
Abstract
Background:
Retinopathy of prematurity (ROP) is one of the most common preventable causes of blindness and impaired vision among children in developed countries. The aim of the study was to compare whole-genome expression in the first month of life in groups of infants with and without ROP.
Methods:
Blood samples were drawn from 111 newborns with a mean gestational age of 27.8 wk on the 5th, 14th, and 28th day of life (DOL). The mRNA samples were evaluated for gene expression with the use of human whole-genome microarrays. The infants were divided into two groups: no ROP (n = 61) and ROP (n = 50).
Results:
Overall, 794 genes were differentially expressed on the 5th DOL, 1,077 on the 14th DOL, and 3,223 on the 28th DOL. In each of the three time points during the first month of life, more genes were underexpressed than overexpressed in the ROP group. Fold change (FC), which was used in analysis of gene expression data, ranged between 1.0 and 1.5 in the majority of genes differentially expressed.
Conclusion:
Pathway enrichment analysis revealed that genes in four pathways related to inflammatory response were consistently downregulated due to the following variables: ROP and gestational age.
Similar content being viewed by others


Main
Retinopathy of prematurity (ROP) is one of the most common preventable causes of blindness and impaired vision among children in developed countries (1,2). Although not all the risk factors for ROP have been identified, the most certain among the identified risk factors is prematurity (3,4,5). Other potential factors reported in the majority of studies are duration of oxygen therapy and mechanical ventilation (6,7,8).
The rationale behind the study was based on the observation that among premature infants of the same gestational age and exposed to the same environmental risk factors only a subgroup will develop ROP. What is more important, infants belonging to the same risk category may develop ROP characterized by different severities. These observations strongly indicate a genetic contribution to the etiology of ROP and provide justification to consider ROP as an example of a complex trait. The use of microarrays has provided a new opportunity to study as many as 20,000 human genes in a single experiment. The greatest advantage to this method is that it enables the assessment of a great number of genetic factors using only a small amount of blood that is necessary for testing, which is very important in preterm neonates.
To explore the pathogenesis of ROP, we carried out genome-wide transcriptional profiling of RNA extracted from peripheral blood mononuclear cells of ROP subjects and no ROP controls, followed by enrichment analysis in an attempt to identify biological pathways that were preferentially associated with ROP.
Results
Characteristics of the Studied Group
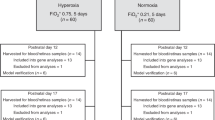
One hundred and twenty newborns were included in the study. The mean birth weight was 1,029 g (SD: 290) and the mean gestational age was 27.8 wk (SD: 2.5). The majority of pregnancies were terminated by abrupt deliveries. Only 50 (42%) of the mothers received antenatal steroids. A considerable proportion of neonates, 49 (41%), were born by vaginal delivery. Sixty-nine newborns (58%) required mechanical ventilation, 20 newborns required noninvasive synchronized nasal ventilation, 25 required nasal continuous positive airway pressure, and 6 required nasal cannulas in the first 3 d of life. Seventy-three (61%) newborns received surfactant treatment (mean number of doses per child was equal to 1.3). During the follow-up period, nine newborns died before the end of the study; therefore, 111 children ( Figure 1 ) were included in the final analysis.

Study flowchart.
Retinopathy of Prematurity
During the follow-up period, ROP was diagnosed in 50 (45%) infants, including 14 (13%) children who had ROP without treatment and 35 (32%) children who underwent laser therapy. Sixty-one babies served as a control group (no ROP). The infants in the ROP group were of lower birth weight and gestational age. As shown in Table 1 , more infants in the ROP group received surfactant therapy.
Whole-Genome Expression
All primary microarray data were submitted to the GEO public repository (accession number GSE32472). A summary of the number of differentially expressed genes between no ROP and ROP patients is presented in Table 2 . Overall, 794 genes were differentially expressed on the 5th day of life (DOL), 1,077 on the 14th DOL, and 3,223 on the 28th DOL. Generally, at each of the three time points during the first month of life, more genes were underexpressed than overexpressed in the ROP group. The difference in expression measured as a fold change (FC) ranged between 1.0 and 1.5 in the majority of genes.
The numbers of significantly differentially expressed genes after multiple testing adjustment are presented in Table 3 . Among 3,223 genes that were differentially expressed on the 28th DOL, only 303 were differentially expressed due to ROP alone. Most of the genes were differentially expressed due to the interaction of two factors: ROP and gestational age. Similar results were obtained from the analysis of genes differentially expressed on the 5th and 14th DOL. The observed differences in gene expression were moderate; only in a few cases were the FC values higher than 1.5.
Supplementary Tables S1–S3 online present the top one hundred genes that were differentially expressed on the 5th, 14th, and 28th DOL, respectively. In the group of overexpressed genes, there were some known factors contributing to the development of ROP, such as growth factors and their receptors ( Table 4 ). In the group of downregulated genes, the majority were involved in the inflammatory response.
Pathway Enrichment Analysis
On the basis of the results presented in Tables 2 and 3 , eight pathway enrichment analyses were performed. A summary of the analyses is presented in Table 5 .
Genes located in four pathways were continuously downregulated in the group of ROP patients. However, the expression of genes depended on the occurrence of both immaturity and disease.
Gene Expression and Rop Severity
To detect the presence of a monotonic trend in gene expression between the ROP severity groups, Spearman’s ρ statistic was used. There were 1,952 genes showing a monotone trend on the 5th DOL, 1,463 on the 14th DOL, and 3,744 on the 28th DOL. Pathway enrichment analysis revealed similar results to those obtained in the two major groups (no ROP and ROP) ( Table 6 ).
Real-Time PCR Validation
The validation procedure did not reveal any significant differences between the results obtained with use of microarrays as compared with real-time PCR technique (P = 0.073) The above analysis is shown in Figure 2 .

Validation of the microarray experiment by means of real-time PCR. White bar, results of microarray evaluation; black bar, results of PCR validation.
Discussion
Currently, there are several publications indicating a strong genetic predisposition in the development of ROP (9). On the basis of a retrospective twin study, the heritability of ROP was found to be as high as 70.1%, which implied a significant contribution of genetic variance to the total phenotype liability (10). Mutations in three genes, Norrin (NDP), frizzled family receptor 4 (FZD4), and low density lipoprotein receptor–related protein 5 (LRP5), that are involved in the Wnt signaling pathways (11) were found in only a small percentage of cases of newborns with advanced-stage ROP. These mutations, however, can help explain unexpected progress to the most severe clinical stages of ROP in some patients or spontaneous regression in others (12).
These observations strongly indicate the genetic contribution to the etiology of ROP and provide justification to consider ROP as an example of a complex trait. During the past decade, the major approach to the identification of the genetic background of complex diseases was based on genome-wide association study and genome-wide linkage analysis. In this article, we propose a novel approach based on whole-genome expression analysis with application of Affymetrix microarrays. We presume that this method will become a breakthrough in the future for identification of genetic background of some common complex traits. It may provide an avenue for identification and dynamic assessment of crucial pathways involving many genes that may interact in the disease pathomechanism. The current knowledge of predefined etiological factors such as immaturity and hyperoxia or hypoxia provide us with the arguments for genetic analysis that will examine gene expression profile rather than gene association study.
We performed whole-genome expression analysis on 5-, 14-, and 28-d-old newborns born before 32 wk of gestation, comparing children who developed ROP with those who did not. The nature of the study was mainly exploratory. We found a large number of differentially expressed genes between ROP and no ROP children. However, the difference in expression measured as an FC in the majority of genes ranged between 1.0 and 1.5. The expression of only a few genes was twofold increased. However, it is a well-known fact that even a 10 or 20% difference in gene expression can be clinically significant. Most of the genes were differentially expressed on the 28th DOL, and only a small number were differentially expressed on the 5th DOL. More genes in the ROP group were underexpressed rather than overexpressed. Detailed analysis of the differentially expressed genes indicated that, for the most part, the cause was lower gestational age in the ROP group. Adjusted analysis revealed that in the majority of cases, the difference in expression was due to a combination of two factors: gestational age and ROP. All these general data are in accordance with our understanding of ROP given that neonate immaturity is a well known and major ROP risk factor.
Detailed analysis of the top differentially expressed genes was performed. Of note, in this group we did not find many of the factors traditionally associated with the development of ROP (inflammatory mediators, growth factors).
Another approach for interpreting genome-wide expression profiles is gene set enrichment analysis. This method can detect transcriptional profiles that are distributed across an entire network of genes, yet are subtle at the level of individual genes. On the basis of this method, we found that genes related to inflammatory response were differentially regulated on the 5th, 14th, and 28th DOL in infants with ROP. These pathways were downregulated in children with ROP. The expression of genes involved in these pathways was related to both maturity and the severity of the ROP. However, this raised an interesting question: Why did pathways involved in the response to oxygen exposure to neonatal tissue not get selected by gene set enrichment analysis? One possible explanation is that the time points designated by protocol for blood sample collection were based on chronological rather than gestational age. The biphasic character of ROP might lead to the diminishment of differences in gene expression. Second, even if oxygen supplementation is considered a continuous stable parameter, it is not necessarily equal to the actual amount of oxygen delivered to the tissues, causing this variable to fluctuate from hyperoxia through normoxia to hypoxia. In certain specific sites, such as the retina, oxygen delivery is a function of perfusion pressure and mechanisms of autoregulation. Perfusion pressure depends on central mechanisms such as cardiac output and hormonal mediators represented by vasopressin, natriuretic peptide, angiotensin II, catecholamines, endothelins, and NO, and the mechanisms of autoregulation depend on an intrinsic mechanism that regulates vascular smooth muscle tone, as well as on blood pH, pCO2, pO2, and prostaglandines.
The introduction of modern protocols for oxygen therapy in preterm infants has demonstrated that we are still unable to define target values for hemoglobin oxygen saturation. On the other hand, efforts directed toward the protection of infants from hyperoxia have shown that oxygen saturation values within the lower range are associated with an increased risk of death. We do not know to what extent even a small rise or drop in oxygen partial pressures influences the stabilization or degradation of transcription factors represented by hypoxia inducible factor (HIF)1 and HIF2, as well as the downstream activation or repression of oxygen-sensing target genes. We can only hypothesize that the delicate equilibrium that exists among HIF1, HIF2, and other growth factors in normoxia may be disturbed following even small fluctuations in oxygen tension. Another question is the duration of this disequilibrium, which may range from a very short to quite a long period of time. Because gene expression profiling was performed at three discrete time points based on the infants’ chronological age, we cannot expect a uniform and unidirectional response in gene transcription. What we obtained was a more or less accurate estimate of an overall trend. To avoid potential bias associated with a more static rather than dynamic assessment of gene expression profiling, we selected two methods. First, we changed the statistical criteria, assuming that acceptance of highly restrictive criteria for FC may lead to the inadvertent skipping of certain differently regulated pathways. Second, we decided to look for gene expression pathways from specially selected candidate genes in the ROP vs. the no ROP groups of newborns ( Table 4 ). Of major interest in this respect was HIF1, which revealed a FC of borderline significance within the three time points that ranged from 1.15 to 1.19. Some experimental studies address the role of HIF in ROP. Morita et al. (13) generated a knockout mouse for HIF1A-like factor that showed a reduction in mRNA expression of 20–80%. After hyperoxia–normoxia treatment, neovascularization was observed in the wild-type mice; however, such was not the case among knockout mice. Knockout mice with abolished function of the RTP801 gene, downstream to HIF1, were resistant to oxygen-induced ROP as compared with wild-type mice (14).
Another well-recognized candidate gene is TGFβ1, which in our ROP group showed significant overexpression at two time points (P = 0.001; P = 0.046 and FC of 1.24 and 1.12 on the 14th and 28th DOL, respectively). Studies conducted in animals have provided proof that TGFβ1 can both stimulate and inhibit vasculogenesis. However, Zhao and Overbeek (15), by using transgenic mice that expressed a self-activating form of TGFβ1 from a lens-specific promoter, showed that in addition to the development of corneal opacities, subcapsular cataracts, and lesions of the iris and ciliary body, these mice presented deficient retinal vascular development and progressive vascular degeneration. An interesting and surprising finding was that vascular endothelial growth factor (VEGF) mRNA expression assessed by in situ hybridization was similar to expression in the wild-type mouse. So was the case with respect to the expression of VEGF receptors. On the basis of these observations, the authors suggest that the inhibitory effect of TGFβ was mediated by the suppression of expression of VEGF or VEGF receptors. The overexpression and underexpression of TGFβ in a dose-dependent manner can lead to serious ocular abnormalities, including abnormal vascularization. In our study, TGFβ was only mildly overexpressed in the ROP group on the 14th and 28th DOL, whereas VEGF expression was not significantly increased on the 5th or 14th DOL. Only on the 28th DOL, was FC equal to 1.16 (P = 0.01), which may signal the beginning of the second phase of the disease, marked by neovascularization. In general, the gene candidate approach did not show any statistically significant trend in gene expression, and observed FC values were subtle without any specific pattern.
Limitations of the Study
On the grounds that hypoxia/hyperoxia influences the organism of the newborn as a whole, we analyzed gene expression within the peripheral blood leukocytes, which are characterized by high mitotic activity, in order to extrapolate these data for other cells. This type of study protocol raises the question of whether gene expression in white blood cells, which are directly exposed to hypoxia/hyperoxia, can be a reflection of gene expression in the retina.
There are very few data that would prove that pathways discovered by us indeed exist in the retina. However articles published by Cabay et al. (16) and El-Asrar et al. (17) show that chemokines are present in the retina in patients with proliferative diabetic retinopathy, proliferative vitreoretinopathy, and rhegmatogenous retinal detachment, although it still to be proven whether these chemokines derive from the retina or come from an exterior source. However, we can speculate that changes in gene expression in leukocytes could indirectly influence retinal metabolism, for example, by immunological, biochemical mechanisms such as the promotion of inflammatory processes or angiogenesis by chemokines in the retina.
It deserves attention that the neonatal intensive care unit in which the study was conducted admits a highly selective group of newborns from primary- and secondary-level centers who require intensive care. Thus, the study focused on a population at a high risk for ROP development. The specific character of the studied population might have influenced the results that were obtained; namely, they were biased toward a statistically significant difference. The prevalence of ROP in the study group may also lead to difficulties in applying the results to the general population and incorporating them into everyday clinical practice.
Another problem relates to the results of the statistical analysis in the case of multiple comparisons. If no correction were applied, multiple testing would increase the probability of a type I error to 1.0 (100%). Bearing this fact in mind, the correction of multiple tests was performed; however, at least a 10% false-discovery rate can still be assumed.
The estimated power of the study to reveal a 1.5 FC difference between the groups was 92.8%. However, the observed differences in expression in most of the studied genes were smaller than those used in our sample size estimation; therefore, we cannot rule out the possibility of a type II error in the studied population. It should be noted, however, that if a difference exists, it is <1.5.
Conclusion
Differential expression of genes in the ROP newborns was in great part the outcome of the relationship between ROP and gestational age. On average in the ROP newborns, more of the differentially expressed genes were underexpressed rather than overexpressed, which might reflect the significant contribution of immaturity related to low gestational age in the development of ROP. Pathway enrichment analysis revealed that genes in four pathways were consistently downregulated due to the following variables: ROP and gestational age. No correlation between gene expression and disease severity was found. Analysis of the expression of selected gene candidates involved in the process of angiogenesis did not show any constant or specific pattern.
Methods
Patients
A prospective study was conducted in the Neonatal Intensive Care Unit of the Department of Pediatrics, Polish-American Children’s Hospital, between 1 September 2008 and 30 November 2010. The entry criteria were preterm birth <32 wk gestational age, birth weight ≤1,500 g, and the need for respiratory support during the first 3 d of life.
The study was approved by the ethics committee of Jagiellonian University, Faculty of Medicine (KBET 27/B/2007).
Microarray Analysis
After informed consent was obtained from the parents, blood samples (0.3 ml) were drawn from all the study participants on the 5th, 14th, and 28th DOL for the assessment of whole-genome expression in peripheral blood leukocytes. Subsequently, Ficoll isopaque gradient centrifugation and RiboPure Blood Kit (Ambion, Life Technologies, Carlsbad, CA) were used for total RNA extraction. RNA concentration was measured with the use of a NanoDrop ND-1000Spectrophotometer (Thermoscientific, Waltham, MA), and RNA quality was determined by a 2100 Bioanalyzer (Agilent, Santa Clara, CA).
Total RNA of 100 ng was used for a single microarray experiment. GeneChip Human Gene 1.0 ST arrays (Affymetrix, Santa Clara, CA) were used. The whole-microarray experiment was performed according to the manufacturer’s protocol (18). The Affymetrix GeneChip Whole Transcript (WT) Sense Target Labeling Assay is designed to generate amplified and biotinylated sense-strand DNA targets from the entire expressed genome. Protocol is optimized for the use with the GeneChip Sense Target (ST) arrays, in which the probes are distributed throughout the entire length of each transcript.
Real-Time PCR Validation
To validate the results obtained by microarray analysis, real-time PCR technique was used. A total of 30 cDNA samples (15 patients with ROP and 15 controls) were used for the validation procedure. Samples were randomly selected from the studied groups. cDNA of 100 ng was used for a single analysis, and the expression level of 14 selected genes was determined by ΔΔCt calculation. Genes chosen for the validation procedure were randomly selected from the group of genes differentially expressed with FC equal to at least 1.5 in the microarray experiment. The following TaqMan Gene Expression Assays (Life Technologies, Applied Biosystems, Foster City, CA) were used: Hs00900055_m1 (VEGF gene), Hs00952786_m1 (AK5 gene), Hs00217864_m1 (OLAH gene), Hs01030384_m1 (ILR2 gene), Hs00187022_m1 (ADAM23 gene), Hs00736937_m1 (DAAM2 gene), Hs00360669_m1 (CD177 gene), Hs00255338_m1 (KLRC4 gene), Hs00171191_m1 (FBN1 gene), Hs00539582_s1 (LRRN3 gene), Hs00196254_m1 (NELL2 gene), Hs00924296_m1 (MPO gene), Hs00541549_m1 (ABCA13 gene), Hs00197437_m1 (OLFM4 gene). GAPDH (Hs02758991_g1) and ACTIN-B (Hs99999903_m1) genes were included as endogenous controls. Each sample was analyzed in duplicate.
Outcome Variables
Ophthalmologic examinations were initiated in the fourth week of life according to the current recommendations of ROP prevention (19) and were performed every 2 wk, or on an individual basis if the severity of the observed changes required it, until the end of the 26th week of life. If at least one eye reached prethreshold or threshold ROP, laser therapy was scheduled within 72 h (20). Ophthalmoscopic examinations and cohort classifications were performed by two independently working experienced ophthalmologists who were not informed of the results of biochemical or molecular studies.
Statistical Analysis
The R package (The R Foundation for Statistical Computing, Vienna, Austria; http://www.r-project.org) sizepower was used to calculate sample size (21). A completely randomized treatment–control design was chosen. The total number of probe sets (genes) on a single microarray was 32,323 (GeneChip Human Gene 1.0 ST Arrays; Affymetrix) and the number of genes that were not differentially expressed but falsely declared by the test procedure to be differentially expressed (number of false positives) was assumed to be about 1%. To simplify calculations, normal distribution of normalized gene expression values, equality of variances, and no interaction among genes were assumed. On the basis of the preliminary data, we anticipated that the variance of log-transformed gene expression levels would be equal to 0.6. The inclusion of 94 newborns (47 in each group) had power equal to 90% for detecting differentially expressed genes if the significant gene expression difference was defined as fold expression change equal or >1.5.
Basic demographic data were compared using the one-way ANOVA or Kruskal–Wallis test as appropriate. Qualitative values (gender, mode of delivery) were compared using the χ2 test.
The microarray data were preprocessed using the R/Bioconductor package aroma.affymetrix (22). For normalization, we used Robust Multiarray Average (23). Quality control was performed by investigating principal component analysis, relative log expression, and normalized unscaled standard error plots. None of the arrays were discarded due to poor quality (data not shown).
Moderated t-tests (24) were performed to detect the probes that were differentially expressed between groups, using the limma package (25) in the R statistical software. Multiple-testing correction using the Benjamini–Hochberg procedure was applied to control the false-discovery rate (26).
The groups determined by the comparisons of interest were not balanced with respect to gestational age, sex, and prenatal steroid treatment. Therefore, we wanted to examine whether differences in gene expression between the groups were due to differences between the groups with respect to these factors. A linear model was fitted to the expression data for each probe, and moderated t-tests and F-statistics were computed for each contrast, i.e., group indicator, gestational age, sex, and steroid treatment. For each probe that was found to be differentially expressed between the groups, i.e., that had a false discovery rate–adjusted P value <0.01 in the first part of the analysis, we tested whether this expression was explained by the group indicator and/or by age, sex, and steroid treatment, using the function classifyTestsF from the limma package.
For the three categories of ROP (no ROP, ROP without treatment, and ROP required laser therapy), we used Spearman’s ρ statistic (27) to test whether there was a monotonic trend in gene expression between the categories.
DAVID functional annotation tools were used to explore which predefined gene sets were significantly enriched in one group as compared with another (28). The Kyoto Encyclopedia of Genes and Genomes (http://www.genome.jp/kegg) and Biocarta pathways (http://www.biocarta.com) were selected for analysis.
The fold-change values representing differences in expression between the ROP group and the control group with regard to each of the 14 selected genes for validation were compared between the microarray and the TaqMan Gene Expression experiments. The Student’s t-test was used for evaluating the statistical significance of observed differences between gene expression.
Statement of Financial Support
This study was supported by unrestricted grants from the Financial Mechanism of European Economic Area (PL0226) and the Polish Ministry of Science (E023/P01/2008/02/85).
Disclosure
The authors declared no other financial ties to products in the study or potential/perceived conflicts of interest.
References
Haines L, Fielder AR, Baker H, Wilkinson AR . UK population based study of severe retinopathy of prematurity: screening, treatment, and outcome. Arch Dis Child Fetal Neonatal Ed 2005;90:F240–4.
Kaiser RS, Trese MT, Williams GA, Cox MS Jr . Adult retinopathy of prematurity: outcomes of rhegmatogenous retinal detachments and retinal tears. Ophthalmology 2001;108:1647–53.
Holmström G, Broberger U, Thomassen P . Neonatal risk factors for retinopathy of prematurity–a population-based study. Acta Ophthalmol Scand 1998;76:204–7.
Karna P, Muttineni J, Angell L, Karmaus W . Retinopathy of prematurity and risk factors: a prospective cohort study. BMC Pediatr 2005;5:18.
Palmer EA . Implications of the natural course of retinopathy of prematurity. Pediatrics 2003;111(4 Pt 1):885–6.
Wheatley CM, Dickinson JL, Mackey DA, Craig JE, Sale MM . Retinopathy of prematurity: recent advances in our understanding. Arch Dis Child Fetal Neonatal Ed 2002;87:F78–82.
Schaffer DB, Palmer EA, Plotsky DF, et al. Prognostic factors in the natural course of retinopathy of prematurity. The Cryotherapy for Retinopathy of Prematurity Cooperative Group. Ophthalmology 1993;100:230–7.
Hutchinson AK, O’Neil JW, Morgan EN, Cervenak MA, Saunders RA . Retinopathy of prematurity in infants with birth weights greater than 1250 grams. J AAPOS 2003;7:190–4.
Shastry BS . Genetic susceptibility to advanced retinopathy of prematurity (ROP). J Biomed Sci 2010;17:69.
Bizzarro MJ, Hussain N, Jonsson B, et al. Genetic susceptibility to retinopathy of prematurity. Pediatrics 2006;118:1858–63.
Clevers H . Wnt/beta-catenin signaling in development and disease. Cell 2006;127:469–80.
Chen J, Stahl A, Hellstrom A, Smith LE . Current update on retinopathy of prematurity: screening and treatment. Curr Opin Pediatr 2011;23:173–8.
Morita M, Ohneda O, Yamashita T, et al. HLF/HIF-2alpha is a key factor in retinopathy of prematurity in association with erythropoietin. EMBO J 2003;22:1134–46.
Brafman A, Mett I, Shafir M, et al. Inhibition of oxygen-induced retinopathy in RTP801-deficient mice. Invest Ophthalmol Vis Sci 2004;45:3796–805.
Zhao S, Overbeek PA . Elevated TGFbeta signaling inhibits ocular vascular development. Dev Biol 2001;237:45–53.
Cabay L, Willermain F, Bruyns C, et al. CXCR4 expression in vitreoretinal membranes. Br J Ophthalmol 2003;87:567–9.
Abu El-Asrar AM, Struyf S, Kangave D, Geboes K, Van Damme J . Chemokines in proliferative diabetic retinopathy and proliferative vitreoretinopathy. Eur Cytokine Netw 2006;17:155–65.
GeneChip Whole Transcript (WT) sense Target Labeling Assay Manual, Version 4. Affymetrix, Santa Clara CA, USA, 2008. (http://media.affymetrix.com/support/downloads/manuals/euk_ds_transcript_protocol_703048_manual.pdf). Accessed 12 February 2013.
An International Committee for the Classification of Retinopathy of Prematurity. The International Classification of Retinopathy of Prematurity Revisited. Arch Ophthalmol 2005;123: 991–9.
Good WV, Hardy RJ, Dobson V et al. Revised indications for the treatment of retinopathy of prematurity - Results of the early treatment for retinopathy of prematurity randomized trial. Arch Ophthalmol 2003;121:1684–96.
Lee M-T. Power and sample size considerations. In: Anonymous Analysis of Microarray Expression Data. Boston, MA: Kluwer Academic, 2004:191–236.
Bengtsson H, Wirapati P, Speed TP . A single-array preprocessing method for estimating full-resolution raw copy numbers from all Affymetrix genotyping arrays including GenomeWideSNP 5 & 6. Bioinformatics 2009;25:2149–56.
Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003;4:249–64.
Smyth GK . Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004;3:Article3.
Smyth GK . Limma: Linear models for microarray data. In: Gentelman R, Carey V, Dudoit S, Irizarry R, Huber W, eds. Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York: Springer, 2005: 397–420.
Benjamini Y, Hochberg Y . Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 1995;57:289–300.
Hollander M, Wolfe DA . Kendall and Spearman Tests. In: Anonymous Nonparametric Statistical Methods. New York: Wiley, 1973:185–94.
Huang da W, Sherman BT, Lempicki RA . Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4:44–57.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Table S1
(DOC 157 kb)
Supplementary Table S2
(DOC 158 kb)
Supplementary Table S3
(DOC 158 kb)
Rights and permissions
About this article
Cite this article
Pietrzyk, J., Kwinta, P., Bik-Multanowski, M. et al. New insight into the pathogenesis of retinopathy of prematurity: assessment of whole-genome expression. Pediatr Res 73, 476–483 (2013). https://doi.org/10.1038/pr.2012.195
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2012.195
This article is cited by
-
Retinopathy of prematurity: contribution of inflammatory and genetic factors
Molecular and Cellular Biochemistry (2022)
-
Short- and long-term impact of hyperoxia on the blood and retinal cells’ transcriptome in a mouse model of oxygen-induced retinopathy
Pediatric Research (2020)
