
Abstract
Mucopolysaccharidosis type IIIA (MPS IIIA; Sanfilippo syndrome) is a lysosomal storage disorder characterized by severe CNS degeneration, resulting in behavioral abnormalities and loss of learned abilities. Early treatment is vital to prevent long-term clinical pathology in lysosomal storage disorders. We have used naturally occurring MPS IIIA mice to assess the effects of long-term enzyme-replacement therapy initiated either at birth or at 6 wk of age. MPS IIIA and normal control mice received weekly i.v. injections of 1 mg/kg recombinant human sulfamidase until 20 wk of age. Sulfamidase is able to enter the brain until the blood-brain barrier completely closes at 10–14 d of age. MPS IIIA mice that were treated from birth demonstrated normal weight, behavioral characteristics, and ability to learn. MPS IIIA mice that were treated from birth performed significantly better in the Morris water maze than MPS IIIA mice that were treated from 6 wk of age or left untreated. A reduction in storage vacuoles in cells of the CNS in MPS IIIA mice that were treated from birth is consistent with the improvements observed. These data suggest that enzyme that enters the brain in the first few weeks of life, before the blood-brain barrier matures, is able to delay the development of behavior and learning difficulties in MPS IIIA mice.
Similar content being viewed by others


Main
Lysosomal storage disorders (LSDs) result from a deficiency of one or more degradative enzymes necessary for normal cell metabolism. Mucopolysaccharidosis type III (MPS III; Sanfilippo syndrome) is an inherited autosomal recessive LSD (1). Four MPS III subtypes result from deficiencies in different lysosomal enzymes that sequentially degrade heparan sulfate: sulfamidase (MPS IIIA), α-N-acetylglucosaminidase (MPS IIIB), acetyl-CoA: α-glucosaminide N-acetyltransferase (MPS IIIC), and glucosamine-6-sulfatase (MPS IIID) (1). MPS III has an estimated incidence of 1 in 66,000 births in Australia, with MPS IIIA the most common (2). MPS III is characterized by severe CNS degeneration, resulting in progressive mental retardation. After a period of seemingly normal development, patients exhibit a range of symptoms, including rapid loss of social skills with hyperactivity and aggressive behavior, loss of learning ability, disturbed sleep patterns, hirsutism, coarse facies, and diarrhea. Death occurs in severely affected children in the mid- to late-teenage years usually as a result of respiratory infection (3). Phenotypic variation in MPS III ranges from severe to intermediate to attenuated, a result of heterogeneity at the molecular level. A total of 62 causative mutations have been identified for MPS IIIA (4).
A naturally occurring mouse model of MPS IIIA has been described (5), a colony of which is established in Adelaide. Disease in the mice results from a base substitution at codon 31 in the sulfamidase gene, altering an aspartic acid to an asparagine (D31N) (6). This aspartic 31 is involved in binding of the divalent metal ion needed for catalytic function (7–9). MPS IIIA mice exhibit widespread intracellular storage in a variety of cell types. Affected mice are also reported to store secondarily gangliosides GM2 and GM3, a feature also reported in two MPS IIIA dog models (10,11), a knock-out MPS IIIB mouse (12), and a caprine MPS IIID model (13). At birth, MPS IIIA mice are indistinguishable from their unaffected litter mates. By 3 wk of age, MPS IIIA pups seem hyperactive when compared with unaffected control pups. The “hyperactive” period lasts until the mice reach sexual maturity at 8–10 wk of age. At this age, male MPS IIIA mice display aggression toward cage mates; this behavior is not observed in control mice at this age. MPS IIIA mice display a coarse, unkempt fur by 20–25 wk of age; also at this age, a distinctly hunched posture and a broader, more flattened face is observed. As the disease progresses, the affected mice become less active; anal prolapse is common in end-stage diseased mice. MPS IIIA mice generally die by 10–12 mo of age. Autopsy generally reveals distended bladders and hepatosplenomegaly. The disease progression observed in MPS IIIA mice is similar to that observed in MPS IIIA patients, making the mice an excellent model to develop and test treatment strategies.
No effective therapies exist for MPS IIIA patients. The concept of treating LSDs by enzyme-replacement therapy (ERT) is widely known; its success relies on the cellular uptake of enzyme by receptor-mediated endocytosis. ERT using macrophage-targeted recombinant β-glucocerebrosidase is successful in treating the nonneuronopathic form of Gaucher disease (14,15). Recently, ERT was approved for use in patients with Fabry disease (16,17) and MPS I (18). Furthermore, ERT clinical trials are under way for MPS II and MPS VI. It is thought that ERT will have only a limited effect in MPS IIIA patients because of the blood-brain barrier (BBB). A preliminary ERT trial performed in one MPS IIID goat kid had no effect on heparan sulfate levels in the brain (19); similarly, a short-term ERT trial in 3-mo-old MPS IIIB mice resulted in enzyme activity in the liver but not in the brain (20). It has been proposed that in mice, the tight junctions between the endothelial cells, which form the barrier, are incompletely formed at birth, rendering the BBB “leaky” to circulating molecules (21). Intravenous administration of β-glucuronidase to newborn MPS VII mice has shown enzyme accumulation to 31% normal levels in the brain (22). Treatment of MPS VII mice with ERT has been investigated extensively and shown to be effective, leading to improvements in behavior and auditory function but only if initiated at birth (23,24).
Human sulfamidase was originally purified and characterized from human liver (25), and subsequently full-length cDNA clones were isolated (26). Stably transfected Chinese hamster ovary (CHO) cells secrete large amounts of recombinant human sulfamidase (rhNS), with a subunit size of 62 kD, which dimerizes to a native size of 115 kD (27). The mannose-6-phosphate receptor was shown to mediate uptake of CHO rhNS in skin fibroblasts by inhibition studies with mannose-6-phosphate (27). Here we show, through an enzyme distribution study, that rhNS administered i.v. to MPS IIIA mice is detected in the brain of newborn mice but not in 6-wk-old mice. Furthermore, MPS IIIA mice that receive weekly i.v. injections of rhNS initiated at birth show improvements in behavior and perform significantly better in a learning and memory test than untreated MPS IIIA mice or MPS IIIA mice that receive weekly i.v. injections of rhNS initiated at 6 wk of age. MPS IIIA mice that are treated from birth also display a persistent reduction in storage vacuoles in CNS cells up to 23 wk of age, when compared with untreated MPS IIIA mice.
METHODS
Enzyme production and purification.
rhNS was purified from the media of transfected CHO cells by a two-column procedure as described previously (27). Specific activity of enzyme was determined regularly during the 20 wk of injections.
Experimental animals.
Homozygous mutant (MPS IIIA, mps/mps) and phenotypically normal (+/mps) mice were obtained from the MPS IIIA mouse colony maintained at the Women's and Children's Hospital. All mice were identified at birth as normal or mutants by obtaining genomic DNA from tissue obtained by a toe clip and amplifying with primers 5′-MSI2 (5′-NNT CTG TCT TCC TCA GCG-3′) and 3′-MS4 (5′-GAT AAG GCT GTG GCT GGA CAG GG-3′) after 3 min at 94°C, 35 cycles of 45 s at 94°C, 45 s at 57°C, 40 s at 72°C, and 4 min final extension at 72°C. PCR products were digested with Aci I at 37°C for 2 h before electrophoresis on a 3.5% agarose gel. Aci I cuts in the presence of the wild type but not in the mutant. Animal studies were approved by the appropriate institutional ethics committee.
rhNS distribution in MPS IIIA mice.
Six MPS IIIA mice received an infusion of 1 mg/kg rhNS into the superficial temporal vein on day 1 of life. One mouse was killed at time points 1, 4, 6, 24, and 48 h and 7 d after injection. Two untreated mice were killed for a time blank. Brain and liver were removed and assayed for rhNS by immunoquantification. Similarly, six MPS IIIA mice each received an infusion of 1 mg/kg rhNS into the tail vein at 6 wk of age. Mice were killed at the same time points, and the brain, liver, heart, kidney, and spleen were removed and assayed for the presence of rhNS by immunoquantification.
Immunoquantification of sulfamidase.
Organs were weighed and homogenized in buffer [0.02 M Tris HCl/0.05 M NaCl (pH 7.2)]. All homogenizations were freeze/thawed six times to disperse cellular membranes and centrifuged at 13,000 ×g for 3 min. Supernatants were assayed for the presence of rhNS. αrhNS polyclonal AB (28) and αrhNS-23.B2 monoclonal AB (28) (which recognizes only human sulfamidase and not mouse sulfamidase) were used in a DELFIA immunoquantification assay as described previously (29).
ERT
Mice that were used in the ERT study were born in the same week and therefore considered age-matched. Five distinct groups of mice were analyzed (Table 1). Mice received weekly injections until they reached 20 wk of age. Mice were weighed before enzyme injection; dose was calculated according to body weight and was diluted in PBS (pH 7.2) to give a concentration of 1 mg of rhNS per kilogram of mouse weight in a volume of 100 μL. Mice that were treated from birth had an initial injection into the superficial temporal vein (30). The second injection was intraperitoneal. The remaining injections were made into the tail vein. Mice that were treated from 6 wk of age received injections into the tail vein.
ANTIBODY TITERS
An ELISA was performed on plasma samples to determine antibody titers to rhNS, as described previously (31).
MORRIS WATER MAZE
Twenty-week-old mice underwent the Morris water maze test (32). A children's inflatable wading pool 143 cm in diameter and 40 cm high was arbitrarily divided into quadrants, northwest (NW), northeast (NE), southwest (SW), and southeast (SE). The pool was filled 30 cm deep with water (21–25°C), and mice were acclimatized to the pool and surroundings on the first day. Testing officially began on the second day. Approximately 500 g of skim milk powder was added to the water and mixed around to give a cloudy appearance. A 10-cm transparent platform was placed 0.5–1.0 cm below the surface of the water and ∼30 cm from the wall in the NW quadrant of the pool. The mice were released into the water facing the wall. The time required for the mice to locate the platform was recorded for each trial. Mice were given four trials each day, from each of the four cardinal points. The mice had 90 s to locate the platform; if this was not achieved, then they were guided to it and allowed to stay for 10 s. The acquisition phase was performed on days 2–6, immediately followed by the probe phase on day 7. The relearning phase was performed on days 8–11, and the reversal phase was performed on days 12–13. Mice were towel dried between each trial and placed on a heating pad. During the 13 d of testing, mice were treated prophylactically with Bactrim (0.8 mg/mL Sulfamethoxazole, 0.16 mg/mL Trimethoprim; Roche Pharmaceuticals).
The time taken for each animal to reach the platform was considered as the dependent variable. A natural log transformation was applied to the time taken data. A repeated measures analysis was used to assess differences in time taken over days, between groups and trials (1–4). Each assessment period (acquisition, relearning, and reversal) was analyzed separately. The analysis was performed with SPSS v10.
HISTOPATHOLOGY
One mouse from each treatment group was killed at 3, 8, 14, 18, and 23 wk of age. The brain was removed, and sections of the cerebral cortex and cerebellum were fixed in 4% (wt/vol) paraformaldehyde/1.25% (wt/vol) glutaraldehyde in PBS, +4% (wt/vol) sucrose (pH 7.2), overnight at 4°C then postfixed in 1% (vol/vol) osmium tetroxide for 1 h; dehydrated through 70, 90, 95, and 100% ethanol; and embedded in epoxy resin. Ultrathin sections were cut and stained with 2% (wt/vol) uranyl acetate/1% (wt/vol) lead citrate and examined with a Phillips 100 Electron Microscope.
RESULTS
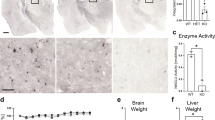
rhNS distribution in MPS IIIA mice.
One hour after injection into the superficial temporal vein of newborn MPS IIIA mice, 180 ng of rhNS was detected in the liver and 90 ng of rhNS was detected in the brain (Fig. 1A). The amount of enzyme detected 4 h after injection into the brain was nearly double that observed at 1 h; 21% of the total amount of enzyme injected was detected in the brain, compared with 42% of total enzyme injected present in the liver 4 h after injection. A small amount of rhNS was detected in the brain of newborn mice 6 h and even 24 h after injection, 60 ng and 45 ng rhNS, respectively. Enzyme was detected in the liver 48 h after injection but not in the brain. Insignificant levels of rhNS were observed in both tissues 7 d after injection.

Distribution of 1 mg/kg rhNS injected i.v. in newborn MPS IIIA mice (A); rhNS was injected into the superficial temporal vein of MPS IIIA mice on day 1 of life. (B) In young adult MPS IIIA mice, rhNS was injected into the tail vein of 6-wk-old MPS IIIA mice. Mice were killed 0, 1, 4, 6, 24, and 48 h and 7 d after injection. Undetectable levels of rhNS were observed in the brain at 48 h and in brain and liver at 7 d in newborn mice and in the brain of adult mice at all time points.
One hour after injection in 6-wk-old MPS IIIA mice, 1,800 ng of rhNS, 10% of the total amount injected, was detected in the liver. Relatively equal amounts were detected in the spleen and kidney; low levels of enzyme were detected in the heart, and undetectable amounts were observed in the brain (Fig. 1B). rhNS content increased in the liver and spleen from 1 to 4 h after injection; 30% of the total amount of injected enzyme was detected in the liver, and significant amounts were also observed in the kidney and to a lesser extent in the heart. Up to 5% of total enzyme injected was detected in the liver 48 h after injection. Except for small amounts in liver, insignificant levels of rhNS were observed in all tissues 7 d after injection. Undetectable levels of rhNS were observed in the brain at all time points.
Disease progression in MPS IIIA mice undergoing ERT.
Five distinct groups of mice were analyzed in the ERT study (Table 1). Male MPS IIIA mice generally displayed aggression toward cage mates by 8–10 wk of age, resulting in the separation of mice into individual cages. In this study, four to six male mice were caged together in a 50 × 20-cm cage. Untreated male MPS IIIA mice began to display lesions on the rump by 10 wk of age, indicating fighting within the group. Similar observations were noted among the male mice in the delayed ERT group. Mice with lesions were removed and caged singly; by ∼15 wk of age, all male mice from these two groups were individually caged. It is interesting that the first sign of fighting among male mice in the birth ERT group was not until 17 wk of age but continued thereafter. Normal male mice did not have to be separated during the study (Table 2).
In general, MPS IIIA mice are heavier than normal mice. Treated MPS IIIA mice displayed body weights close to that of normal mice (Fig. 2).

Body weights of male mice in ERT study. Birth ERT and delayed ERT MPS IIIA mice displayed body weights closer to that of normal mice throughout the 20-wk study.▪, normal mice (n = 6);□, untreated MPS IIIA mice (n = 6);□, birth ERT MPS IIIA mice (n = 10);□, delayed ERT MPS IIIA mice (n = 10).
Antibody response.
MPS IIIA mice that were treated from birth had no obvious adverse reactions throughout the study. This was also observed in the delayed ERT MPS IIIA mice, although one mouse in this group had a mild reaction immediately after injection at 10 wk of age. The reaction involved labored breathing and lethargy, which resolved in ∼1 h.
Serum was analyzed by ELISA for the presence of rhNS antibodies. Serum from delayed ERT MPS IIIA mice displayed the highest titer of 1,280, which was considered to be relatively insignificant. Birth ERT MPS IIIA mice and normal treated mice displayed low titers of 640. As expected, untreated mice displayed no titer.
Morris water maze.
Three days after the final injection at 20 wk of age, at least 10 male mice from each of the five treatment groups underwent the Morris water maze test. MPS IIIA mice show a marked decline in spatial learning abilities by 20 wk of age compared with normal mice (data not shown). On day 1 of the acquisition phase (platform located in the NW quadrant), no difference was observed between the groups in the time it took to locate the platform, and most mice required guidance to the platform (Fig. 3A). On day 2 of the acquisition phase, the search time for normal mice (treated and untreated) and birth ERT MPS IIIA mice had reduced by almost half for the normal mice and one third for the birth ERT MPS IIIA mice. On days 3, 4, and 5, a significant decrease was observed in search time of the treated/untreated normal mice and birth ERT MPS IIIA mice compared with the delayed ERT and untreated MPS IIIA mice (Fig. 3A). Similarly, no significant difference was observed between treated and untreated normal mice.

Morris water maze. The latencies to find the platform during the acquisition phase, days 1–5 (A), the relearning phase, days 7–10 (C), and reversal phase, days 11–12 (D), of treated (♦; n = 10) and untreated (□; n = 10) normal mice and birth ERT (▵; n = 11), delayed ERT (▴; n = 12), and untreated (×; n = 10) MPS IIIA mice are shown. Significant differences (*p <0.01) were observed in the latency between delayed ERT and untreated MPS IIIA mice when compared with birth ERT MPS IIIA mice. (B) The probe phase was performed on day 6 immediately after the acquisition phase. The time spent in the target quadrant is shown.▪, average time spent in the target quadrant;□, average time spent in each of the remaining quadrants.
In the probe phase, the platform was removed from the pool and the time spent in each quadrant was recorded. Treated and untreated normal mice and birth ERT MPS IIIA mice spent most of their time in the target quadrant (NW), where they had learned the platform to be from the acquisition phase. The time that these groups spent in the target quadrant was almost double that spent in the nontarget quadrants (Fig. 3B). In comparison, untreated MPS IIIA mice and delayed ERT MPS IIIA mice spent their time equally in all four quadrants, suggesting that they had not learned/remembered the location of the platform from the acquisition phase (Fig. 3B).
No significant difference was observed in search time between delayed ERT and untreated MPS IIIA mice in the relearning phase (platform replaced to the NW quadrant), taking these groups ∼30% longer to find the platform than the other three groups (Fig. 3C). Treated and untreated normal mice went straight to the platform in each trial and located it within ∼10 s on each day of this phase. Birth ERT MPS IIIA mice performed notably worse during this phase compared with the last 2 d of the acquisition phase. However, although the birth ERT mice were distinct from the treated and untreated normal mice, this group performed significantly better than the untreated MPS IIIA mice and delayed ERT MPS IIIA mice.
There was no significant difference on the first day of platform relocation (platform relocated to the SE quadrant) between any of the five groups (Fig. 3D). By day 2, the search times of the treated and untreated normal mice and the birth ERT MPS IIIA mice had significantly decreased. In comparison, the search time in the other two groups was the same as that on day 1. In fact, the search times of the untreated MPS IIIA mice and delayed ERT MPS IIIA mice remained fairly constant, at an average of 50 s on the last 2 d of the acquisition phase, the 4 d of the relearning phase, and the 2 d of the reversal phase.
Histopathology.
Sulfamidase-deficient mice exhibit glycosaminoglycan storage vacuoles in neuronal cells from day 1 of life, which increases with age. Between 8 and 14 wk of age, the storage vacuoles in MPS IIIA mice contain granular, lipid-like material as a result of the secondary storage of gangliosides GM2 and GM3, which are reported to accumulate in this model (5). Cerebellum and cerebral cortex sections were assessed from mice in each of the treatment groups at ages ranging from 3 to 23 wk.
In birth ERT MPS IIIA mice, a dramatic reduction of storage to almost negligible levels was observed in cortical cells by 3 wk of age (Fig. 4B), compared with untreated MPS IIIA mice (Fig. 4A). Small to medium cytoplasmic inclusions were observed in cerebral Purkinje cells of untreated MPS IIIA mice (Fig. 4D); these were cleared from the cerebral Purkinje cells of birth ERT mice (Fig. 4E). Similarly, perivascular cells of untreated MPS IIIA mice contained numerous electron lucent vacuoles (Fig. 4F), which were absent in birth ERT mice (Fig. 4G).

Histopathology in 3-wk-old mice. Lysosomal distension is observed in cortical neurons from an untreated MPS IIIA mouse (A). The lysosomal distension is greatly reduced from a similar region of the cerebral cortex in a birth ERT MPS IIIA mouse (B). Lysosomal storage was not observed in a treated normal mouse (C). Cytoplasmic inclusions are observed in Purkinje cells from an untreated MPS IIIA mouse (D) but are absent from Purkinje cells of a birth ERT MPS IIIA mouse (E). Numerous large, electron lucent vacuoles are observed in perivascular cells of untreated MPS IIIA mice (F) but have been cleared from perivascular cells of birth ERT MPS IIIA mice (G). Magnifications: ×1,100 in A–C, uranyl acetate/lead citrate; ×3,400; in D–G, uranyl acetate/lead citrate. Scale bar A–C = 20 μM; D–G = 2 μM.
By 8 wk of age, storage vacuoles had started to return to cortical cells in birth ERT MPS IIIA mice (Fig. 5C), with the extent of vacuolation being moderately less than that seen in untreated MPS IIIA mice at 3 wk of age. The number of cells with storage vacuoles in delayed ERT MPS IIIA mice (Fig. 5B) was comparable to untreated MPS IIIA mice (Fig. 5A), with most neuronal cells containing medium to large empty inclusions. Neurons in untreated MPS IIIA mice and delayed ERT MPS IIIA mice appeared swollen and distended compared with those from normal mice (Fig. 5D) and birth ERT MPS IIIA mice, which displayed a normal cell architectural shape.

Histopathology of cortical neurons. Eight-week mice: numerous large, electron lucent vacuoles are observed in untreated (A) and delayed ERT (B) MPS IIIA mice. In comparison, much reduced electron lucent vacuoles are observed in a birth ERT MPS IIIA mouse (C), and no lysosomal inclusions are present from a treated normal mouse (D). Twenty-three-week mice: untreated (E) and delayed ERT (F) MPS IIIA mice display abundant storage vacuoles with whorl-like inclusions. Birth ERT MPS IIIA mice contain lysosomal inclusions with a whorl-like appearance (G) but are storing to a lesser extent then in E or F. No lysosomal inclusions were evident in a treated normal mouse (H). Magnifications: ×4,600 in A–D, uranyl acetate/lead citrate; ×3,400 in E–H, uranyl acetate/lead citrate. Scale bar = 2 μM.
Membranous inclusions of whorls and stacks observed in cortical neurons of 14-wk-old untreated MPS IIIA mice were also present at 14 wk of age in delayed ERT MPS IIIA mice. In contrast, cortical neurons from birth ERT MPS IIIA mice contained electron lucent storage vacuoles similar to that observed in untreated MPS IIIA mice at 3–8 wk of age. However, by 18 wk of age, lipid-containing storage vacuoles were apparent in neuronal cells in birth ERT MPS IIIA mice. However, in the cortical neurons at least, the majority of cells contained a mixture of open and flocculent vacuoles. By 23 wk of age, birth ERT MPS IIIA mice were storing membranous lipid-containing vacuoles in their neurons (Fig. 5G), although to a much lesser extent than in untreated or delayed treated MPS IIIA mice (Fig. 5E and F, respectively). No obvious morphologic abnormalities were observed in the brain of treated and untreated normal mice.
DISCUSSION
MPS IIIA results in progressive neurologic deterioration. The effect of active sulfamidase on modifying behavioral and neurologic problems in MPS IIIA mice had not previously been established. This study evaluates and compares the efficacy of weekly ERT from birth or from 6 wk of age in MPS IIIA mice until 20 wk of age at a dose rate of 1 mg/kg rhNS.
As mentioned earlier, enzyme administered i.v. to newborn mice should access the CNS because of a “leaky” BBB in the first 2 wk of life. Enzyme distribution studies in MPS IIIA mice demonstrate the presence of rhNS in the brain of newborn mice but not in the brain of 6-wk-old mice. MPS IIIA mice have only ∼3% of normal control mouse sulfamidase activity (5). It has been proposed that a correction of ∼10% of normal sulfamidase activity is required to avoid an MPS IIIA clinical phenotype (33). One hour after i.v. injection of rhNS in newborn MPS IIIA mice, enzyme was detected in the brain, with higher levels in the liver; enzyme levels in the brain had doubled by 4 h after injection, resulting in 21% of total enzyme injected being targeted to the brain. In comparison, 4 h after injection in 6-wk-old MPS IIIA mice, widespread tissue uptake of enzyme had occurred, with highest levels observed in the liver; 30% of total enzyme injected was detected in the liver, followed by spleen, kidney, and heart. No enzyme was detected in the adult brain.
Severe behavioral disturbances are noted in MPS IIIA children around the ages of 3–7 y; this is generally the most difficult phase of the disease. Patients are aggressive and have frequent temper tantrums; the normal growth and well-developed muscular strength of an affected child makes them hard to control (3). An aggressive phenotype is also observed in the MPS IIIA mice at ∼9–12 wk of age. Weekly rhNS injections from birth were successful in delaying the onset of aggression until 17 wk of age; no difference was noted in the delayed ERT MPS IIIA mice. This improvement in behavior correlates with a reduction in storage material in neurons of the cerebral cortex in MPS IIIA mice that were treated from birth.
Correction of storage in cerebellar Purkinje cells in birth ERT MPS IIIA mice was achieved up to at least 3 wk of age. It is interesting that cytoplasmic vacuolation was not reduced in Purkinje cells of MPS VII mice that received ERT from birth (34).
Both treatment groups of MPS IIIA mice were tolerant to enzyme. No significant antibody titers were observed, and generally no side effects to the enzyme were noted. Overall, a decrease in body weight approaching normal was observed in both the birth and delayed ERT MPS IIIA mice.
At 20 wk of age, birth ERT mice performed significantly better than untreated or delayed ERT mice in every phase of the Morris water maze test. The performance of birth ERT mice was similar to normal mice during the acquisition phase, but this diminished slightly in the relearning phase. It is likely that only the first two to three enzyme injections reached the CNS of mice that were treated from birth, resulting in decreased lysosomal storage. Negligible storage was demonstrated in these mice at 3 wk of age, and a persistent reduction in storage vacuoles in cortical and cerebral cells was observed up to 23 wk of age. However, the accumulation of lipid-like material, possibly GM2 and GM3 gangliosides, did appear in the cortical cells of these mice by 18 wk of age, although the extent was minimal compared with that observed in 14-wk-old untreated MPS IIIA mice. The gangliosides may be secondarily stored as a result of glycosaminoglycan interference with ganglioside catabolism (35,36). It has been proposed that ganglioside accumulation is the possible cause of neurologic deficits observed in LSDs that secondarily store these compounds (37). The initiation of lipid/ganglioside accumulation may be responsible for the slight decline in performance of the birth ERT MPS IIIA mice during the relearning phase, when compared with normal mice in the Morris water maze.
No difference in performance was observed between treated and untreated normal mice during the Morris water maze, suggesting that excess sulfamidase is not deleterious to cognitive function in mice. In contrast, excess β-glucuronidase was found to induce spatial learning deficits in normal mice when assessed via the Morris water maze (24).
ERT was effective in restoring (or preventing) cognitive deficits characteristic of MPS IIIA mice but only when initiated at birth. ERT did not alter the neurologic impairment in mice that were treated from 6 wk of age. The limited number of injections resulting in enzyme entering the brain only in the first 2 wk of life and the remarkable improvements observed in the Morris water maze at 20 wk of age suggest that infrequent doses of enzyme into the CNS may be needed to significantly improve MPS IIIA patients' quality of life. Preliminary experiments whereby rhNS was injected directly into the brain of MPS IIIA mice at 6, 12, and 18 wk of age demonstrated a reduction of lysosomal vacuolization and gliosis. These injections were also successful in delaying the onset of ubiquitin-positive neurodegenerative changes in widespread areas of the MPS IIIA brain, when assessed at 24 wk of age (Hemsley K, Savas P, Hopwood JJ, unpublished observations). Thus, methods to achieve direct injection of sulfamidase into the CNS of MPS IIIA patients should be considered. Direct injections of enzyme into the cerebrospinal fluid is considered to be a feasible method for supplying active enzyme to the CNS and is being trialed in the MPS I dog and MPS IIIA mouse. Obviously, from this study in mice, these injections may be needed only every 3 mo to maintain storage at nonpathologic levels. A combination of ERT and substrate deprivation therapy using inhibitors of glycosphingolipid synthesis (38) to reduce the accumulation of gangliosides may have a more lasting effect on maintaining cognitive function.
Abbreviations
- BBB:
-
blood-brain barrier
- ERT:
-
enzyme-replacement therapy
- LSD:
-
lysosomal storage disorder
- MPS:
-
mucopolysaccharidosis
- rhNS:
-
recombinant human sulfamidase
References
Neufeld EF, Muenzer J 2001 The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill, New York, pp 3421–3452.
Meikle PJ, Hopwood JJ, Clague AE, Carey WF 1999 Prevalence of lysosomal storage disorders. JAMA 281: 249–254.
Cleary MA, Wraith JE 1993 Management of mucopolysaccharidosis type III. Arch Dis Child 69: 403–406.
Yogalingam G, Hopwood JJ 2001 Molecular genetics of mucopolysaccharidosis type IIIA and IIIB: diagnostic, clinical and biological implications. Hum Mutat 18: 264–281.
Bhaumik M, Muller VJ, Rozaklis T, Johnson L, Dobrenis K, Bhattacharyya R, Wurzelmann S, Finamore P, Hopwood JJ, Walkley SU, Stanley P 1999 A mouse model for mucopolysaccharidosis type IIIA (Sanfilippo syndrome). Glycobiology 9: 1389–1396.
Bhattacharyya R, Gliddon B, Beccari T, Hopwood JJ, Stanley P 2001 A novel missense mutation in lysosomal sulfamidase is the basis of MPS IIIA in a spontaneous mouse mutant. Glycobiology 11: 99–103.
Bond CS, Clements PR, Ashby SJ, Collyer CA, Harrop SJ, Hopwood JJ, Guss JM 1997 Structure of a human lysosomal sulfatase. Structure 5: 277–289.
Waldow A, Schmidt B, Dierks T, von Bulow R, von Figura K 1999 Amino acid residues forming the active site of arylsulfatase A. Role in catalytic activity and substrate binding. J Biol Chem 274: 12284–12288.
Hopwood JJ, Ballabio A 2001 The Metabolic and Molecular Bases of Inherited Diseases. McGraw-Hill, New York, pp 3725–3774.
Fischer A, Carmichael KP, Munnel JF, Jhabvala P, Thompson JN, Matalon R, Jezyk PF, Wang P, Giger U 1998 Sulfamidase deficiency in a family of Dachshunds: a canine model of mucopolysaccharidosis IIIA (Sanfilippo A). Pediatr Res 44: 74–82.
Jolly RD, Allan FJ, Collett MG, Rozaklis T, Muller VJ, Hopwood JJ 2000 Mucopolysaccharidosis IIIA (Sanfilippo syndrome) in a New Zealand Huntaway dog with ataxia. N Z Vet J 48: 144–148.
Li HH, Yu W-H, Rozengurt N, Zhao H-Z, Lyons KM, Anagnostaras S, Fanselow MS, Suzuki K, Vanier MT, Neufeld EF 1999 Mouse model of Sanfilippo syndrome type B produced by targeted disruption of the gene encoding α-N-acetylglucosaminidase. Proc Natl Acad Sci U S A 96: 14505–14510.
Jones MZ, Alroy J, Boyer PJ, Cavanagh KT, Johnson K, Gage D, Vorro J, Render JA, Common RS, Leedle RA, Lowrie C, Sharp P, Liour S-S, Levene B, Hoard H, Lucas R, Hopwood JJ 1998 Caprine mucopolysaccharidosis-IIID: clinical, biochemical, morphological and immunohistochemical characteristics. J Neuropathol Exp Neurol 57: 148–157.
Altarescu G, Hill S, Wiggs E, Jeffries N, Kreps C, Parker CC, Brady RO, Barton NW, Schiffmann R 2001 The efficacy of enzyme replacement therapy in patients with chronic neuronopathic Gaucher's disease. J Pediatr 138: 539–547.
Altarescu G, Schiffmann R, Parker CC, Moore DF, Kreps C, Brady RO, Barton NW 2000 Comparative efficacy of dose regimens in enzyme replacement therapy of type I Gaucher disease. Blood Cells Mol Dis 26: 285–290.
Schiffmann R, Kopp JB, Austin HA 3rd, Sabnis S Morre DF Weibel T Balow JE Brady RO 2001 Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 285: 2743–2749.
Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, Caplan L, Linthorst GE, Desnick RJ 2001 Safety and efficacy of recombinant human α-galactosidase A replacement therapy in Fabry's disease. N Engl J Med 345: 9–16.
Kakkis ED, Muenzer J, Tiller GE, Waber L, Belmont J, Passage M, Izykowski B, Phillips J, Doroshow R, Walot I, Hoft R, Neufeld EF 2001 Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med 344: 182–188.
Downs-Kelly E, Jones MZ, Alroy J, Cavanagh KT, King B, Lucas RE, Baker JC, Kraemer SA, Hopwood JJ 2000 Caprine mucopolysaccharidosis IIID: a preliminary trial of enzyme replacement therapy. J Mol Neurosci 15: 251–262.
Yu W-H, Zhao K-W, Ryazantsev S, Rozengurt N, Neufeld EF 2000 Short-term enzyme replacement in the murine model of Sanfilippo syndrome type B. Mol Genet Metab 71: 573–580.
Stewart PA, Hayakawa EM 1987 Interendothelial junctional changes underlie the developmental ‘tightening’ of the blood-brain barrier. Dev Brain Res 32: 271–281.
Vogler C, Sands M, Higgins A, Levy B, Grubb J, Birkenmeier EH, Sly WS 1993 Enzyme replacement with recombinant beta-glucuronidase in the newborn mucopolysaccharidosis type VII mouse. Pediatr Res 34: 837–840.
Sands MS, Vogler C, Kyle JW, Grubb JH, Levy B, Galvin N, Sly WS, Birkenmeier EH 1994 Enzyme replacement therapy for murine mucopolysaccharidosis type VII. J Clin Invest 93: 2324–2331.
O'Connor LH, Erway LC, Vogler CA, Sly WS, Nicholes A, Grubb J, Holmberg SW, Levy B, Sands MS 1998 Enzyme replacement therapy for murine mucopolysaccharidosis type VII leads to improvements in behavior and auditory function. J Clin Invest 101: 1394–1400.
Freeman C, Hopwood JJ 1986 Human liver sulphamate sulphohydrolase. Determinations of native protein and subunit Mr values and influence of substrate aglycone structure on catalytic properties. Biochem J 234: 83–92.
Scott HS, Blanch L, Guo XH, Freeman C, Orsborn A, Barker E, Sutherland GR, Morris CP, Hopwood JJ 1995 Cloning of the sulphamidase gene and identification of mutations in Sanfilippo A syndrome. Nat Genet 11: 465–467.
Bielicki J, Hopwood JJ, Melville EL, Anson DS 1998 Recombinant human sulphamidase: expression, amplification, purification and characterisation. Biochem J 329: 145–150.
Perkins KJ, Byers S, Yogalingam G, Weber B, Hopwood JJ 1999 Expression and characterisation of wild type and mutant recombinant human sulfamidase. Implications for Sanfilippo (mucopolysaccharidosis IIIA) syndrome. J Biol Chem 274: 37193–37199.
Whitfield PD, Nelson P, Sharp PC, Bindloss CA, Dean C, Ravenscroft EM, Fong BA, Fietz MJ, Hopwood JJ, Meikle PJ 2002 Correlation among genotype, phenotype, and biochemical markers in Gaucher disease: implications for the prediction of disease severity. Mol Genet Metab 45: 46–55.
Sands MS, Barker JE 1999 Percutaneous intravenous injection in neonatal mice. Lab Anim Sci 49: 328–330.
Turner CT, Hopwood JJ, Brooks DA 2000 Enzyme replacement therapy in mucopolysaccharidosis I: altered distribution and targeting of alpha-L-iduronidase in immunized rats. Mol Genet Metab 69: 277–285.
Morris R 1984 Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11: 47–60.
Perkins KJ, Muller V, Weber B, Hopwood JJ 2001 Prediction of Sanfilippo phenotype severity from immunoquantification of heparan-N-sulfamidase in cultured fibroblasts from mucopolysaccharidosis type IIIA patients. Mol Genet Metab 73: 306–312.
Sands MS, Vogler C, Torrey A, Levy B, Gwynn B, Grubb J, Sly WS, Birkenmeier EH 1997 Murine mucopolysaccharidosis type VII: long term therapeutic effects of enzyme replacement followed by bone marrow transplantation. J Clin Invest 99: 1596–1605.
Baumkotter J, Cantz M 1983 Decreased ganglioside neuraminidase activity in fibroblasts from mucopolysaccharidosis patients: inhibition of the activity in vitro by sulfated glycosaminoglycans and other compounds. Biochim Biophys Acta 761: 163–170.
Avila JL, Convit J 1975 Inhibition of leucocytic lysosomal enzymes by glycosaminoglycans in vitro. Biochem J 152: 57–64.
Walkley SU 1998 Cellular pathology of lysosomal storage disorders. Brain Pathol 8: 175–193.
Platt FM, Jeyakumar M, Andersson U, Priestman DA, Dwek RA, Butters TD, Cox TM, Lachmann RH, Hollack C, Aerts JM, Van Weely S, Hrebicek M, Moyses C, Gow I, Elstein D, Zimran A 2001 Inhibition of substrate synthesis as a strategy for glycolipid lysosomal storage disease therapy. J Inherit Metab Dis 24: 275–290.
Acknowledgements
We gratefully acknowledge the animal care staff at the WCH for daily care of the mouse colony. We thank Peter Clements, Liz Melville, and Vivienne Muller for supplying rhNS and substrate. Allison Crawley is gratefully acknowledged for assistance with histologic analysis and Mark Sands for expert guidance regarding the Morris water maze and superficial temporal vein injections. Assistance from Elaine Ravenscroft is greatly appreciated, as is technical assistance from Lyn Waterhouse and Craig Hirte. The original MPS IIIA mice were given by Pamela Stanley from the Albert Einstein Institute.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by a Sam Lister PhD scholarship to B.G. and the National Health and Medical Research Council of Australia.
Rights and permissions
About this article
Cite this article
Gliddon, B., Hopwood, J. Enzyme-Replacement Therapy from Birth Delays the Development of Behavior and Learning Problems in Mucopolysaccharidosis Type IIIA Mice. Pediatr Res 56, 65–72 (2004). https://doi.org/10.1203/01.PDR.0000129661.40499.12
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000129661.40499.12
This article is cited by
-
Glycosaminoglycan levels and structure in a mucopolysaccharidosis IIIA mice and the effect of a highly secreted sulfamidase engineered to cross the blood-brain barrier
Metabolic Brain Disease (2017)
-
Mucopolysaccharidosis IIIB confers enhanced neonatal intracranial transduction by AAV8 but not by 5, 9 or rh10
Gene Therapy (2016)
-
Liver Production of Sulfamidase Reverses Peripheral and Ameliorates CNS Pathology in Mucopolysaccharidosis IIIA Mice
Molecular Therapy (2012)
-
Lessons learnt from animal models: pathophysiology of neuropathic lysosomal storage disorders
Journal of Inherited Metabolic Disease (2010)
-
Mannitol-facilitated CNS entry of rAAV2 vector significantly delayed the neurological disease progression in MPS IIIB mice
Gene Therapy (2009)
