
Abstract
Due to the lack of safe carriers for the delivery of small interfering RNA (siRNA), clinical applications of nucleotide-based therapeutics have been limited. In this study, biodegradable amphiphilic triblock copolymers with tailored molecular weights for each block composed of methoxy poly(ethylene glycol) (2000 g/mol), poly(L-lysine) (1300 g/mol) and poly(D,L-lactic acid) (1800 g/mol) (mPEG45-α-PLL10-PLA25) were synthesized and fully characterized. The peptide synthesis was carried out on a solid phase to limit the presence of cationic charges. The arrangement and availability of cationic amino groups within a micellar vector were investigated to determine the colloidal stability as well as the predisposition of these systems to vectorize siRNAs in addition to their already known ability to improve the solubility of hydrophobic compounds. For this purpose, a triblock copolymer containing an epsilon poly(L-lysine) was synthesized similarly. Accordingly, the arrangement of the cationic segment modifies the rigidity involving a complexation constraint due to limited cationic charges available on the surface, which can compromise the efficiency of delivery into cells. In addition, the two vectors were biocompatible in different human cell lines.
Similar content being viewed by others

Introduction
The commitment to manufacture low-molecular-weight amphiphilic block copolymers with precisely controlled architecture for assembly into nanoscale vectors has driven remarkable progress in the field of nanomedicine research and development. This holds especially true for the synthesis of PEGylated block copolymers, which can be formulated into polymeric micelles and appear to be the most advanced nanoparticulate systems for clinical trials, with particular strength in solubilizing hydrophobic anticancer drugs [1,2,3]. At the forefront, Genexol-polymeric micelles (PM), a paclitaxel-encapsulated formulation, are based on a combination of synthetic polymers, including poly(D,L-lactic acid) (PLA) of a molecular weight (Mw) of 1750 g/mol and methoxy poly(ethylene glycol) (mPEG), Mw 2000 g/mol [4]. This Cremophor® EL-free delivery system has been approved as a first-line therapy for recurrent and metastatic breast cancer and non-small-cell lung cancer (NSCLC) in South Korea. With a higher maximum tolerated dose than standard paclitaxel, Genexol-PM exhibited favorable clinical efficacy with well-tolerated toxicities [5,6,7].
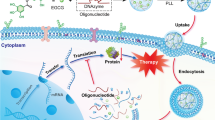
Regarding treatments of advanced solid tumors, a promising step forward may be achieved by a combination of therapeutic strategies involving a different mechanism of action, such as RNAi, to cooperatively impede tumor growth. Considerations for the design of polymeric micelles [8] as well as choices of supporting materials [9] suitable for the delivery of siRNAs have been recently described (Marquet and Borchard, submitted), highlighting potential systems with specific core-shell structures used for the delivery of anticancer drugs and siRNAs.
Poly(L-lysine) (PLL) has been known to effectively condense DNA since the mid-1960s [10]. Accordingly, PLL is one of the first and most common polymers used as a transfection agent and possesses an acceptable degree of biodegradability, low immunogenicity and relative safety according to the number of cationic charges [11]. Indeed, nonviral transfection efficacy is known to be associated with toxicity, especially if the system is based on polycations with high molecular weight [12,13,14], which ultimately limits their clinical application.
Nevertheless, PEGylated catiomers containing short segments of PLL generate high interest [15]. PLL has been widely used as an oligopeptide in the preparation of triblock copolymeric carriers [16,17,18,19,20]. Usually, PLL synthesis is performed by the ring-opening polymerization (ROP) of α-amino acid N-carboxyanhydrides [21]. In this study, we aimed to develop a novel synthetic route to intercalate a 10-mer of lysine into an mPEG-PLA (2000–1800 g/mol) polymeric system. We have previously established in silico that the use of 10-mer oligocations can enable the stable complexation of siRNA [22]. Molecular dynamics simulations showed that the optimal length for the cationic part is given in the presence of 10 lysines. Therefore, to control the number of amino groups and at the same time reduce cytotoxicity due to superfluous positive charges, the synthesis of a 10-mer lysine was carried out using solid-phase peptide synthesis. This opens the possibility of a fascinating synthetic procedure for the incorporation of rationally designed peptides into amphiphilic mPEG-PLA copolymers.
Herein, we report for the first time the synthesis of two tailored triblock copolymers based on poly(L-lysine) with different orientations of lysine in either the alpha or epsilon position. These materials were then formulated into polymeric micelles and compared in terms of size, charge, molecular mass and biocompatibility against different human cell lines.
Materials and methods
Materials
2-Chlorotrityl chloride resin (100–200 mesh, 1% DVB, 1.0–1.6 mmol Cl/g) was obtained from Abcr Swiss AG (Zug, Switzerland). Oxyma pure and Fmoc-L-Lys-(Boc)-OH were purchased from Iris Biotech GmbH (Marktredwitz, Germany). N,N,N′,N′-Tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) was provided by Fluorochem (Hadfield, UK) and dimethylformamide (DMF) was obtained from VWR (Leuven, Belgium). Acetone was supplied by Thommen-Furler (Büren, Switzerland). Alpha-cyano-4-hydroxycinnamic acid (CHCA), dichloromethane (DCM), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluoro-phosphate (HATU), N,N-diisopropylethylamine (DIPEA), 1-methyl-2-pyrrolidinone (NMP), 3,6-dimethyl-1,4-dioxane-2,5-dione (D,L-lactide), 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU), formic acid (FA) 98–100%, methoxy poly(ethylene glycol) (mPEG) (Mn 2000) and Nile red (Nile blue A oxazone), minimum essential Eagle medium with Earle’s salts, L-glutamine and sodium bicarbonate (MEM) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Piperidine, potassium hydroxide 85% (KOH), toluene 99.8%, extra dry, over molecular sieves, triethylamine 99% pure, and trifluoroacetic acid (TFA) were purchased from Acros Organics (Geel, Belgium). Chlorobenzene 99% (PhCl) was purchased from Alfa Aesar (Karlsruhe, Germany). Acetonitrile HPLC grade (ACN), diethylether, tetrahydrofuran HPLC grade (THF) and methanol (MeOH) were purchased from Fisher Chemicals (Loughborough, UK). Sodium chloride (NaCl) for analysis was obtained from Applichem GmbH (Darmstadt, Germany). Methoxy polyethylene glycol amine (mPEG-NH2, Mn 2000) was provided by Creative PEG Work (Chapel Hill, NC, USA). Boc-protected epsilon-poly(L-lysine) 10-mer was obtained from ChinaPeptides (Shanghai, China). Deuterated dimethylsulfoxide 99.9% (DMSO-d6) was purchased from Cambridge Isotope Laboratories (Andover, MA, USA). Human lung carcinoma (A549, ATCC CCL-185) cells, human mammary gland adenocarcinoma (MCF-7, ATCC HTB-22) and human prostate cancer (PC-3, ATCC CRL-1435) cells were distributed by LGC Standards (Molsheim, France). Noncancerous primary fibroblasts (007-F) were collected from the interior leg skin of an 86-year-old male at the University Hospital Lausanne (CHUV) with patient consent. The regulations were approved by the Legal Affairs Unit (CHUV), reviewed and approved on 14.07.2016 by the Operational Center for Biobanks and Registers of the Hospital. Ham’s F-12 nutrient mixture Kaighn’s modification (F-12K) with L-glutamine, Dulbecco’s modified Eagle’s medium (DMEM) with 4.5 g/l D-glucose, L-glutamine, Dulbecco’s phosphate-buffered saline (DPBS) without calcium and magnesium, nonessential amino acid solution (100x), sodium pyruvate (100 mM), 0.25% trypsin-EDTA (5x), fetal bovine serum (FBS), penicillin 100 UI/ml and streptomycin 100 µg/ml (Pen-Strep) were provided by Gibco™ Life Technologies (Thermo Fisher Scientific Inc., Loughborough, UK). The WST-1 proliferation assay was obtained from Roche (Mannheim, Germany). SDS solution 10% (w/v) was purchased from Bio-Rad Laboratories, Inc. (Hercules, CA, USA). All chemicals were used without further purification unless mentioned.
The general synthetic procedure to obtain triblock copolymers (consisting of four different steps described below) is summarized in Fig. 1.
Synthesis of Boc-protected α-poly(L-lysine)
Dry 2-chlorotrityl chloride resin (100–200 mesh) (220 mg, 0.7 mmol, 1.0 eq) was swelled in DCM (5 ml) for 10 min in a glass funnel. The resin was filtered, and a solution of Nα-Fmoc-Nε-Boc-L-lysine (235 mg, 0.5 mmol, 2.0 eq) and DIPEA (174 µl, 1.0 mmol, 4.0 eq) was dissolved in 2 ml of DCM:DMF (1:1), shaken at room temperature (RT) for 1 h to attach the first amino acid on the resin and subsequently filtered. After washing the resin with DMF (3 × 2 min with 3 ml) and DCM (3 × 2 min with 3 ml), excess 2-chlorotrityl groups were capped with methanol by adding 10 ml of DCM:MeOH:DIPEA (16:3:1). The reaction was allowed to proceed for 10 min under shaking before the resin was drained. The resin was washed several times with DMF and DCM and subsequently vacuum-dried over KOH pellets.
An automated solid-phase peptide synthesis of poly(Nε-tert-butyloxycarbonyl-L-lysine) (9 units) was performed with a Liberty microwave peptide synthesizer (CEM, Kamp-Lintfort, Germany) to form a 10-mer polylysine with final Fmoc deprotection.
For coupling, Nα-Fmoc-Nε-Boc-L-lysine (2.44 g, 5.2 mmol, 5.0 eq relative to resin) was activated with 0.5 M Oxyma/HBTU in DMF (4.0 eq relative to Nα-Fmoc-Nε-Boc-L-lysine) and 2 M DIPEA in NMP (8.0 eq relative to Nα-Fmoc-Nε-Boc-L-lysine) for 20 min. For the removal of N-terminal Fmoc groups, treatment with 20% piperidine in NMP was performed for 10 min.
The linear, side-chain-protected peptide was cleaved from the resin using 20 ml of 0.8% TFA in DCM (4 × 3 min with 5 ml) while bubbling nitrogen through the suspension in a glass vessel. The TFA solution was filtered off from the resin into a flask and neutralized to pH 7 by adding a few drops of triethylamine. The filtrate was concentrated on a rotary evaporator (Rotavap, Büchi, Flawil, Switzerland) to remove TFA and DCM until 2 ml remained. Then, the peptide was precipitated in ice-cold water (50 ml) and lyophilized, yielding 180 mg of peptide (7.4%) as a white powder.
Boc-protected epsilon-poly(L-lysine) 10-mer was obtained by solid-phase peptide synthesis from ChinaPeptides (Shanghai, China).
Synthesis of Boc-protected mPEG-b-PLL copolymer (generic pathway)
PEGylation of polylysine was performed according to a published procedure [23, 24]. Briefly, Boc-protected α-poly(L-lysine) (poly(Nε-tert-butyloxycarbonyl-L-lysine)) (115 mg; 0.05 mmol, 1.0 eq) and HATU (20.91 mg, 0.055 mmol, 1.1 eq) were dissolved in 2 ml of DMF for 15 min. Then, mPEG-NH2 (120 mg, 0.06 mmol, 1.2 eq) was added to the reaction flask with DIPEA (43.5 µl, 0.25 mmol, 5.0 eq) and stirred at RT for 24 h. The copolymer was precipitated 3 times in ice-cold diethylether under vigorous stirring. The excess mPEG-NH2 was removed by dialysis against Milli-Q water for 72 h using a Spectra/Por 7 regenerated cellulose membrane with a 1 kDa cutoff Spectra/Por 7, SpectrumLabs (Repligen, Breda, The Netherlands). The copolymer was then lyophilized, yielding 180 mg (76.6% yield) of mPEG-α-PLL (Boc) as a white powder.
The same procedure was carried out using Boc-protected epsilon-poly(L-lysine) 10-mer and yielded 160 mg (68% yield) of mPEG-ε-PLL (Boc).
Synthesis of Boc-protected mPEG-b-PLL-b-PLA triblock copolymer (generic pathway synthesis)
Ring-opening polymerization (ROP) of D,L-lactide initiated by the mPEG-PLL macroinitiator was performed using DBU as the catalyst [25]. mPEG-PLL (131.84 mg, 0.03 mmol, 1.0 eq) and D,L-lactide (91.90 mg, 0.64 mmol, 20.81 eq) were added to the reaction flask and lyophilized for 24 h. The reaction flask was then dried overnight under vacuum and conditioned under an argon atmosphere before starting the reaction. DBU (4.7 µl, 0.03 mmol, 1 eq) dissolved in 1 ml of PhCl was added to the flask, and the reaction ran for 6 h at 90 °C. The triblock copolymer was precipitated three times in ice-cold diethylether, purified by dialysis for 36 h, and then lyophilized for 48 h, yielding 140 mg (63% yield) of mPEG-α-PLL(Boc)-PLA as a white powder.
Ninety milligrams (40%) of mPEG-ε-PLL(Boc)-PLA was obtained using the same procedure.
Synthesis of mPEG-b-PLL-b-PLA
For Boc deprotection, mPEG-α-PLL(boc)-PLA (120 mg, 0.02 mmol) was dissolved in 1 ml of DCM and conditioned in a reaction flask under nitrogen. Then, 1 ml of TFA was injected into the flask. The mixture was stirred at RT for 20 min. After completion of the reaction, the solution was neutralized with a few drops of triethylamine and co-evaporated 3 times with MeOH, resolubilized in DMF, dialyzed for 48 h and lyophilized, yielding 90 mg of mPEG-α-PLL-PLA (75% yield) as a white powder.
Fifty-seven milligrams (71%) of mPEG-ε-PLL-PLA was obtained as the final product.
Polymer characterization
Chemical structures and reactions were drawn using ChemDraw Professional version 15.1. (PerkinElmer, Inc., Waltham, MA, USA). The polymerization conversions, molecular weights of each block and purity were determined by 1H NMR analysis. The 1H NMR spectra (16 scans) and 13C NMR spectra (2048 scans) were recorded in DMSO-d6 at 298 K with a Bruker Avance III HD NMR spectrometer equipped with a SampleJet automated sample changer (Bruker, BioSpin, Rheinstetten, Germany). Recorded NMR spectra were treated with MestReNova (Mnova) software V10.0 (Mestrelab, Santiago de Compostela, Spain). All chemical shifts were reported as parts per million (ppm).
Selective coupling between mPEG-NH2 and Boc-protected poly(L-lysine) 10-mer was controlled by a high-performance liquid chromatograph (HPLC) coupled to an evaporative light scattering detector (ELSD). Samples were prepared at a concentration of 5 mg/ml in ACN:H2O (3:1) and 0.1% FA and separated on a C18 column (250 × 4.6 mm, 10 μm) (Interchim, Montluçon, France) mounted on a 1260 Infinity Binary HPLC (Agilent Technologies, Santa Clara, CA, USA) coupled to a Sedex 85 ELSD (Sedere, Alfortville, France) at a flow rate of 1 ml/min. A gradient mode over 1 h starting with 5% (MeOH, 0.1% FA), 95% (H2O, 0.1% FA) to 100% (MeOH, 0.1% FA) was used.
Molecular weights were determined by a MALDI-TOF MS Axima CFR+ (Shimadzu, Kyoto, Japan) operating in linear mode. Samples were prepared in ACN:H2O, 0.1% TFA (1:1, v/v) at a concentration of 5 mg/ml. The CHCA matrix (10 mg/ml) and samples (5 mg/ml) were prepared in ACN:H2O, 0.1% TFA (1:1). A series of dilutions were carried out to obtain final concentrations of 0.5 and 0.05 mg/ml. Then, 0.5 µl of the matrix was deposited onto a stainless steel MALDI target and allowed to air dry, followed by 1 µl of sample and 0.5 µl of matrix. Spectra were obtained in the positive ion mode.
Molecular weights and polydispersity (PDI) were determined by gel permeation chromatography (GPC) on a Shimadzu chromatographer (Kyoto, Japan) mounted with three Styragel HR 1, 3, 4 columns (300 × 7.8 mm, 5 μm) (Waters, Dublin, Ireland) and connected to a Shimadzu differential refractometer. THF was the continuous phase at a flow rate of 1 ml/min at 40 °C, and polystyrenes of known molecular weights, 1050, 1310, 1990, 2970, 3950, 4490, 5030, 6930, and 10700 g/mol, were used as calibration standards (Shimadzu Schweiz GmbH, Reinach, Basel).
Preparation of polymeric micelles
The micelles were prepared by a solvent-evaporation technique adapted from [26]. The purified triblock copolymer (4 mg) was dissolved in 400 µl (acetone:MeOH) (1:1). The solution was added dropwise into 1 ml of Milli-Q water (prefiltered at 0.1 µm) and alternatively sonicated for 5 s pulsation at a 10% amplitude for 3 min using a digital sonicator (Branson, Danbury, CT, USA). The organic solvents were removed under reduced pressure to reach a typical micelle concentration of 4 mg/ml.
Critical micellar concentration (CMC) determination
Fluorescence measurements were performed with Nile red to determine the CMC. Different dilutions were prepared from a 4 mg/ml stock solution of micelles to obtain samples of concentrations ranging from 0.001 to 1 mg/ml. Fluorescence measurements were performed using a Synergy Mx microplate reader (BioTeK, Winooski, VT, USA) at an excitation wavelength of 550 nm, as previously described by Trimaille et al. [26]. Emission spectra were recorded from 560 to 750 nm. Methoxy poly(ethylene glycol) 2000 Da was used as a negative control.
Dynamic (DLS) and static light scattering (SLS) and zeta potential
The DLS measurements were performed using a Zeta-sizer Nano (Malvern, Worcestershire, UK). The scattered light of a 4 mW (max. output power) HeNe linear polarized laser (633 nm) was measured at a backscatter detection angle of 173°. The samples were maintained at 25 ± 0.2 °C after an equilibration counting time of 60 s using disposable polystyrene cuvettes.
Samples were prepared in Milli-Q water at a concentration of 0.4 mg/ml (dilution factor 10x) and filtered through a 0.22 µm pore size membrane prior to loading into the cuvette, as recommended by NIST-NCL [27]. Three samples were analyzed in three measurements. The hydrodynamic diameter (dH) of the polymeric micelles was calculated from the translational diffusion coefficient using the Stokes-Einstein Eq. (1):
where k is Boltzmann’s constant, T is the absolute temperature, η is the viscosity (0.89 mPaּs) and D is the translational diffusion coefficient of the polymeric micelles.
SLS measurements were carried out at a scattering angle of 173°. The intensity of scattered light (KC/Rϴ) at various concentrations (0.1, 0.2, 0.25. 0.5, and 1 mg/ml) was compared to the scattering produced from toluene used as a standard. The molecular weight (Mw) of the triblock copolymer micelles was estimated by Eqs. (2) and (3):
where K is the optical constant as defined in (3), \(R_\theta\) is the ratio of scattered light to incident light of the sample, \(A_2\) is the second virial coefficient, and the polymer concentration c is given in g/ml. The differential refractive increment (∂η/∂c) was determined to be 0.099 ml/g at ambient temperature by measuring the polymer solution in Milli-Q water at different precisely prepared concentrations.
The average density (ρ) was estimated by Eq. (4) as:
where NA is Avogadro’s number.
Zeta potential (ZP) measurement was based on laser Doppler electrophoresis using the Smoluchowski approximation. Electrophoretic determinations of ZP were performed at 0.4 mg/ml and a moderate electrolyte concentration of 1 mM NaCl. Replicates were composed of 10–100 measurements.
Transmission electron microscopy (TEM) dry stage
Samples were prepared in Milli-Q water (0.1 µm) at a concentration of 0.4 mg/ml (dilution factor 10x) and filtered through a 0.22 µm pore size hydrophilic PVDF membrane. Three microliters of the sample solution were deposited onto a hexagonal 300 mesh copper-coated grid and dried in a desiccator for 48 h at RT before analysis. Images were taken at a high voltage of 80 kV using a Tecnai G2 12 transmission electron microscope (FEI Company, Eindhoven, The Netherlands). Particle size and morphology were analyzed from TEM images by an iTEM soft imaging system (Olympus Soft Imaging Solutions GmbH, Münster, Germany).
Cell proliferation assay
The biocompatibility of triblock copolymers was evaluated by a WST-1-based colorimetric assay for the quantification of cell proliferation and viability. Prostate cancer cells (PC-3) and primary fibroblasts (007-F) were seeded in DMEM on 96-well plates at initial seeding densities of 10,000 and 20,000 cells/well, respectively. Breast adenocarcinoma cells (MCF-7) were seeded in MEM at a density of 20,000 cells/well. Lung carcinoma cells (A549) were seeded in F-12K cell culture medium at an initial seeding density of 15,000 cells/well. Cell incubation was performed at 37 °C in an atmosphere containing 5% CO2. The total concentration of living cells was determined with a Countess II FL (Life Technology, Singapore). After 24 h, cells were treated with synthesized copolymers at concentrations ranging from 0.01 to 1000 µg/ml and incubated for 24 h. In addition, primary fibroblasts were seeded for 48 h to obtain an appropriate confluence for this assay. SDS 0.02% was used as a positive control for cytotoxicity. Cell viability was determined by normalization to untreated cells, which were used as a negative control. A directly soluble tetrazolium reagent (WST-1) was incubated. Absorbance was measured at two different wavelengths (450 and 690 nm) with a microplate reader (BioTeK, Winooski, VT, USA).
Results and discussion
Synthesis of mPEG-b-PLL-b-PLA
Herein, an mPEG-b-α-PLL-b-PLA triblock copolymer was synthesized according to the protocol shown (Fig. 1).

Synthesis of mPEG45-b-PLL10-b-PLA25 (Resin: 2-chlorotrityl chloride)
The structure, purity, molecular weight and selective coupling of mPEG to 10-mer lysine to form mPEG-b-α-PLL-b-PLA were confirmed by 1H NMR, 13C NMR spectroscopy, HPLC-ELSD, MALDI-TOF mass spectrometry and GPC chromatography (Fig. 2, Table 1, Fig. S1, Fig. S2, Fig. S3 and Fig. S4).

1H NMR spectra (600 MHz) in DMSO-d6 of (A) Boc protected α-PLL10 peptide, (B) Boc-protected mPEG45-b-α-PLL10, (C) Boc-protected mPEG45-b-α-PLL10-b-PLA25, (D) mPEG45-b-α-PLL10-b-PLA25
The integration of protons by NMR confirms the appropriate length of each block. Similarly, a triblock copolymer containing 10 lysines in epsilon orientation (mPEG-b-ε-PLL-b-PLA) was synthesized. The final product was confirmed by 1H NMR, 13C NMR spectroscopy and GPC (Fig. 3, Table 1 and Fig. S5), which all showed the same chain length of each block and high purity. The main intention of creating a triblock based on an epsilon structure was primarily to confirm the opening of the D,L-lactide ring by the primary amine to ensure the repeatability of the polymerization initiated by a macroinitiator.

(A) 1H NMR spectrum (600 MHz) in DMSO-d6 of mPEG45-b-ε-PLL10-b-PLA25 and (B) 13C NMR spectrum (150 MHz) in DMSO-d6 of mPEG45-b-ε-PLL10-b-PLA25
The degree of polymerization (DP) of the PLL block was calculated to be 10 based on the peak intensity ratios of the methoxy protons present in the NMR spectra: peak a, mPEG (CH3-O-CH2-CH2-, δ = 3.24 ppm); peak b, characteristic of aliphatic –CH2— from mPEG present at 3.51 ppm; peaks c, e and g, the α, γ and ε of the methylene protons of the lysine (-CH-CH2-CH2-CH2-CH2-), represented by (c, δ = 4.19 ppm and 3.80 ppm respectively, e, δ = 1.25-1.27 ppm and g, δ = 2.85-2.86 and 2.97 ppm respectively) (Fig. 2 and Fig. 3A). Integrals corresponding to peaks c, e and g were used to calculate the DP of the PLL block due to the mismatch in integration for peaks d and f. The degree of polymerization of the PLA block was calculated to be 25 based on the peak intensity ratios of the methoxy protons a of mPEG to the methyl group l of PLA (CH-CH3, δ = 1.46 ppm). Peaks assignment was confirmed by 13C NMR (Fig. S1 and Fig 3B). For both triblock copolymers, the conversion rate of D,L-lactide to poly(D,L-lactic acid) (PLA conv.) was determined to be >80%. The results of polymerization are summarized in Table 1.
To the best of our knowledge, the triblock copolymer mPEG-b-ε-PLL-b-PLA with an epsilon orientation of lysine has never been produced before. In addition, this polymer may have interesting antibacterial properties arising from the presence of ε-PLL [28,29,30], which are worth further evaluation. The micellar formulations of these polymeric systems were also investigated for potential anticancer treatment.
Micellar formation and characterization
Nile red fluorescent probe was incorporated into the PLA segment of the amphiphilic triblock copolymers to characterize the critical concentration of micellar formation through a high-sensitivity technique. In fact, CMC is one of the most important parameters for micelle formation. The increase in polymer concentration results in an increase in fluorescence intensity (Fig. 4A). The CMC value can be seen as the onset of the fluorescence intensity increase, as previously mentioned by Coutinho et al. [31]. Accordingly, Fig. 4B shows a hypsochromic shift of the emission maxima with increasing concentrations of amphiphilic triblock copolymers.

Plot of the fluorescence emission intensity (A) of Nile red vs. copolymer concentration for mPEG45-b-ε-PLL10-b-PLA25 (○) and mPEG45-b-α-PLL10-b-PLA25 (▪) (B) Maximum emission wavelength of Nile red vs. copolymer concentration, mean ± SD; n = 3
The fluorescence of this probe is strongly quenched by the polarity of its immediate environment, as Nile red forms aggregates in aqueous media that can be prevented by encapsulation in a hydrophobic space [32]. The CMC was determined at the inflection point of the plot representing the maximum emission wavelength as a function of the copolymer concentration (Fig. 4B). The linear arrangement of ε-poly(L-lysine) results in a twofold lower CMC, or 0.05 mg/ml compared to 0.1 mg/ml. This can be explained by the influence of the orientation of the polycationic segment on the number of amphiphilic polymers per micelle. An epsilon orientation leads to stronger fluorescence at a lower concentration due to a better loading capacity of hydrophobic compounds. This decrease in physical stress suggests a hardcore phenomenon favoring the rigidity of the polymeric core given by an increase in the density of biomaterials. The polymeric micelles obtained were thoroughly characterized.
The morphology and size of polymeric micelles were investigated by TEM imaging (Fig. 5).

TEM images of (A) mPEG45-b-α-PLL10-b-PLA25 and (B) mPEG45-b-ε-PLL10-b-PLA25
For both formulations, amphiphilic triblock copolymers were self-assembled into isolated micelles, spherical in shape with a particle size of ~60–65 nm in diameter (nparticle = 60). More prolate deformations were observed to occur in the drying step of the samples, as shown by the ellipsoid micelle shape (Fig. 5A). In comparison, dense core polymeric micelles formed mostly flattened spheroids. The results of size, zeta potential and molecular weight measured by scattered light are summarized in Table 2.
The mean size determined by DLS with the advantage of measuring millions of polymeric micelles was in the nanoscale range (82 and 90 nm), as defined as a size threshold of 1–100 nm [33]. PDI values below 0.13 are perfectly acceptable, suggesting a nearly monomodal distribution of the micelles. Moreover, TEM observations (Fig. 5) agreed with the DLS results (Fig. S6) summarized in Table 2. As TEM size measurement is a number-based technique, the numerically estimated size distribution corroborated the values determined by TEM images. In contrast, there remained a great discrepancy regarding the molecular weight determined by static light scattering (Fig. 6). These results were used to estimate the densities of polymeric micelles at RT, according to Eq. (3). Although the number of cationic charges can be tightly controlled by the use of solid-phase peptide synthesis, the architectural organization of the cationic segment impacts self-assembly by creating micelles with different flexible peripheries. An epsilon orientation therefore promotes better colloidal stability by a lower CMC and a reduction in the zeta potential. This reduction in cationic charge density could also represent a constraint for the possible complexation of hydrophilic compounds such as siRNAs. Due to the flexibility of the peripheral region, the alpha micelles show a better deployment of charges, forming a hollow polymeric shell with an increased cationic charge density suitable for the complexation of hydrophilic payloads. The properties of CMC and cationic charge density lead to a dilemma and an open debate.

Debye plot of mPEG45-b-α-PLL10-b-PLA25 micelles (□) and of mPEG45-b-ε-PLL10-b-PLA25 (▲) obtained by SLS. Estimated molecular weights were 4830 ± 27.1 kDa and 31700 ± 1170 kDa respectively
The absence of rigidity has been recognized to play a role in clearance by the immune system, as supported by in vitro and in vivo studies [34, 35]. Greater deformation allows soft nanoparticles (NPs) to be less readily absorbed and filtered, resulting in an increase in blood circulation time. However, the relationship of rigidity to uptake by cancer cells remains disputed and unclear. Therefore, it is still arduous to define biological performance in terms of tumor penetration. Indeed, an elasticity adjustment by freeze-drying increased the Young’s modulus from 165 to 260 kPa for polymeric micelles formed from mPEG-PLA [36]. Stern et al. demonstrated better penetration of tumor spheroids from pancreatic BxPC3 cells with rigid micelles 86 nm in diameter. An elasticity of 260 kPa remains a relatively flexible value compared to much more solid materials such as silica nanoparticles [34, 37, 38]. It is interesting to consider that the anticancer drug delivery systems currently on the market are based on “soft” materials (e.g., liposomes, albumin NPs and polymeric micelles) [34].
Cell viability assay
The WST-1 assay measures the change in activity of a mitochondrial enzyme in cells in vitro during incubation with a test compound. This is interpreted as the effect of the compound on cellular viability. Human lung carcinoma cells (A549) and human breast adenocarcinoma cells (MCF-7) are considered adequate cell line targets for the therapeutic application of micelles. Human prostate adenocarcinoma cells (PC-3) and human primary fibroblasts (007-F) were used herein for secondary assessments of biocompatibility with cancerous and noncancerous cell lines.
No differences in cell viability were detected in the A549, MCF-7, PC-3 and 007-F cell lines in the presence or absence of the polymers (Fig. 7). Mitochondrial activity was above 80% for each polymer solution at concentrations between 0.01 and 1000 µg/ml. This demonstrates that the synthesized copolymers are non-cytotoxic according to the WST-1 assay. Drug delivery assays can be performed in these concentration intervals. A wide concentration range has been used to encompass the biocompatibility of polymers in free form (below CMC) and in self-assembled form (above CMC). A viability above 80% is generally considered to indicate that the test compound is not toxic. By tailoring the number of cationic charges, biocompatible triblock copolymers can be obtained, as micellar formation is always an equilibrium state between assembled and free polymers. Particle properties can be drastically different at a low concentration, leading to aggregates or non-assembled copolymers. Sun et al. revealed by molecular dynamics simulation that the decreased cellular uptake of soft NPs stemmed from their deformation during internalization [39]. Moreover, different binding strengths have been evaluated between polymeric micelles and the plasma membrane [40], showing that fusion to the membrane could lead to cytotoxicity. More investigation regarding safety would be worth considering.

Mitochondrial activity of (A) A549, (B) MCF-7, (C) PC-3 and (D) 007-F cells incubated for 24 h with different concentrations (0.01, 0.1, 1, 10, 100, 1000 µg/ml) of mPEG45-b-ε-PLL10-b-PLA25 (square-patterned) and mPEG45-b-α-PLL10-b-PLA25 (black-patterned), mean ± SD; n = 3
Conclusions
In summary, we designed self-assembled triblock copolymeric micelles. The synthesis of the cationic group in the alpha position was performed on an automated solid-phase peptide synthesizer to allow for better control of cationic charges inside the delivery system. We demonstrated a direct ring-opening polymerization of D,L-lactide initiated by mPEG-PLL as a macroinitiator. Furthermore, the impact of lysine orientation by synthesizing a vector containing lysine in alpha and epsilon orientation was investigated. The orientation of lysine in the alpha position allows better exposure of cationic charges with a higher zeta potential and a lower density of polymers per micelle. We conclude that the structural properties through the geometry of the polycationic part affect the physical stiffness parameter of micelles, which modifies the cationic charge density of these systems as a consequence. By the novelty of the synthetic approach, we provide herein a new toolkit for gene delivery. The choice between these two polymers is an impossible balance and depends on what we wish to further incorporate as a purpose. In this respect, we emphasize that the properties of micelles may be adjusted by using copolymers of alpha, epsilon or a mixture of both. The limited number of cationic charges displayed by the two types of copolymers enables improved safety of the material with no acute cytotoxicity to different human cell lines. By applying different characterization techniques combined with in vitro assays, we addressed issues related to characterization and safety as a strategic approach for mitigating the risks of nanotoxicity. The timelines of cancerous pathologies, the transient nature of RNAi activity, and the unregulated replication of cancer cells involve frequent and permanent changes in the administration of cationic nanomaterials.
References
Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20–37. https://doi.org/10.1038/nrc.2016.108.
Cabral H, Kataoka K. Progress of drug-loaded polymeric micelles into clinical studies. J Control Release. 2014;190:465–76. https://doi.org/10.1016/j.jconrel.2014.06.042.
Ghosh B, Biswas S. Polymeric micelles in cancer therapy: state of the art. J Control Release. 2021;332:127–47. https://doi.org/10.1016/j.jconrel.2021.02.016.
Kim SC, Kim DW, Shim YH, Bang JS, Oh HS, Wan Kim S. et al. In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. J Control Release. 2001;72:191–202. https://doi.org/10.1016/s0168-3659(01)00275-9.
Park IH, Sohn JH, Kim SB, Lee KS, Chung JS, Lee SH. et al. An open-label, randomized, parallel, phase III trial evaluating the efficacy and safety of polymeric micelle-formulated paclitaxel compared to conventional cremophor EL-based paclitaxel for recurrent or metastatic HER2-negative breast cancer. Cancer Res Treat. 2017;49:569–77. https://doi.org/10.4143/crt.2016.289.
Shi M, Gu A, Tu H, Huang C, Wang H, Yu Z. et al. Comparing nanoparticle polymeric micellar paclitaxel and solvent-based paclitaxel as first-line treatment of advanced non-small-cell lung cancer: an open-label, randomized, multicenter, phase III trial. Ann Oncol. 2021;32:85–96. https://doi.org/10.1016/j.annonc.2020.10.479.
Lee SW, Kim YM, Cho CH, Kim YT, Kim SM, Hur SY. et al. An open-label, randomized, parallel, phase II trial to evaluate the efficacy and safety of a cremophor-free polymeric micelle formulation of paclitaxel as first-line treatment for ovarian cancer: a Korean gynecologic oncology group study (KGOG-3021). Cancer Res Treat. 2018;50:195–203. https://doi.org/10.4143/crt.2016.376.
Amjad MW, Kesharwani P, Amin MCIM, Iyer AK. Recent advances in the design, development, and targeting mechanisms of polymeric micelles for delivery of siRNA in cancer therapy. Prog Polym Sci. 2017;64:154–81. https://doi.org/10.1016/j.progpolymsci.2016.09.008.
Mokhtarzadeh A, Alibakhshi A, Hashemi M, Hejazi M, Hosseini V, de la Guardia M. et al. Biodegradable nano-polymers as delivery vehicles for therapeutic small non-coding ribonucleic acids. J Control Release. 2017;245:116–26. https://doi.org/10.1016/j.jconrel.2016.11.017.
Tsuboi M, Matsuo K, Ts’o PO. Interaction of poly-L-lysine and nucleic acids. J Mol Biol. 1966;15:256–67. https://doi.org/10.1016/s0022-2836(66)80225-5.
Nimesh S. Poly-L-lysine nanoparticles. Gene therapy: potential applications of nanotechnology. Cambridge: Woodhead Publishing; 2013. p. 147–62. https://doi.org/10.1533/9781908818645.147.
Hunter AC. Molecular hurdles in polyfectin design and mechanistic background to polycation induced cytotoxicity. Adv Drug Deliv Rev. 2006;58:1523–31. https://doi.org/10.1016/j.addr.2006.09.008.
Lin CW, Jan MS, Kuo JH, Hsu LJ, Lin YS. Protective role of autophagy in branched polyethylenimine (25K)- and poly(L-lysine) (30–70K)-induced cell death. Eur J Pharm Sci. 2012;47:865–74. https://doi.org/10.1016/j.ejps.2012.09.007.
Monnery BD, Wright M, Cavill R, Hoogenboom R, Shaunak S, Steinke JHG. et al. Cytotoxicity of polycations: relationship of molecular weight and the hydrolytic theory of the mechanism of toxicity. Int J Pharm. 2017;521:249–58. https://doi.org/10.1016/j.ijpharm.2017.02.048.
Watanabe S, Hayashi K, Toh K, Kim HJ, Liu X, Chaya H. et al. In vivo rendezvous of small nucleic acid drugs with charge-matched block catiomers to target cancers. Nat Commun. 2019;10:1894.https://doi.org/10.1038/s41467-019-09856-w.
Fu C, Sun X, Liu D, Chen Z, Lu Z, Zhang N. Biodegradable tri-block copolymer poly(lactic acid)-poly(ethylene glycol)-poly(l-lysine)(PLA-PEG-PLL) as a non-viral vector to enhance gene transfection. Int J Mol Sci. 2011;12:1371–88. https://doi.org/10.3390/ijms12021371.
Liu P, Yu H, Sun Y, Zhu M, Duan Y. A mPEG-PLGA-b-PLL copolymer carrier for adriamycin and siRNA delivery. Biomaterials. 2012;33:4403–12. https://doi.org/10.1016/j.biomaterials.2012.02.041.
Zheng C, Zheng M, Gong P, Deng J, Yi H, Zhang P. et al. Polypeptide cationic micelles mediated co-delivery of docetaxel and siRNA for synergistic tumor therapy. Biomaterials. 2013;34:3431–8. https://doi.org/10.1016/j.biomaterials.2013.01.053.
Zhu M-Q, Xiang L, Yang K, Shen L-J, Long F, Fan J-B. et al. Synthesis and characterization of biodegradable amphiphilic triblock copolymers methoxy-poly(ethylene glycol)-b-poly(L-lysine)-b-poly(L-lactic acid). J Polym Res. 2012;19:9808. https://doi.org/10.1007/s10965-011-9808-y.
Lim C, Sim T, Hoang NH, Oh KT. A stable nanoplatform for antitumor activity using PEG-PLL-PLA triblock co-polyelectrolyte. Colloids Surf B Biointerfaces. 2017;153:10–8. https://doi.org/10.1016/j.colsurfb.2017.01.027.
Kricheldorf HR. Polypeptides and 100 years of chemistry of alpha-amino acid N-carboxyanhydrides. Angew Chem Int Ed Engl. 2006;45:5752–84. https://doi.org/10.1002/anie.200600693.
Grasso G, Deriu MA, Patrulea V, Borchard G, Moller M, Danani A. Free energy landscape of siRNA-polycation complexation: elucidating the effect of molecular geometry, polymer flexibility, and charge neutralization. PLoS ONE. 2017;12:e0186816. https://doi.org/10.1371/journal.pone.0186816.
Bouilloux J, Yushchenko O, Dereka B, Boso G, Zbinden H, Vauthey E. et al. Cyclopeptidic photosensitizer prodrugs as proteolytically triggered drug delivery systems of pheophorbide A: part I - self-quenched prodrugs. Photochem Photobio Sci. 2018;17:1728–38. https://doi.org/10.1039/c8pp00317c.
Bouilloux J, Yushchenko O, Dereka B, Boso G, Babic A, Zbinden H. et al. Cyclopeptidic photosensitizer prodrugs as proteolytically triggered drug delivery systems of pheophorbide A: part II - co-loading of pheophorbide A and black hole quencher. Photochem Photobio Sci. 2018;17:1739–48. https://doi.org/10.1039/c8pp00318a.
Alba A, du Boullay OT, Martin-Vaca B, Bourissou D. Direct ring-opening of lactide with amines: application to the organo-catalyzed preparation of amide end-capped PLA and to the removal of residual lactide from PLA samples. Polym Chem. 2015;6:989–97. https://doi.org/10.1039/C4PY00973H.
Trimaille T, Mondon K, Gurny R, Moller M. Novel polymeric micelles for hydrophobic drug delivery based on biodegradable poly(hexyl-substituted lactides). Int J Pharm. 2006;319:147–54. https://doi.org/10.1016/j.ijpharm.2006.03.036.
Hackley VA, Clogston JD. Measuring the hydrodynamic size of nanoparticles in aqueous media using batch-mode dynamic light scattering. In: McNeil SE, editor. Characterization of nanoparticles intended for drug delivery. Totowa, NJ: Humana Press; 2011. p. 35–52. https://doi.org/10.1007/978-1-60327-198-1_4.
Ye R, Xu H, Wan C, Peng S, Wang L, Xu H. et al. Antibacterial activity and mechanism of action of epsilon-poly-L-lysine. Biochem Biophys Res Commun. 2013;439:148–53. https://doi.org/10.1016/j.bbrc.2013.08.001.
Tan Z, Shi Y, Xing B, Hou Y, Cui J, Jia S. The antimicrobial effects and mechanism of ε-poly-lysine against Staphylococcus aureus. Bioresour Bioprocess. 2019;6:11.https://doi.org/10.1186/s40643-019-0246-8.
Li Y-Q, Han Q, Feng J-L, Tian W-L, Mo H-Z. Antibacterial characteristics and mechanisms of ɛ-poly-lysine against Escherichia coli and Staphylococcus aureus. Food Control. 2014;43:22–7. https://doi.org/10.1016/j.foodcont.2014.02.023.
Coutinho PJG, Castanheira EMS, Céu Rei M, Real Oliveira MECD. Nile red and DCM fluorescence anisotropy studies in C12E7/DPPC mixed systems. J Phys Chem B. 2002;106:12841–6. https://doi.org/10.1021/jp026479u.
Ray A, Das S, Chattopadhyay N. Aggregation of Nile red in water: prevention through encapsulation in beta-cyclodextrin. ACS Omega. 2019;4:15–24. https://doi.org/10.1021/acsomega.8b02503.
Rauscher H, Roebben G, Mech A, Gibson N, Kestens V, Linsinger TPJ, et al. An overview of concepts and terms used in the European Commission’s definition of nanomaterial, EUR 29647 EN, Publications Office of the European Union, Ispra, 2019, ISBN 978-92-79-99660-3. https://doi.org/10.2760/459136.
Hui Y, Yi X, Hou F, Wibowo D, Zhang F, Zhao D. et al. Role of nanoparticle mechanical properties in cancer drug delivery. ACS Nano. 2019;13:7410–24. https://doi.org/10.1021/acsnano.9b03924.
Anselmo AC, Mitragotri S. Impact of particle elasticity on particle-based drug delivery systems. Adv Drug Deliv Rev. 2017;108:51–67. https://doi.org/10.1016/j.addr.2016.01.007.
Stern T, Kaner I, Zer NL, Shoval H, Dror D, Manevitch Z. et al. Rigidity of polymer micelles affects interactions with tumor cells. J Control Release. 2017;257:40–50. https://doi.org/10.1016/j.jconrel.2016.12.013.
Teng Z, Wang C, Tang Y, Li W, Bao L, Zhang X. et al. Deformable hollow periodic mesoporous organosilica nanocapsules for significantly improved cellular uptake. J Am Chem Soc. 2018;140:1385–93. https://doi.org/10.1021/jacs.7b10694.
Hui Y, Wibowo D, Liu Y, Ran R, Wang HF, Seth A. et al. Understanding the effects of nanocapsular mechanical property on passive and active tumor targeting. ACS Nano. 2018;12:2846–57. https://doi.org/10.1021/acsnano.8b00242.
Sun J, Zhang L, Wang J, Feng Q, Liu D, Yin Q. et al. Tunable rigidity of (polymeric core)-(lipid shell) nanoparticles for regulated cellular uptake. Adv Mater. 2015;27:1402–7. https://doi.org/10.1002/adma.201404788.
Guan Z, Wang L, Lin J. Interaction pathways between plasma membrane and block copolymer micelles. Biomacromolecules. 2017;18:797–807. https://doi.org/10.1021/acs.biomac.6b01674.
Acknowledgements
This study was supported by the Swiss National Science Foundation project “Novel micellar drug carrier systems for gene therapies” (project ID 200021_157033). Dr. Viorica Patrulea is recipient of a Swiss National Science Foundation grant (project ID: P400PM_194482). The authors thank Dr. Tina Riedel for assistance in peptide synthesis, Mr. Hervé Blanchard for technical assistance with transmission electron microscopy, Dr. Laurence Marcourt for obtaining the NMR spectra and Mrs. Emmanuelle Sublet for support in performing cell culture. We are also grateful to Dr. Lee Ann Laurent-Applegate for her generous donation of human fibroblasts and Dr. Olivier Jordan for useful discussions related to this research.
Funding
Open Access funding provided by Université de Genève.
Author information
Authors and Affiliations
Contributions
FM: Resources, methodology, investigation, visualization, writing original draft preparation. manuscript review and editing. VP: conceptualization, validation, manuscript review and editing. GB: Supervision, project administration, manuscript review and editing, and funding acquisition.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marquet, F., Patrulea, V. & Borchard, G. Comparison of triblock copolymeric micelles based on α- and ε-poly(L-lysine): a Cornelian choice. Polym J 54, 199–209 (2022). https://doi.org/10.1038/s41428-021-00552-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-021-00552-5
