells104. Similarly, antibody-modified siRNAs have extended the relatively short half-life of GalNAc−siRNAs from about two hours to more than ten hours in serum105. Chemically engineering oligonucleotides with lipophilic moieties can alter accumulation in extrahepatic tissues, enabling gene modulation in a broad range of tissues including heart, lung and muscle106,107. Recent developments demonstrate that nucleic acid bioconjugates are an emerging class with clinical utility (Table 2). The major advancements through directly conjugating chemical moieties—for example, sugars, lipids, antibodies or aptamers—to ASOs or siRNAs have been reviewed elsewhere108,109.
Following successful development of the first RNAi drug Onpattro, LNP technology is now facilitating mRNA applications for protein replacement therapy and vaccine development. Harnessing efficient ApoE-mediated hepatocyte transfection, LNP formulations of mRNA can convert the liver into a ‘bioreactor’ for therapeutic protein production110. Simultaneously, LNP-mRNA-encoding antigens are being used for subcutaneous, intramuscular or intradermal immunizations. Compared to virus- or DNA-based vaccines, mRNA has several advantages: it is non-infectious, non-integrating and requires only cytoplasmic delivery103. This approach has shown potential in the development of a broadly protective influenza virus vaccine111, complete protection from Zika virus in mice and non-human primates112, and, most recently, a vaccine against P. falciparum113, a causative agent of malaria. Similarly, two LNP-mRNA vaccines are also leading the fight against SARS-CoV-23,114,115,116,117,118 with one formulation administered to the first subject only 63 days after the viral genome was published. The formulation named mRNA-1273 achieved a 94.1% efficacy in a phase 3 trial involving 30,420 volunteers (randomized as 1:1 ratio of vaccine-to-placebo)119. The second formulation, BNT162b2, conferred a 95% protection against viral infection in a phase 3 trial that included 43,448 participants120. As of February 2021, more than 76 million doses of these vaccines have been administered121.
Comparable strategies are also being used to generate personalized vaccines for cancer immunotherapy122. This approach involves sequencing a patient’s healthy and tumour tissues, then producing mRNA vaccines encoding individual cancer-specific neoepitopes122. These vaccines have promising therapeutic effects in preclinical cancer models123,124 and are currently in the early stages of clinical evaluation for breast cancer (NCT02316457) and melanoma (NCT02410733). The transient protein expression induced by LNP-mRNA vaccines is well suited to combating infections and cancer, but other applications, such as protein replacement therapies, require repeated or chronic administration. Beyond vaccines, however, LNP formulations are administered intravenously, which creates a substantial burden on healthcare systems and limits therapeutic application to rare or life-threatening disorders.
Platform technologies for delivering large nucleic acid payloads also enable clinical translation of gene editing-based treatments, mediated by nuclease-induced double-stranded breaks or single-base DNA mutations. These approaches hold great promise, because unlike conventional gene transfer techniques, gene editing can treat disorders caused by dominant negative mutations. Furthermore, gene editing allows precise engineering so that gene insertion or correction is controlled by an endogenous promotor and probably provides physiologically accurate gene expression85. In addition to ex vivo approaches, several in vivo gene editing therapies, enabled by LNP or AAV vector technology, to treat retinal dystrophies, hATTR and metabolic disorders (NCT03041324, NCT02702115, NCT04601051, NCT03872479) are undergoing clinical translation85,125.
Advanced genome editing tools, including base and prime editors, have been shown to further enhance target specificity in preclinical studies, as described elsewhere126. Whereas the limited cargo capacity of AAVs demands the use of a split protein delivered in two separate vectors, LNP technology allows editor mRNA and guide RNA to be formulated into a single particle. Both platform technologies have recently been used for in vivo genome base editing in mice to correct the metabolic liver disease phenylketonuria94,127 and ex vivo modification of human hematopoietic stem cells128.
Generally, the various approved platform technologies are well tolerated, but nanoparticles, macromolecules and biologics often require pre- or co-medications to suppress undesirable infusion- or immune-related reactions. For example, patients receiving Onpattro also take a combination of dexamethasone, acetaminophen and antihistamines at least sixty minutes prior to treatment79. For Glybera, patients receive oral cyclosporine and mycophenolate mofetil three days prior and methylprednisolone thirty minutes prior to infusion129. Since the majority of approved nucleic acid therapeutics treat rare or monogenic disorders that would otherwise have few or no available treatments, the clinical outcomes justify the risks associated with potential adverse reactions.
With nucleic acid therapeutics now demonstrating clinical benefits in patients, their socioeconomic impact related to treatment pricing (Table 1) has come under debate. Do clinical results justify high costs? This complex calculus depends on many factors, including the duration and magnitude of the therapeutic effect, the rarity of the disease, its associated severity or lifetime morbidity, the costs of alternative treatment options, healthcare and insurance models, and, ultimately, the overall improvement in patients’ quality of life.
Clearly, for nucleic acid therapeutics that induce long-lasting or curative effects—for example, Zolgensma, Strimvelis or Luxturna—high drug prices seem justified, especially when taking into account the healthcare resources required for ongoing treatment and the economic costs of losing productive life years. With regard to drugs such as Spinraza, Tegsedi and Onpattro that induce significant but transient therapeutic effects, current treatment pricing may not appropriately reflect long-term therapeutic benefits for patients130. Furthermore, since a number of the currently approved nucleic acid therapeutics were developed for ultra-rare diseases, demand for these treatments is very limited. This is exemplified by the first approved gene therapy, Glybera: although highly effective, it was withdrawn from the market owing to limited access caused by low demand and high treatment costs ($US1 million in 2012). Several genetic drug manufacturers have implemented outcome-based agreements, whereby payment depends on treatment success or duration. For example, short-term efficacy and long-term durability measures have been established to determine paying rebates for Luxturna, and similar arrangements have been set up for other nucleic acid therapeutics as well, for example, for Onpattro. Other controversial practices, like a global ‘lottery’ system, are also in place. For example, 100 doses of Zolgensma are donated each year to children under two who are diagnosed with SMA and live outside the USA131. Pricing has also been the subject of debate in light of vaccine development during the current pandemic. While the price of mRNA vaccines is higher (US$30−40) than conventional or viral vector-based vaccines (US$2−5), it is eclipsed by the direct and indirect costs resulting from the global COVID-19 outbreak. In addition, the unprecedented speed by which mRNA vaccines were developed (one year) will contribute to reopening economies.
To conclude, the four platform technologies described in this Review have helped to transform nucleic acid therapeutics from intriguing theory into clinical reality. Whereas most currently approved nucleic acid therapeutics aim to treat orphan diseases, their delivery technologies are now being leveraged for more broadly applicable genetic drugs and have enabled rapid vaccine development in times of a pandemic. In addition, these platforms are facilitating the clinical translation of novel approaches, such as gene-editing therapeutics. Although their widespread implementation still faces several challenges, including manufacturing, toxicity and socioeconomic issues, it is clear that nucleic acid therapeutics are poised to have a revolutionary impact on many diseases that previously had limited or no treatment options.
Change history
30 June 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41565-021-00937-w
References
Friedmann, T. & Roblin, R. Gene therapy for human genetic disease? Science 175, 949–955 (1972).
Anderson, E. J. et al. Safety and immunogenicity of SARS-CoV-2 mRNA-1273 vaccine in older adults. New Engl. J. Med. 383, 2427−2438 (2020).
Sahin, U. et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T-cell responses. Nature 586, 594−599 (2020).
Rossi, J. J. & Rossi, D. Oligonucleotides and the COVID-19 pandemic: a perspective. Nucleic Acid Ther. 30, 129–132 (2020).
Aiuti, A. et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 360, 447–458 (2009).
June, C. H. et al. CAR T cell immunotherapy for human cancer. Science 359, 1361–1365 (2018).
Hammond, S. M., Bernstein, E., Beach, D. & Hannon, G. J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 404, 293–296 (2000).
Southwell, A. L., Skotte, N. H., Bennett, C. F. & Hayden, M. R. Antisense oligonucleotide therapeutics for inherited neurodegenerative diseases. Trends Mol. Med. 18, 634–643 (2012).
Doench, J. G. & Sharp, P. A. Specificity of microRNA target selection in translational repression. Genes Dev. 18, 504–511 (2004).
Grimson, A. et al. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell 27, 91–105 (2007).
Jackson, A. L. et al. Widespread siRNA ‘off-target’ transcript silencing mediated by seed region sequence complementarity. RNA 12, 1179–1187 (2006).
Holt, J. T., Redner, R. L. & Nienhuis, A. W. An oligomer complementary to c-myc mRNA inhibits proliferation of HL-60 promyelocytic cells and induces differentiation. Mol. Cell. Biol. 8, 963–973 (1988).
Stephenson, M. L. & Zamecnik, P. C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 75, 285–288 (1978).
Zamecnik, P. C. & Stephenson, M. L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 75, 280–284 (1978).
Goodchild, J., Kim, B. & Zamecnik, P. C. The clearance and degradation of oligodeoxynucleotides following intravenous injection into rabbits. Antisense Res. Dev. 1, 153–160 (1991).
Davis, S., Lollo, B., Freier, S. & Esau, C. Improved targeting of miRNA with antisense oligonucleotides. Nucleic Acids Res. 34, 2294–2304 (2006).
Liang, X.-h, Vickers, T. A., Guo, S. & Crooke, S. T. Efficient and specific knockdown of small non-coding RNAs in mammalian cells and in mice. Nucleic Acids Res. 39, e13 (2010).
Liang, X.-h et al. Antisense oligonucleotides targeting translation inhibitory elements in 5′ UTRs can selectively increase protein levels. Nucleic Acids Res. 45, 9528–9546 (2017).
Liang, X. H. et al. Translation efficiency of mRNAs is increased by antisense oligonucleotides targeting upstream open reading frames. Nat. Biotechnol. 34, 875–880 (2016).
Lim, K. R., Maruyama, R. & Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 11, 533–545 (2017).
Finkel, R. S. et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 377, 1723–1732 (2017).
Agrawal, S., Temsamani, J. & Tang, J. Y. Pharmacokinetics, biodistribution, and stability of oligodeoxynucleotide phosphorothioates in mice. Proc. Natl. Acad. Sci. USA 88, 7595–7599 (1991).
Summerton, J. Morpholino antisense oligomers: the case for an RNase H-independent structural type. 1489, 141–158 (1999).
Manoharan, M. 2′-Carbohydrate modifications in antisense oligonucleotide therapy: importance of conformation, configuration and conjugation. 1489, 117–130 (1999).
Shen, W., Liang, X.-H., Sun, H. & Crooke, S. T. 2′-Fluoro-modified phosphorothioate oligonucleotide can cause rapid degradation of P54nrb and PSF. Nucleic Acids Res. 43, 4569–4578 (2015).
Henry, S. et al. Chemically modified oligonucleotides exhibit decreased immune stimulation in mice. J. Pharmacol. Exp. Ther. 292, 468−479 (2000).
Pp, S. et al. Design, synthesis and evaluation of constrained methoxyethyl (cMOE) and constrained ethyl (cEt) nucleoside analogs. Nucleic Acids Symp. Ser. 52, 553−554 (2008).
Singh, S. K., Nielsen, P., Koshkin, A. A. & Wengel, J. LNA (locked nucleic acids): synthesis and high-affinity nucleic acid recognition. Chem. Commun. 455–456 (1998).
Yahara, A. et al. Amido-bridged nucleic acids (AmNAs): synthesis, duplex stability, nuclease resistance, and in vitro antisense potency. ChemBioChem 13, 2513–2516 (2012).
Wada, F. et al. Cholesterol-GalNAc dual conjugation strategy for reducing renal distribution of antisense oligonucleotides. Nucleic Acid Ther. 28, 50–57 (2018).
Monia, B. P. et al. Evaluation of 2′-modified oligonucleotides containing 2′-deoxy gaps as antisense inhibitors of gene expression. J. Biol. Chem. 268, 14514–14522 (1993).
Benson, M. D. et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N. Engl. J. Med. 379, 22–31 (2018).
SPINRAZA (US Food and Drug Administration, 2016); https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/209531lbl.pdf
Lefebvre, S. et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165 (1995).
Lorson, C. L., Hahnen, E., Androphy, E. J. & Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 96, 6307–6311 (1999).
Lefebvre, S. et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 16, 265–269 (1997).
Wirth, B. et al. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum. Genet. 119, 422–428 (2006).
Chiriboga, C. A. et al. Results from a phase 1 study of nusinersen (ISIS-SMN Rx) in children with spinal muscular atrophy. Neurology 86, 890–897 (2016).
Mercuri, E. et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 378, 625–635 (2018).
Geary, R. S. et al. Pharmacokinetic properties of 2′-O-(2-methoxyethyl)-modified oligonucleotide analogs in rats. J. Pharmacol. Exp. Ther. 296, 890–897 (2001).
Springer, A. D. & Dowdy, S. F. GalNAc-siRNA conjugates: leading the way for delivery of RNAi therapeutics. Nucleic Acid Ther. 28, 109–118 (2018).
Nair, J. K. et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 136, 16958–16961 (2014).
Stockert, R. J. The asialoglycoprotein receptor: relationships between structure, function, and expression. Physiol. Rev. 75, 591−609 (1995).
Spiess, M. The asialoglycoprotein receptor: a model for endocytic transport receptors. Biochemistry 29, 10009–10018 (1990).
Lundquist, J. J. & Toone, E. J. The cluster glycoside effect. Chem. Rev. 102, 555–578 (2002).
Lee, Y. C. et al. Binding of synthetic oligosaccharides to the hepatic Gal/GalNAc lectin. Dependence on fine structural features. J. Biol. Chem. 258, 199–202 (1983).
Willoughby, J. L. S. et al. Evaluation of GalNAc-siRNA conjugate activity in pre-clinical animal models with reduced asialoglycoprotein receptor expression. Mol. Ther. 26, 105–114 (2018).
Foster, D. J. et al. Advanced siRNA designs further improve in vivo performance of GalNAc-siRNA conjugates. Mol. Ther. 26, 708–717 (2018).
Nair, J. K. et al. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc-siRNA conjugates. Nucleic Acids Res. 45, 10969–10977 (2017).
Brown, C. R. et al. Investigating the pharmacodynamic durability of GalNAc–siRNA conjugates. Nucleic Acids Res. 48, 11827–11844 (2020).
Schlegel, M. K. et al. Chirality dependent potency enhancement and structural impact of glycol nucleic acid modification on siRNA. J. Am. Chem. Soc. 139, 8537–8546 (2017).
Kumar, P. et al. Chimeric siRNAs with chemically modified pentofuranose and hexopyranose nucleotides: altritol-nucleotide (ANA) containing GalNAc–siRNA conjugates: in vitro and in vivo RNAi activity and resistance to 5′-exonuclease. Nucleic Acids Res. 48, 4028–4040 (2020).
Mikami, A. et al. Synthesis, chirality-dependent conformational and biological properties of siRNAs containing 5′-(R)- and 5′-(S)-C-methyl-guanosine. Nucleic Acids Res. 48, 10101–10124 (2020).
Zimmermann, T. S. et al. Clinical proof of concept for a novel hepatocyte-targeting GalNAc-siRNA conjugate. Mol. Ther. 25, 71–78 (2017).
FDA approves first treatment for inherited rare disease. US Food and Drug Administration https://www.fda.gov/news-events/press-announcements/fdaapproves-first-treatment-inherited-rare-disease (2019).
Chan, A. et al. Preclinical development of a subcutaneous ALAS1 RNAi therapeutic for treatment of hepatic porphyrias using circulating RNA quantification. Mol. Ther. Nucleic Acids 4, e263–e263 (2015).
Sardh, E. et al. Phase 1 trial of an RNA interference therapy for acute intermittent porphyria. N. Engl. J. Med. 380, 549–558 (2019).
Balwani, M. et al. Phase 3 trial of RNAi therapeutic givosiran for acute intermittent porphyria. N. Engl. J. Med. 382, 2289–2301 (2020).
Felgner, P. L. et al. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 84, 7413–7417 (1987).
Jayaraman, M. et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. 51, 8529–8533 (2012).
Akinc, A. et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. J. Am. Soc. Gene Ther. 18, 1357–1364 (2010).
Chen, S. et al. Influence of particle size on the in vivo potency of lipid nanoparticle formulations of siRNA. J. Control. Release 235, 236−244 (2016).
Kulkarni, J. A., Cullis, P. R. & van der Meel, R. Lipid nanoparticles enabling gene therapies: from concepts to clinical utility. Nucleic Acid Ther. 28, 146–157 (2018).
Belliveau, N. M. et al. Microfluidic synthesis of highly potent limit-size lipid nanoparticles for in vivo delivery of siRNA. Mol. Ther. Nucleic Acids 1, e37 (2012).
Witzigmann, D. Lipid nanoparticle technology for therapeutic gene regulation in the liver. Adv. Drug Deliv. Rev. 344–363 (2020).
Senior, J. H., Trimble, K. R. & Maskiewicz, R. Interaction of positively-charged liposomes with blood: implications for their application in vivo. Biochim. Biophys. Acta Membranes 1070, 173–179 (1991).
Zimmermann, T. S. et al. RNAi-mediated gene silencing in non-human primates. Nature 441, 111−114 (2006).
Kumar, V. et al. Shielding of lipid nanoparticles for siRNA delivery: Impact on physicochemical properties, cytokine induction, and efficacy. Mol. Ther. Nucleic Acids 3, e210 (2014).
Akinc, A. et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 14, 1084–1087 (2019).
Kulkarni, J. A. Fusion-dependent formation of lipid nanoparticles containing macromolecular payloads. Nanoscale 11, 9023–9031 (2019).
Harvie, P., Wong, F. M. P. & Bally, M. B. Use of poly(ethylene glycol)–lipid conjugates to regulate the surface attributes and transfection activity of lipid–DNA particles. J. Pharm. Sci. 89, 652–663 (2000).
Judge, A., McClintock, K., Phelps, J. R. & Maclachlan, I. Hypersensitivity and loss of disease site targeting caused by antibody responses to PEGylated liposomes. Mol. Ther. J. Am. Soc. Gene Ther. 13, 328–337 (2006).
Mui, B. L. et al. Influence of polyethylene glycol lipid desorption rates on pharmacokinetics and pharmacodynamics of siRNA lipid nanoparticles. Mol. Ther. Nucleic Acids 2, e139–e139 (2013).
Maurer, N. et al. Spontaneous entrapment of polynucleotides upon electrostatic interaction with ethanol-destabilized cationic liposomes. Biophys. J. 80, 2310–2326 (2001).
Jeffs, L. B. et al. A scalable, extrusion-free method for efficient liposomal encapsulation of plasmid DNA. Pharm. Res. 22, 362–372 (2005).
Kulkarni, J. A. et al. On the formation and morphology of lipid nanoparticles containing ionizable cationic lipids and siRNA. ACS Nano 12, 4787–4795 (2018).
Henrcikson, A. et al. Density matching multi-wavelength analytical ultracentrifugation to measure drug loading of lipid nanoparticle formulations. ACS Nano 15, 5068–5076 (2021).
ONPATTRO (US Food and Drug Administration, 2018); https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210922s000lbl.pdf
Suhr, O. B. et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J. Rare Dis. 10, 109 (2015).
Butler, J. S. et al. Preclinical evaluation of RNAi as a treatment for transthyretin-mediated amyloidosis. Amyloid 23, 109–118 (2016).
Coelho, T. et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N. Engl. J. Med. 369, 819–829 (2013).
Adams, D. et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 379, 11–21 (2018).
Lin, H., Merkel, M., Hale, C. & Marantz, J. L. Experience of patisiran with transthyretin stabilizers in patients with hereditary transthyretin-mediated amyloidosis. Neurodegen. Dis. Manag. 10, 289−300 (2020).
Wilson, J. M. Lessons learned from the gene therapy trial for ornithine transcarbamylase deficiency. Mol. Genet. Metab. 96, 151–157 (2009).
Dunbar, C. E. et al. Gene therapy comes of age. Science 359, eaan4672 (2018)
Naldini, L. Gene therapy returns to centre stage. Nature 526, 351–360 (2015).
Wang, D., Tai, P. W. L. & Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 18, 358–378 (2019).
Tenney, R. M., Bell, C. L. & Wilson, J. M. AAV8 capsid variable regions at the two-fold symmetry axis contribute to high liver transduction by mediating nuclear entry and capsid uncoating. Virology 454–455, 227–236 (2014).
Govindasamy, L. et al. Structurally mapping the diverse phenotype of adeno-associated virus serotype 4. J. Virol. 80, 11556–11570 (2006).
Bartlett, J. S., Wilcher, R. & Samulski, R. J. Infectious entry pathway of adeno-associated virus and adeno-associated virus vectors. J. Virol. 74, 2777–2785 (2000).
Nicolson, S. C. & Samulski, R. J. Recombinant adeno-associated virus utilizes host cell nuclear import machinery to enter the nucleus. J. Virol. 88, 4132–4144 (2014).
Korbelin, J. et al. Optimization of design and production strategies for novel adeno-associated viral display peptide libraries. Gene Ther. 24, 470–481 (2017).
Burger, C. et al. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol. Ther. J. Am. Soc. Gene Ther. 10, 302–317 (2004).
Villiger, L. et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 24, 1519–1525 (2018).
Ehrhardt, A., Xu, H. & Kay, M. A. Episomal persistence of recombinant adenoviral vector genomes during the cell cycle in vivo. J. Virol. 77, 7689–7695 (2003).
Calcedo, R., Vandenberghe, L. H., Gao, G., Lin, J. & Wilson, J. M. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J. Infect. Dis. 199, 381–390 (2009).
Russell, S. et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 390, 849–860 (2017).
Acland, G. M. et al. Gene therapy restores vision in a canine model of childhood blindness. Nat. Genet. 28, 92–95 (2001).
Cideciyan, A. V. et al. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc. Natl. Acad. Sci. USA 110, E517–E525 (2013).
Bennett, J. et al. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: a follow-on phase 1 trial. Lancet 388, 661–672 (2016).
Jacobson, S. G. et al. Improvement and decline in vision with gene therapy in childhood blindness. N. Engl. J. Med. 372, 1920–1926 (2015).
Bainbridge, J. W. et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N. Engl. J. Med. 372, 1887–1897 (2015).
Pardi, N., Hogan, M. J., Porter, F. W. & Weissman, D. mRNA vaccines—a new era in vaccinology. Nat. Rev. Drug Discov. 17, 261–279 (2018).
Prakash, T. P. et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 42, 8796–8807 (2014).
Nanna, A. R. et al. Generation and validation of structurally defined antibody–siRNA conjugates. Nucleic Acids Res. 48, 5281–5293 (2020).
Biscans, A. et al. Diverse lipid conjugates for functional extra-hepatic siRNA delivery in vivo. Nucleic Acids Res. 47, 1082–1096 (2018).
Prakash, T. P. et al. Fatty acid conjugation enhances potency of antisense oligonucleotides in muscle. Nucleic Acids Res. 47, 6029–6044 (2019).
Roberts, T. C., Langer, R. & Wood, M. J. A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 19, 673–694 (2020).
Benizri, S. et al. Bioconjugated oligonucleotides: recent developments and therapeutic applications. Bioconjugate Chem. 30, 366–383 (2019).
Pardi, N. et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun. 8, 14630 (2017).
Pardi, N. et al. Nucleoside-modified mRNA immunization elicits influenza virus hemagglutinin stalk-specific antibodies. Nat. Commun. 9, 3361 (2018).
Pardi, N. et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 543, 248 (2017).
Raj, D. K. et al. Anti-PfGARP activates programmed cell death of parasites and reduces severe malaria. Nature 582, 104–108 (2020).
Thanh Le, T. et al. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 19, 305–306 (2020).
Lurie, N., Saville, M., Hatchett, R. & Halton, J. Developing Covid-19 vaccines at pandemic speed. N. Engl. J. Med. 382, 1969–1973 (2020).
Corbett, K.S. et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 586, 567–571 (2020).
McKay, P. F. et al. Self-amplifying RNA SARS-CoV-2 lipid nanoparticle vaccine candidate induces high neutralizing antibody titers in mice. Nat. Commun. 11, 3523–3523 (2020).
Mulligan, M. J. et al. Phase 1/2 study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 586, 589–593 (2020).
Baden, L. R. et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 384, 403−416 (2021).
Polack, F. P. et al. Safety and efficacy of the BNT162b2 mRNA covid-19 vaccine. N. Engl. J. Med. 383, 2603–2615 (2020).
Ritchie, H. et al. Coronavirus (COVID-19) vaccinations. Our World In Data https://ourworldindata.org/covid-vaccinations (2021).
Sahin, U. & Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 359, 1355–1360 (2018).
Kranz, L. M. et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534, 396–401 (2016).
Kreiter, S. et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 520, 692–696 (2015).
Sheridan, C. Sangamo’s landmark genome editing trial gets mixed reception. 36, 907–908 (2018).
Anzalone, A. V., Koblan, L. W. & Liu, D. R. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38, 824–844 (2020).
Villiger, L. et al. In vivo cytidine base editing of hepatocytes without detectable off-target mutations in RNA and DNA. Nat. Biomed. Eng. 5, 179–189 (2021).
Gaudelli, N. M. et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 38, 892–900 (2020).
Ferreira, V., Petry, H. & Salmon, F. Immune responses to AAV-vectors, the glybera example from bench to bedside. Front. Immunol. 5, https://doi.org/10.3389/fimmu.2014.00082 (2014).
Mickle, K. et al. The effectiveness and value of patisiran and inotersen for hereditary transthyretin amyloidosis. J. Manag. Care. Spec. Pharm. 25, 10–15(2019).
Chapman, M. Up to 100 free Zolgensma treatments going out worldwide in 2020 under access program. SMA News Today https://smanewstoday.com/newsposts/2019/12/28/novartis-global-access-program-for-zolgensma/ (2019).
Khvorova, A. & Watts, J. K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 35, 238–248 (2017).
Crooke, S. T., Wang, S., Vickers, T. A., Shen, W. & Liang, X.-H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 35, 230–237 (2017).
Keeler, C. E. Gene therapy. J. Hered. 38, 294–298 (1947).
Wagner, R. R. Influenza virus infection of transplanted tumors. Cancer Res. 14, 377–385 (1954).
Viruses in treatment of cancer. Br. Med. J. 2, 1481–1482 (1957).
Wolff, J. A. et al. Direct gene transfer into mouse muscle in vivo. Science 247, 1465–1468 (1990).
Neufeld, E. F., Sweeley, C. C., Rogers, S., Friedmann, T. & Roblin, R. Gene therapy for human genetic disease? Science 178, 648–649 (1972).
Stec, W. J., Zon, G., Egan, W. & Stec, B. Automated solid-phase synthesis, separation, and stereochemistry of phosphorothioate analogues of oligodeoxyribonucleotides. J. Am. Chem. Soc. 106, 6077–6079 (1984).
Miller, P. S. et al. Control of ribonucleic acid function by oligonucleoside methylphosphonates. Biochimie 67, 769–776 (1985).
Matsukura, M. et al. Phosphorothioate analogs of oligodeoxynucleotides: inhibitors of replication and cytopathic effects of human immunodeficiency virus. Proc. Natl Acad. Sci. USA 84, 7706–7710 (1987).
Agrawal, S. et al. Oligodeoxynucleoside phosphoramidates and phosphorothioates as inhibitors of human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 85, 7079–7083 (1988).
Raal, F. J. et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet 375, 998–1006 (2010).
Finkel, R. S. et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 388, 3017–3026 (2016).
Fraley, R., Subramani, S., Berg, P. & Papahadjopoulos, D. Introduction of liposome-encapsulated SV40 DNA into cells. J. Biol. Chem. 255, 10431–10435 (1980).
Filion, M. C. & Phillips, N. C. Toxicity and immunomodulatory activity of liposomal vectors formulated with cationic lipids toward immune effector cells. Biochim. Biophys. Acta 1329, 345–356 (1997).
Lappalainen, K., Jaaskelainen, I., Syrjanen, K., Urtti, A. & Syrjanen, S. Comparison of cell proliferation and toxicity assays using two cationic liposomes. Pharm. Res. 11, 1127–1131 (1994).
Bailey, A. L. & Cullis, P. R. Modulation of membrane fusion by asymmetric transbilayer distributions of amino lipids. Biochemistry 33, 12573–12580 (1994).
Sambrook, J., Westphal, H., Srinivasan, P. R. & Dulbecco, R. The integrated state of viral DNA in SV40-transformed cells. Proc. Natl. Acad. Sci. USA 60, 1288–1295 (1968).
Rogers, S. & Pfuderer, P. Use of viruses as carriers of added genetic information 219, 749−751 (Academic Press, 1968).
Mulligan, R. C., Howard, B. H. & Berg, P. Synthesis of rabbit β-globin in cultured monkey kidney cells following infection with a SV40 β-globin recombinant genome. Nature 277, 108–114 (1979).
Hamer, D. H. & Leder, P. Expression of the chromosomal mouse βmaj-globin gene cloned in SV40. Nature 281, 35–40 (1979).
Miller, A. D., Jolly, D. J., Friedmann, T. & Verma, I. M. A transmissible retrovirus expressing human hypoxanthine phosphoribosyltransferase (HPRT): gene transfer into cells obtained from humans deficient in HPRT. Proc. Natl. Acad. Sci. USA 80, 4709–4713 (1983).
Samulski, R. J., Berns, K. I., Tan, M. & Muzyczka, N. Cloning of adeno-associated virus into pBR322: rescue of intact virus from the recombinant plasmid in human cells. Proc. Natl Acad. Sci. USA 79, 2077–2081 (1982).
Rosenberg, S. A. et al. Gene transfer into humans–immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N. Engl. J. Med. 323, 570–578 (1990).
Blaese, R. M. et al. T lymphocyte-directed gene therapy for ADA-SCID: initial trial results after 4 years. Science 270, 475–480 (1995).
High-dose AAV gene therapy deaths. Nat. Biotechnol. 38, 910(2020).
Wilson, J. M. Gendicine: the first commercial gene therapy product. Hum. Gene Ther. 16, 1014–1015 (2005).
Morrison, C. $1-million price tag set for Glybera gene therapy. Nat. Biotechnol. 33, 217–218 (2015).
Senior, M. After Glybera’s withdrawal, what’s next for gene therapy? Nat. Biotechnol. 35, 491–492 (2017).
Hoggatt, J. Gene therapy for ‘bubble boy’ disease. Cell 166, 263–263 (2016).
Acknowledgements
This work was supported by a Foundation grant (FDN 148469) from the Canadian Institutes of Health Research and a British Columbia Innovation Council Ignite grant. J.A.K. acknowledges support from the NanoMedicines Innovation Network (NMIN). D.W. acknowledges support from the Swiss National Science Foundation (number 183923). B.R.L. acknowledges support from Brain Canada, Huntington Society of Canada, Weston Brain Institute, and NMIN. S.B.T. acknowledges support from the Huntington Society of Canada. The work of R.v.d.M. is supported by the Netherlands Research Council (NWO: ZonMW Vici grant number 016.176.622 to W. J. M. Mulder). We acknowledge the use of Servier Medical Art (https://smart.servier.com) templates, which are licensed under a Creative Commons Attribution 3.0 Unported License, for compiling the figures.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
P.R.C. is a co-founder and shareholder of Acuitas Therapeutics and Precision Nanosystems and Scientific Director and CEO of the NMIN. B.R.L. is a co-founder, shareholder and CEO of Incisive Genetics Inc. The authors declare no other competing financial interests.
Additional information
Peer review information Nature Nanotechnology thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kulkarni, J.A., Witzigmann, D., Thomson, S.B. et al. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 16, 630–643 (2021). https://doi.org/10.1038/s41565-021-00898-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41565-021-00898-0
This article is cited by
-
A novel microglia-targeting strategy based on nanoparticle-mediated delivery of miR-26a-5p for long-lasting analgesia in chronic pain
Journal of Nanobiotechnology (2024)
-
Injectable hydrogel-based combination therapy for myocardial infarction: a systematic review and Meta-analysis of preclinical trials
BMC Cardiovascular Disorders (2024)
-
Novel STAT3 oligonucleotide compounds suppress tumor growth and overcome the acquired resistance to sorafenib in hepatocellular carcinoma
Acta Pharmacologica Sinica (2024)
-
Strategies to reduce the risks of mRNA drug and vaccine toxicity
Nature Reviews Drug Discovery (2024)
-
Nucleic acid therapeutics as differentiation agents for myeloid leukemias
Leukemia (2024)
