
Abstract
Vinyl polymers are typically synthesized through the addition polymerization of corresponding vinyl compounds. However, the polymerization ability significantly depends on the substituent on the vinyl moiety, resulting in various synthetic limitations in the molecular structure of vinyl polymers. Given the increasing societal demand for enhanced properties and functions of polymer materials, innovative synthetic technologies are required for developing next-generation polymers through flexible molecular design. The author has made considerable efforts to overcome these limitations in polymer synthesis by employing alkenyl boronates as monomers for radical polymerization. The resulting polymers bear boron on the main chain, allowing the replacement of boron side chains with other elements through the cleavage of carbon–boron bonds in postpolymerization transformations. This strategy, based on “side-chain replacement,” has enabled the synthesis of various polymers that were previously inaccessible.
Similar content being viewed by others
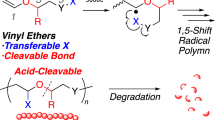


Vinyl polymers are typically synthesized through chain-growth polymerization of the corresponding vinyl compounds. Polymerization systems are classified into radical, anionic, and cationic polymerizations based on the active species, and the appropriate system varies depending on the element or functional group attached to the vinyl moiety in the monomer [1]. For example, styrene and acrylates are typical vinyl monomers that exhibit high radical polymerization ability, and the growing radical species are moderately stabilized by phenyl or carbonyl groups (Fig. 1a). On the other hand, vinyl ethers, in which an oxygen atom is directly bonded to the vinyl group, generally have poor radical polymerization ability but readily form high polymers through cationic polymerization. The active species in cationic polymerization is stabilized by the lone pair on the oxygen atom (note that recent research has revealed that specific vinyl ethers can indeed undergo radical polymerization [2, 3]). Some vinyl compounds, such as isopropenyl acetate, are difficult to polymerize using any polymerization system [4, 5]. Such limitations are also encountered in the copolymerization of multiple monomers. Although the radical copolymerization of acrylate and styrene occurs efficiently, the synthesis of a copolymer of vinyl acetate and styrene is not straightforward (Fig. 1b). Despite the high radical polymerization ability of both monomers, the attempt to copolymerize them results in the formation of a styrene homopolymer [6,7,8,9]. These limitations, which depend on monomer structures and their combinations, highlight the challenges in the flexible molecular design of polymer materials. Polymer reactions, also known as postpolymerization transformations or modifications, provide opportunities to synthesize polymers that are otherwise inaccessible [10,11,12,13]. A representative example is the synthesis of poly(vinyl alcohol)s (PVAs), which are typically prepared through the radical polymerization of vinyl acetate followed by saponification due to the instability of the vinyl alcohol monomer caused by keto-enol tautomerization. N-Hydroxysuccinimide and 2,3,4,5,6-pentafluorophenol-bearing acrylate monomers have recently garnered significant attention due to their high reactivity for aminolysis and alcoholysis in polymer reactions [14,15,16,17]. However, the transformation of elements or functional groups directly attached to the main chain (e.g., oxygen in vinyl ethers and carbonyl groups in acrylates), which directly affect polymerizability, is generally challenging in conventional polymer reactions. Discovering a strategy that enables the replacement of elements attached to the main chain during the postpolymerization transformation step could overcome the restrictions in vinyl polymer design imposed by the polymerizability of the vinyl compound (Fig. 1c).

Synthetic limitations in vinyl polymer synthesis through homopolymerization (a) and copolymerization (b) of vinyl compounds. Concept of “side-chain replacement” for novel polymer synthesis (c). The idea of using alkenyl boronate-type compounds as monomers to realize polymer synthesis through “side-chain replacement” (d)
In the field of organic synthesis, organoboron compounds are recognized as synthetically useful molecules due to the versatility in the transformation of the carbon–boron bond [18]. The oxidation of boron compounds is known as a reliable route for accessing various alcohol products [19, 20]. Suzuki-Miyaura cross-coupling is another famous transformation for the introduction of various aryl groups [21]. Additionally, amination and protonation involving the cleavage of carbon–boron bonds are also helpful for the preparation of valuable compounds [22, 23]. The high transformability of the boryl group has also been utilized in polymer reactions. There are numerous examples of boron-containing monomers in which the structure is designed by introducing boron to the side-chain terminus embedded in common monomers such as styrene and acrylamide [24,25,26,27]. Conversely, alkenylboronic acid esters, where boron is directly attached to the vinyl group, have rarely been used as monomers for addition polymerization. The radical polymerization of vinylboronic acid without a protecting group on boron was attempted in 1966 [28], but the chemical structure of the resulting polymer was not discussed, likely due to the structural complexity arising from the condensation of boronic acid. In 1982, the radical polymerization ability of vinylboronic acid dibutyl ester was also investigated [29]. However, the instability of the dibutyl ester in the presence of moisture interfered with the identification of the polymer structure. To improve the stability of boron pendants, some research groups have focused on the aromaticity of the azaborine group, which is based on the double-bond nature of the boron-nitrogen bond [30,31,32]. Vinyl azaborine-type compounds were found to be polymerizable under radical conditions, affording polymers with well-defined structures. The polymerization behavior is similar to that of styrene due to the aromaticity of the azaborine pendant. Notably, Klausen’s group reported that postpolymerization oxidation of the azaborine pendant can provide poly(vinyl alcohol) (PVA) copolymers [33]. Although the aromatic azaborine pendant is useful for suppressing side reactions during polymerization or preventing reactions between boron pendants that cause structural complexity, its high stability interrupts the application of versatile boron transformation reactions developed in synthetic organic chemistry.
To balance both stability and transformability, the author noticed the potential of alkenylboronic acid pinacol esters, which are commercially available as substrates for organic reactions such as Suzuki-Miyaura cross-coupling. At the beginning of the author’s research, these compounds have not been used as monomers for addition polymerization, despite the popularity of bulky pinacol protection for organoboron substrates due to their high stability and transformability. In this focus review, the author’s discovery of the radical polymerization ability of alkenylboronic acid pinacol esters and a detailed investigation of the polymerization behavior are summarized, together with the postpolymerization transformation of the boron pendants on the resulting polymers (Fig. 1d). Isopropenyl- and vinylboronic acid pinacol esters showed high radical polymerization ability under typical free radical polymerization conditions. Copolymerizations with common vinyl monomers (e.g., styrene, methacrylate, acrylonitrile, etc.) were also possible, and the copolymerizability with conjugated-type monomers was especially high. Reversible addition-fragmentation chain transfer (RAFT) polymerization using commercially available chain transfer agents (CTAs) enables control of the molecular weight and molecular structure of chain-end groups. The author revealed that the unique elemental properties of boron were essential for its compatibility with common monomers in copolymerization and the choice of appropriate CTAs for high-level polymerization control. The resulting polymers have boron pendants attached to the main chain, and the transformation of the carbon–boron bond enables “side-chain replacement” type polymer reactions. Oxidation enabled the transformation of the boron unit to vinyl alcohol and its derivatives, leading to the synthesis of vinyl alcohol-based (co)polymers, which are inaccessible due to limitations in the (co)polymerizability of vinyl acetate-type monomers. Amination and protonation also allowed access to isopropenylamine- or ethylene-containing (co)polymers, and thus, the side-chain replacement-based strategy was found to be useful for overcoming the limitations in chain-growth polymerization ascribed to the polymerization ability of vinyl compounds. The author believes that the concept of boron chemistry is helpful for expanding the potential of polymer science toward developing novel polymer materials to address societal demands.
Discovery of the radical polymerization ability of isopropenylboronic acid pinacol ester
The author initially performed radical homopolymerization of isopropenylboronic acid pinacol ester (IPBpin), which is commercially available as a substrate for Suzuki-Miyaura cross-coupling (Fig. 2a) [34]. The author applied typical free radical polymerization conditions using 2,2’-azobisisobutyronitrile (AIBN) as the initiator at 60 °C. The conversion of IPBpin proceeded smoothly and reached 69% in 24 h. The molecular weight of the resulting polymer was greater than 10,000, and structural analysis by 1H and 13C NMR supported that the addition polymerization occurred as expected. The polymerization ability of 2-methylnonene and isopropenyl acetate (IPOAc), which have vinylidene-attached carbon or oxygen atoms instead of boron, was verified under the same polymerization conditions for comparison, and the conversion of these monomers was quite low. Thus, vinylidene-attached boron was found to play an essential role in the high radical polymerization ability of IPBpin. A density functional theory (DFT)-based investigation was then conducted to elucidate the role of boron in the radical polymerization process. For monomers with a methyl group at the α-position, the chain-growth reaction can be inhibited by a degenerate chain transfer reaction, which results in the formation of a relatively stable allyl radical species due to hydrogen abstraction by the growing radical species (Fig. 2b). The poor polymerization ability of IPOAc is known to be attributed to such degenerative chain transfer [4, 5], which is consistent with the total energy change estimated to be exothermic (−35.7 kJ/mol) based on DFT calculations. In contrast, for isopropenylboronic acid pinacol ester (IPBpin), this side reaction was found to be endothermic (+3.0 kJ/mol), suggesting a relatively low frequency of side reactions. The suppression of the side reaction is likely due to the presence of a vacant p-orbital of boron, which can stabilize the neighboring carbon radical; the formation of an allyl radical is thus unfavorable because of the relatively stable chain-growth radical. Methyl methacrylate (MMA), which has an α-methyl group, is a typical vinyl monomer for radical polymerization, and the calculation results indicated that the carbonyl group plays a crucial role in stabilizing the chain-growth radical to suppress the side reaction (+8.5 kJ/mol), suggesting that the roles of boron in IPBpin are similar to those of the carbonyl group in MMA.

a Comparison of size exclusion chromatography (SEC) traces in the radical polymerization of IPBpin, 2-methyl-1-nonene, and isopropenyl acetate ([Alkenyl compound]/[AIBN] = 4000 mM/40 mM in toluene at 60 °C). b DFT-based calculation of the total energy change in degenerative chain transfer dependent on the monomer structure [(U)B3LYP/6-31 G(d)]. c Side-chain replacement of poly(IPBpin) for the synthesis of poly(α-methyl vinyl alcohol) and poly(α-methyl vinyl amine) (Reproduced with permission from Ref. [34]. Copyright 2019 Wiley-VCH)
The radical polymerization of IPBpin afforded a unique vinyl polymer bearing boron on the main chain. The author then performed “side-chain replacement” in the postpolymerization transformation step through the cleavage of the carbon–boron bond. The transformation of boron into a hydroxy group is known as one of the most reliable reactions for the synthesis of valuable alcohol molecules in organic chemistry. When oxidation using aqueous H2O2 and sodium hydroxide was applied to poly(IPBpin), the boron pendant was quantitatively converted to a hydroxy group, affording poly(α-methyl vinyl alcohol) (PMVA), as confirmed by 1H, 13C NMR, and FT-IR measurements (Fig. 2c). Given that the synthesis of PMVA is not straightforward due to the poor polymerization ability of the corresponding monomer (i.e., IPOAc) [35], a synthetic route based on the boron monomer is valuable for overcoming the limitations of chain-growth polymerization. Furthermore, amination of the boron pendant afforded poly(α-methyl vinyl amine) using methoxyamine and tert-BuOK as reagents for postpolymerization transformation [22]. Thus, the author demonstrated that the concept of side-chain replacement is helpful for novel polymer synthesis in a versatile fashion.
Radical copolymerization with various common monomers leading to elucidation of the monomer character
The author’s interest was then directed to the radical copolymerization of IPBpin with common vinyl monomers [36]. In the case of 1:1 copolymerization with styrene (St), both monomers were consumed over time, although the conversion of IPBpin was slower than that of St, yielding a copolymer with an average molecular weight of approximately 10,000 (Fig. 3a). IPBpin can also be copolymerized with other typical conjugated-type monomers, such as MMA, methyl acrylate (MA), and acrylonitrile (AN). In contrast, nonconjugated isobutyl vinyl ether (IBVE) was consumed less during copolymerization, and the molecular weight of the resulting polymer was low. Interestingly, copolymerization with electron-deficient N-ethylmaleimide (EMI) proceeded rapidly; the consumption of both monomers exceeded 85% in 2 hours, resulting in a high-molecular-weight copolymer. The monomer reactivity ratios (r1, r2) for each copolymerization were then investigated (Fig. 3b, M1 = IPBpin and M2 = comonomer). During copolymerization with conjugated monomers (e.g., St, MMA, and MA), r1 was smaller than r2, resulting in fewer consecutive sequences of IPBpin units in the copolymer. When IBVE was used as the comonomer, r1 was greater than r2. The use of the EMI comonomer resulted in both r1 and r2 being close to zero, suggesting alternating-rich copolymerization. To determine the effect of boron on copolymerization behavior, the properties of the radical species derived from IPBpin or other common monomers were compared based on DFT calculations (Fig. 3c). The author focused on SOMO energy levels, known as the index of the electron-rich nature of radical species [37, 38]. IPBpin showed a relatively high SOMO energy level among the monomers, suggesting that the IPBpin radical behaves as an electron-rich species compared to acrylate or maleimide, which are known as electron-deficient monomers. The electron-rich nature is ascribed to the low electronegativity of boron; σ–bond donation by boron increases the electron density of the radical-centered carbon. The electronegativity-based discussion is applicable not only to carbon-centered radicals of chain-growth species but also to the alkene moiety in the monomer. Efficient alternating copolymerization with EMI is likely due to the large difference in the electronic density of the alkene moieties between IPBpin and EMI. The spatial distribution of SOMO is also informative for investigating the role of the side chain in stabilizing radical species, and the SOMO shape of the IPBpin radical was compared with that of MMA, a typical conjugated monomer (Fig. 3d). In both cases, the SOMO was found to be delocalized from the radical center to the side chain (i.e., boron or carbonyl group). This suggested the contribution of the side chain to the stabilization of the chain-growth radical, which is attributed to the vacant p-orbital of boron or the antibonding orbital of the carbonyl. Therefore, IPBpin can be classified as a conjugated monomer similar to MMA, corresponding to the high copolymerizability of IPBpin with styrene. Thus, IPBpin was found to be a relatively electron-rich and conjugated-type monomer in the vinyl monomer family for radical polymerization.

a Radical copolymerization of IPBpin with common vinyl monomers. b Monomer reactivity ratios. c SOMO energy levels of the radical species corresponding to each monomer based on DFT calculations. d Spatial distribution of SOMO for IPBpin and MMA [(U)B3LYP/6-31 G(d)] (Reproduced with permission from Ref. [36]. Copyright 2020 American Chemical Society)
RAFT-based controlled polymerization and synthesis of end-functionalized polymers through orthogonal transformation of boron pendants
Reversible deactivation radical polymerization (RDRP) is an important technique for controlling primary polymer structures such as molecular weight and chain-end functionality [39,40,41]. Reversible addition-fragmentation chain transfer (RAFT) polymerization can be applied to various monomers by selecting an appropriate chain transfer agent (CTA) [42,43,44]. The author investigated suitable CTAs for the controlled polymerization of IPBpin to achieve precise synthesis of boron-containing polymers (Fig. 4a) [45]. Among commercially available CTAs, dithiobenzoate- and trithiocarbonate-type CTAs were found to be effective; the resulting polymer peak in SEC clearly shifted to a higher molecular weight as IPBpin was consumed. A relatively narrow molecular weight distribution (Mw/Mn < 1.4) was maintained during the polymerization. In contrast, the use of a dithiocarbamate-type CTA resulted in poor polymerization control, as indicated by the broad SEC peak (Mw/Mn > 2.0). Dithiobenzoate and trithiocarbonate-based CTAs are known to be effective for the controlled polymerization of conjugated monomers such as St and MMA, whereas dithiocarbamate CTAs are typically applied to nonconjugated monomers such as vinyl acetate and N-vinyl-2-pyrrolidone [44]. These results suggest that IPBpin behaves as a conjugated monomer due to the effect of the vacant p-orbital of boron, which is consistent with the trend observed in the copolymerization section. The compatibility of IPBpin and CTA was further investigated through DFT calculations (Fig. 4b). To achieve high-level polymerization control, the growing radical species must frequently and reversibly react with the CTA, and this reaction was analyzed from the perspective of the total energy change. When IPBpin was combined with a trithiocarbonate-type CTA, the energy difference before and after the reaction was very small (−3.2 kJ/mol), suggesting that chain transfer to the CTA occurs frequently in a reversible manner. This calculation result corresponds to the successfully controlled polymerization observed with trithiocarbonate as the CTA. For dithiocarbamates, where polymerization was not well controlled, the reaction with the CTA was found to be endothermic (+26.3 kJ/mol). Consequently, dithiocarbamate cannot effectively participate in the radical polymerization process of IPBpin, resulting in the free radical polymerization of IPBpin despite the presence of the CTA. The larger energy barrier (44.9 kJ/mol) for chain transfer to the dithiocarbamate CTA compared to that of the trithiocarbonate (30.3 kJ/mol) should also be a reason why dithiocarbamate cannot be involved in the polymerization process.

a SEC traces of the radical polymerization of IPBpin in the presence of a chain transfer agent for RAFT. b Energy diagrams of the reversible degradative chain transfer of carbon-centered radical species from IPBpin on the basis of DFT calculations [(U)B3LYP/6-31 g(d)] (Reproduced with permission from Ref. [45]. Copyright 2021 Nature Springer)
One of the advantages of controlled polymerization is the precise control of the terminal structure of vinyl polymers, and RAFT polymerization affords polymers bearing CTA-derived groups at the ω-terminus. The CTA group of PMMA at the ω-terminus can be removed using a cobalt catalyst, resulting in end-olefinated PMMA [46]. Given the presence of the α-methyl group in IPBpin similar to MMA, the author applied cobalt-catalyzed olefination to poly(IPBpin) prepared by RAFT polymerization using a trithiocarbonate-type CTA. Structural analysis of the product polymer by 1H NMR and MALDI-TOF MS indicated that the reaction proceeded quantitatively as expected. The resulting polymer has a C(sp²)–B bond at the terminus and C(sp³)–B bonds in the repeating units. Suzuki-Miyaura cross-coupling (SMC), which is one of the most famous boron transformation reactions, shows high reactivity for C(sp²)–B bonds relative to C(sp³)–B bonds [47]. This led to the concept of end-selective postpolymerization transformation using SMC to synthesize end-functionalized polymers by orthogonal C–B bond transformation (Fig. 5). End-selective Suzuki-Miyaura cross-coupling (SMC) with a catalytic amount of PdCl₂(dppf) was performed using methyl p-bromobenzoate as a substrate (Fig. 6a) [48]. The structure of the resulting polymer was analyzed by 1H NMR spectroscopy, where selective arylation was demonstrated by the appearance of phenylene peaks (g) in the aromatic region and a peak derived from the methyl ester (h), with reasonable peak areas. The author also detected a complete shift of the olefin peak (a → a’), suggesting the quantitative conversion of the terminal C(sp²)–B bond. The shape of the peaks derived from the Bpin side chain in the repeating structure remained unchanged, indicating selective conversion in the terminal Bpin pendant. MALDI-TOF MS measurements also supported this conversion. This end-selective SMC allowed the quantitative terminal introduction of various functional groups, such as trifluoromethyl, amide, nitro, and fluoro/methoxy groups, as well as esters (Fig. 6b).

Concept of end-functionalized polymer synthesis through controlled polymerization and orthogonal C–B bond transformation based on Suzuki-Miyaura cross-coupling (Reproduced with permission from Ref. [48]. Copyright 2021 American Chemical Society)

a 1H NMR-based analysis of the terminus-selective Suzuki-Miyaura cross-coupling of the end-olefinated boron polymer. b Introducing aryl groups to the polymer terminus. c MALDI-TOF-MS-based analysis of the oxidation of repeating units for the synthesis of end-functionalized poly(α-methyl vinyl alcohol) (Reproduced with permission from Ref. [48]. Copyright 2021 American Chemical Society)
The transformation of the boron side chains in the repeating structure was then performed. Oxidation of poly(IPBpin) bearing a fluoro/methoxy end group by hydrogen peroxide and sodium hydroxide was conducted to synthesize end-functionalized poly(α-methyl vinyl alcohol)s. According to MALDI-TOF MS measurements (Fig. 6c), the peak interval changed from a value corresponding to the molecular weight of IPBpin to that of α-methyl vinyl alcohol through pendant oxidation, and the peak positions were in good agreement with the values calculated based on the assumed end structures. The success of side-chain replacement was also supported by the change in the 1H NMR spectrum; the disappearance of the pinacol-derived peak was accompanied by the formation of a broad peak derived from the hydroxy group, and the small peaks derived from the terminal aryl group were clearly retained. As already mentioned, isopropenyl acetate, a possible precursor monomer for PMVA, has extremely low polymerization ability [4, 5], so there have been no examples of the synthesis of end-functional PMVA. The author believes that this achievement demonstrates the significance of polymer reactions based on C–B bond transformation with high efficiency and selectivity.
Vinylboron monomers for radical (co)polymerization enabling the synthesis of various inaccessible (co)polymers
The high radical polymerization ability of IPBpin and the impact of boron pendants on the (co)polymerization behavior encouraged the author to further explore boron-based monomers. Vinylboronic acid pinacol ester (VBpin), which does not have an α-methyl group, is also commercially available, and the author recognized it as a potential vinyl alcohol precursor monomer due to the high transformability of boron into hydroxy groups. Poly(vinyl alcohol) is typically synthesized through the radical polymerization of vinyl acetate followed by saponification, as vinyl alcohol itself is unstable and undergoes rapid keto-enol tautomerization. However, vinyl acetate is difficult to copolymerize with common conjugated monomers such as St due to its nonconjugated nature, making the copolymer of vinyl alcohol and styrene inaccessible by conventional methods [6,7,8,9]. As mentioned earlier, the vacant p-orbital of boron is the reason why IPBpin exhibits copolymerization behavior similar to that of conjugated monomers. The author therefore hypothesized that VBpin would have better copolymerization ability with St than vinyl acetate, allowing for the synthesis of a vinyl alcohol-St copolymer through the replacement of the boron pendant with a hydroxy group in the postreaction. The author found that the monomer reactivity ratios in the copolymerization of VBpin with styrene were as follows: rVBpin = 0.25 and rSt = 3.77 (Fig. 7a) [49]. These values clearly demonstrate the greater copolymerization ability compared to the combination of vinyl acetate and styrene (rVOAc = 0.01, rSt = 58.3) [7], as the author expected. When the copolymer of VBpin and St was oxidized using aqueous H2O2 and sodium hydroxide, the IR spectra of the resulting polymer exhibited a broad peak at 3500 cm−1 corresponding to the formation of hydroxy pendants (Fig. 7b). The quantitative transformation of boron pendants to hydroxy groups was confirmed by 1H NMR spectroscopy in DMSO-d6; the disappearance of the pinacol peak was accompanied by the appearance of a hydroxy peak (c’) at approximately 4.0 ppm with the expected peak area. The peak shape in the aromatic region derived from the St unit was almost unchanged, suggesting the preservation of the St unit after the postreaction. The composition ratio of the resulting PVA-St copolymer was tunable in the range from 11/89 to 71/29 (VA/St) by varying the injection ratio of VBpin to St in copolymerization, although the molecular weight decreased with increasing VBpin ratio. Notably, the composition ratio had a considerable impact on the basic properties of the copolymer, such as its solubility in polar solvents and thermal properties. For example, PVA-rich copolymers can be easily dissolved in DMSO and methanol.

a Synthesis of poly(vinyl alcohol-co-styrene) through radical copolymerization of VBpin and styrene followed by oxidation and comparison with the synthesis using VOAc as the vinyl alcohol precursor monomer. b IR- and c 1H NMR-based analysis of the transformation of boron pendants into hydroxy groups (Reproduced with permission from Ref. [49]. Copyright 2021 Royal Society of Chemistry)
The author’s interest was then directed toward another transformation of the boron pendant to overcome limitations in polymer synthesis. The protodeboronation reaction reported in organic chemistry [23] suggested the possibility of using the vinylboron unit as a precursor for ethylene units. Ethylene-containing copolymers are recognized as valuable materials due to their crystallinity and solvent resistance [50]. Although ethylene-acrylate copolymer is a typical example, copolymerization is not straightforward; coordination copolymerization faces challenges due to the need for novel catalyst design to suppress the catalyst-poisoning behavior of acrylate monomers [51,52,53], and radical copolymerization is difficult because of the low conversion rate of nonactivated ethylene [54,55,56]. The author then attempted postpolymerization protodeboronation of the VBpin-acrylate copolymer (Fig. 8a). The reaction proceeded to a certain extent, but it was unfortunately not quantitative. The author speculated that the steric effect of the bulky main chain lowered the reactivity of the Bpin pendant compared to that of low-molecular-weight substrates. To enhance the transformation ability, boron-protecting group chemistry was found to be useful. In the field of organic synthesis, various protecting groups for boron have been developed to tune the reactivity. The author designed new boron monomers by introducing various protecting groups instead of pinacol. Anthranilamide, which was originally used for boron protection by Suginome’s group [57, 58], was found to be valuable for quantitative protodeboronation in polymer reactions (Fig. 8b) [59]. The protodeboronation was performed using Mn(OAc)3 as the oxidant, tetrabutylammonium fluoride as the boron activator, and tert-butyl catechol as the proton source. The successful synthesis of the ethylene-acrylate copolymer was confirmed by 1H, 13C NMR, and IR measurements. In particular, the 13C NMR spectrum strongly supported the quantitative transformation. The author found the peak series corresponding to each triad composed of ethylene and acrylate units. In addition to the peaks of the homotriad of tert-butyl acrylate (A-A-A), the peak series of ethylene-containing triads (A-E-E and A-E-A) were clearly observed, indicating the superior transformability of the VBaam unit for replacement with protons relative to that of VBpin.

a Comparison of the transformability of VBpin and VBaam units into ethylene units through the replacement of boron with protons using 4-tert-butylpyrocatechol (TBC) as the proton source. b Synthesis of poly(ethylene-co-tert-butyl acrylate) via radical copolymerization of VBaam and tert-butyl acrylate and postpolymerization protonation and characterization of the product by 13C NMR (Reproduced with permission from Ref. [59]. Copyright 2022 Royal Society of Chemistry)
Summary and perspective
In this focus review, the author demonstrated the radical polymerization ability of vinylboronic acid monomers and described its impact on polymer synthesis. The author discovered that commercially available IPBpin and VBpin could be used as monomers for radical (co)polymerization, with the vacant p-orbital of boron being essential for their behavior as conjugated-type monomers. The boron pendants in the resulting polymer could be transformed into another element in polymer reactions, allowing for the replacement of side-chain elements that dominate the polymerization behavior of the corresponding monomers. This “side-chain replacement”-type polymer reaction enabled us to overcome synthetic limitations in the chain-growth polymerization of vinyl monomers. For example, the author succeeded in synthesizing poly(α-methyl vinyl alcohol), poly(α-methyl vinyl amine), and vinyl alcohol-styrene copolymers, which are typically inaccessible due to their poor (co)polymerization ability. An ethylene-acrylate copolymer was obtained through side-chain replacement based on the enhanced transformability of the anthranilamide-protected boron pendant. The author believes this transformation provides a new synthetic route for the copolymer, which is typically synthesized by coordination copolymerization using carefully designed transition metal complexes [51,52,53]. Additionally, IPBpin was usable for RAFT-based controlled polymerization using commercially available CTAs, allowing us to design terminal groups through cobalt-catalyzed end-group olefination and functionalization by Suzuki-Miyaura cross-coupling. Importantly, the high selectivity of the coupling reaction to terminal C(sp2)–B bonds enabled the orthogonal transformation of boron pendants at the terminus and repeating units, leading to the synthesis of end-functionalized poly(α-methyl vinyl alcohol)s. Thus, the unique reactivity and transformability of organoboron compounds can be utilized to broaden the chemical scope of polymer chemistry. The versatility of C–B bond-cleaving side-chain replacement is superior to that of other polymer reaction methods, which can transform the element attached to the main chain [60,61,62,63]. Furthermore, boron-containing compounds are known to be useful not only as synthetic intermediates but also as functional materials because of their Lewis acidity and stimuli-responsive properties. The author believes that the design of vinylboron-containing (co)polymers can contribute to the development of unique polymer functions.
References
Odian, G., editor. Principles of polymerization. 4th ed. Hoboken: Wiley; 2004.
Sugihara S, Kawamoto Y, Maeda Y. Direct radical polymerization of vinyl ethers: reversible addition—fragmentation chain transfer polymerization of hydroxy-functional vinyl ethers. Macromolecules. 2016;49:1563–74.
Sugihara S, Yoshida A, Kono T, Takayama T, Maeda Y. Controlled radical homopolymerization of representative cationically polymerizable vinyl ethers. J Am Chem Soc. 2019;141:13954–61.
Kuwae Y, Kamachi M, Nozakura S. Kinetic and electron spin resonance studies on radical polymerization of isopropenyl acetate. Macromolecules. 1986;19:2912–5.
Gaylord NG, Eirich FR. Allyl polymerization. III. Kinetics of polymerization of allyl esters1,2. J Am Chem Soc. 1952;74:337–42.
Mayo FR, Walling C, Lewis FM, Hulse W. Copolymerization. V.1 Some copolymerizations of vinyl acetate. J Am Chem Soc. 1948;70:1523–5.
Nakata T, Otsu T, Imoto M. Vinyl polymerization. XCI. Polymerization of styrene initiated by nickel peroxide. J Polym Sci Part A Gen Pap. 1965;3:3383–97.
Bevington JC, Johnson M. Radical polymerizations involving esters of vinyl alcohol—II. Copolymerizations. Eur Polym J. 1968;4:669–75.
Brar AS, Charan S. Sequence determination of vinyl acetate–methyl acrylate copolymers by NMR spectroscopy. J Appl Polym Sci. 1994;53:1813–22.
Gauthier MA, Gibson MI, Klok H-A. Synthesis of functional polymers by post-polymerization modification. Angew Chem Int Ed. 2009;48:48–58.
Günay KA, Theato P, Klok H-A. Standing on the shoulders of Hermann Staudinger: post-polymerization modification from past to present. J Polym Sci Part A Polym Chem. 2013;51:1–28.
Hawker CJ, Wooley KL. The convergence of synthetic organic and polymer chemistries. Science. 2005;309:1200–5.
Nishikawa T, Ouchi M. Recent development in polymer reactions for overcoming synthetic limitations in chain-growth polymerization. Chem Lett. 2021;50:411–7.
Batz HG, Franzmann G, Ringsdorf H. Model reactions for synthesis of pharmacologically active polymers by way of monomeric and polymeric reactive esters. Angew Chem Int Ed. 1972;11:1103–4.
Ferruti P, Bettelli A, Feré A. High polymers of acrylic and methacrylic esters of N-hydroxysuccinimide as polyacrylamide and polymethacrylamide precursors. Polymer. 1972;13:462–4.
Eberhardt M, Mruk R, Zentel R, Théato P. Synthesis of pentafluorophenyl(meth)acrylate polymers: new precursor polymers for the synthesis of multifunctional materials. Eur Polym J. 2005;41:1569–75.
Eberhardt M, Théato P. RAFT polymerization of pentafluorophenyl methacrylate: preparation of reactive linear diblock copolymers. Macromol Rapid Commun. 2005;26:1488–93.
Sandford C, Aggarwal VK. Stereospecific functionalizations and transformations of secondary and tertiary boronic esters. Chem Commun. 2017;53:5481–94.
Brown HC, Jadhav PK, Mandal AK. Asymmetric syntheses via chiral organoborane reagents. Tetrahedron. 1981;37:3547–87.
Brown HC, Zweifel G. Hydroboration as a convenient procedure for the asymmetric synthesis of alcohols of high optical purity. J Am Chem Soc. 1961;83:486–7.
Miyaura N, Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem Rev. 1995;95:2457–83.
Edelstein EK, Grote AC, Palkowitz MD, Morken JP. A protocol for direct stereospecific amination of primary, secondary, and tertiary alkylboronic esters. Synlett. 2018;29:1749–52.
Rasappan R, Aggarwal VK. Synthesis of hydroxyphthioceranic acid using a traceless lithiation–borylation–protodeboronation strategy. Nat Chem. 2014;6:810–4.
Brooks WLA, Sumerlin BS. Synthesis and applications of boronic acid-containing polymers: from materials to medicine. Chem Rev. 2016;116:1375–97.
Kitano S, Koyama Y, Kataoka K, Okano T, Sakurai Y. Novel drug delivery system utilizing a glucose responsive polymer complex between poly(vinyl alcohol) and poly(N-vinyl-2-pyrrolidone) with a phenylboronic acid moiety. J Control Release. 1992;19:161–70.
He C, Pan X. MIDA boronate stabilized polymers as a versatile platform for organoboron and functionalized polymers. Macromolecules. 2020;53:3700–8.
Vancoillie G, Hoogenboom R. Synthesis and polymerization of boronic acid containing monomers. Polym Chem. 2016;7:5484–95.
Kato S, Kimura K, Nagata K, Tsuzuki Y. Organoboron compounds. XI. The polymerization of vinylboronic acid. Bull Chem Soc Jpn. 1966;39:2018–20.
Mulvaney JE, Ottaviani RA, Laverty JJ. Preparation of vinyl boronate copolymers and reactions. J Polym Sci Polym Chem Ed. 1982;20:1949–52.
Wan W-M, Baggett AW, Cheng F, Lin H, Liu S-Y, Jakle F, et al. Synthesis by free radical polymerization and properties of BN-polystyrene and BN-poly(vinylbiphenyl). Chem Commun. 2016;52:13616–9.
Thiedemann B, Gliese PJ, Hoffmann J, Lawrence PG, Sönnichsen FD, Staubitz AA. High molecular weight poly(N-methyl -B-vinylazaborine) - a semi-inorganic B-N polystyrene analogue. Chem Commun. 2017;53:7258–61.
van de Wouw HL, Lee JY, Klausen RS. Gram-scale free radical polymerization of an azaborine vinyl monomer. Chem Commun. 2017;53:7262–5.
van de Wouw HL, Lee JY, Awuyah EC, Klausen RS. A BN aromatic ring strategy for tunable hydroxy content in polystyrene. Angew Chem Int Ed. 2018;57:1673–7.
Nishikawa T, Ouchi M. An alkenyl boronate as a monomer for radical polymerizations: boron as a guide for chain growth and as a replaceable side chain for post-polymerization transformation. Angew Chem Int Ed. 2019;58:12435–9.
Nishino T, Kitamura N, Murotani K. High-pressure-synthesis of poly(isopropenyl alcohol) and its biocompatibilities. J Polym Sci Part A Polym Chem. 2009;47:754–61.
Makino H, Nishikawa T, Ouchi M. Elucidating monomer character of an alkenyl boronate through radical copolymerization leads to copolymer synthesis beyond the limitation of copolymerizability by side-chain replacement. ACS Macro Lett. 2020;9:788–93.
Fischer H, Radom L. Controlling the addition of carbon-centered radicals to alkenes- an experimental and theoretical perspective. Angew Chem Int Ed. 2001;40:1340–71.
Mishima E, Yamago S. Controlled random and alternatingcopolymerization of (meth)acrylates, acrylonitrile, and (meth)- acrylamides with vinyl ethers by organotellurium-, organostibine-, and organobismuthine-mediated living radical polymerization reactions. J Polym Sci Part A Polym Chem. 2012;50:2254–64.
Kamigaito M, Ando T, Sawamoto M. Metal-catalyzed living radical polymerization. Chem Rev. 2001;101:3689–746.
Ouchi M, Terashima T, Sawamoto M. Transition metal-catalyzed living radical polymerization: toward perfection in catalysis and precision polymer synthesis. Chem Rev. 2009;109:4963–5050.
Matyjaszewski K, Xia JH. Atom transfer radical polymerization. Chem Rev. 2001;101:2921–90.
Chiefari J, Chong YKB, Ercole F, Krstina J, Jeffery J, Le TP, et al. Living free-radical polymerization by reversibleaddition-fragmentation chain transfer: the RAFT process. Macromolecules. 1998;31:5559–62.
Moad G, Rizzardo E, Thang SH. Radical addition—fragmentation chemistry in polymer synthesis. Polymer. 2008;49:1079–131.
Keddie DJ, Moad G, Rizzardo E, Thang SH. RAFT agent design and synthesis. Macromolecules. 2012;45:5321–42.
Kanazawa T, Nishikawa T, Ouchi M. RAFT polymerization of isopropenyl boronate pinacol ester and subsequent terminal olefination: precise synthesis of poly(alkenyl boronate)s and evaluation of their thermal properties. Polym J. 2021;53:1167–74.
Soeriyadi AH, Boyer C, Burns J, Becer CR, Whittaker MR, Haddleton DM, et al. High fidelity vinyl terminated polymers by combining RAFT and cobaltcatalytic chain transfer (CCT) polymerization methods. Chem Commun. 2010;46:6338–640.
Crudden CM, Ziebenhaus C, Rygus JPG, Ghozati K, Unsworth PJ, Nambo M, et al. Iterative protecting group-free cross-coupling leading to chiral multiply arylated structures. Nat Commun. 2016;7:11065.
Kanazawa T, Nishikawa T, Ouchi M. Orthogonal C–B bond transformation as an approach for versatile synthesis of end-functionalized polymers. ACS Macro Lett. 2022;11:706–10.
Makino H, Nishikawa T, Ouchi M. Vinylboronic acid pinacol ester as a vinyl alcohol-precursor monomer in radical copolymerizations with styrene. Chem Commun. 2021;57:7410–3.
Keyes A, Basbug Alhan HE, Ordonez E, Ha U, Beezer DB, Dau H, et al. Olefins and vinyl polar monomers: bridging the gap for next generation materials. Angew Chem Int Ed. 2019;58:12370–91.
Ittel SD, Johnson LK, Brookhart M. Late-metal catalysts for ethylene homo- and copolymerization. Chem Rev. 2000;100:1169–204.
Nakamura A, Ito S, Nozaki K. Coordination−insertion copolymerization of fundamental polar monomers. Chem Rev. 2009;109:5215–44.
Nakamura A, Anselment TMJ, Claverie J, Goodall B, Jordan RF, Mecking S, et al. Ortho-phosphinobenzenesulfonate: a superb ligand for palladium-catalyzed coordination–insertion copolymerization of polar vinyl monomers. Acc Chem Res. 2013;46:1438–49.
Liu S, Elyashiv S, Sen A. Copper-mediated controlled copolymerization of methyl acrylate with 1-alkenes under mild conditions. J Am Chem Soc. 2001;123:12738–9.
Liu S, Sen AJ. Living/controlled copolymerization of acrylates with nonactivated alkenes. Polym Sci Part A Polym Chem. 2004;42:6175–92.
Buback M, Dietzsch H. High-pressure free-radical copolymerization of ethene and methyl methacrylate. Macromol Chem Phys. 2001;202:1173–81.
Ihara H, Koyanagi M, Suginome M. Anthranilamide: a simple, removable ortho-directing modifier for arylboronic acids serving also as a protecting group in cross-coupling reactions. Org Lett. 2011;13:2662–5.
Yamamoto T, Ishibashi A, Koyanagi M, Ihara H, Eichenauer N, Suginome M. C–H activation-based transformation of naphthalenes to 3-Iodo-2-naphthylboronic acid derivatives for use in iterative coupling synthesis of helical oligo(naphthalene-2,3-diyl)s. Bull Chem Soc Jpn. 2017;90:604–6.
Suzuki H, Nishikawa T, Makino H, Ouchi M. Anthranilamide-protected vinyl boronic acid: rational monomer design for improved polymerization/transformation ability providing access to conventionally inaccessible copolymers. Chem Sci. 2022;13:12703–12.
Chapman R, Melodia D, Qu JB, Stenzel MH. Controlled poly(olefin)s via decarboxylation of poly(acrylic acid). Polym Chem. 2017;8:6636–43.
Frech S, Molle E, Hub C, Theato P. Decarboxylation of poly[N-(acryloyloxy)phthalimide] as a versatile tool for postpolymerization modification. Macromol Rapid Commun. 2022;43:2200068.
Frech S, Molle E, Butzelaar AJ, Theato P. Ethylene-free synthesis of polyethylene copolymers and block copolymers. Macromolecules. 2021;54:9937–46.
Garrison JB, Hughes RW, Young JB, Sumerlin BS. Photoinduced SET to access olefin-acrylate copolymers. Polym Chem. 2022;13:982–8.
Acknowledgements
The author sincerely thanks Prof. Makoto Ouchi for his kind advice.
Funding
This work was partially supported by JSPS [KAKENHI grants 18H05975, 19K15622, and 22K14724], the Foundation for the Promotion of Ion Engineering, the Mazda Foundation, the Kyoto Technoscience Center, the Masuya Memorial Foundation of the Promotion of Basic Research, the Hattori Hokokai Foundation, and the Tokuyama Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nishikawa, T. Radical polymerization of alkenyl boronates and C–B bond transformation: polymer synthesis through side-chain replacement for overcoming synthetic limitations. Polym J (2024). https://doi.org/10.1038/s41428-024-00935-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41428-024-00935-4
