
Abstract
Although it is a widely held thought that direct hormone action on peripheral tissues is sufficient to mediate the control of nutrient handling, the role of the central nervous system in certain aspects of metabolism has long been recognized. Furthermore, recent findings have suggested a more general role for the central nervous system in metabolic control, and have revealed the importance of a number of cues and hypothalamic circuits. The brain's contributions to metabolic control are more readily revealed and play a crucial part in catabolic states or in hormone deficiencies that mimic starvation.
Similar content being viewed by others
Main
The survival of multicellular organisms depends on the appropriate uptake and release of nutrients by major metabolic tissues. In the absence of continuous feeding, the availability of metabolic fuels (for example, glucose, fatty acids and amino acids) for use in tissues is maintained by storing nutrients, which are later released at the appropriate time and rate. Hormones that are secreted by the pancreatic islets of Langerhans modulate important aspects of nutrient uptake and storage (insulin) or their release into the circulation (glucagon). They do this partly by acting directly on the tissues that are the main reservoirs for these nutrients (for example, liver, adipose and muscle tissue).
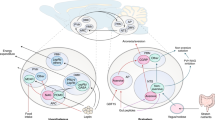
Although the direct actions of these hormones on metabolic tissues are crucial for whole-body metabolic homeostasis, several central nervous system (CNS)-regulated systems have long been recognized to control important aspects of metabolism. For instance, the elaboration of catecholamines by the sympathetic nervous system (SNS), along with hypothalamically controlled hormones (such as glucocorticoids and thyroid hormone), functions in concert with glucagon to mediate the counter-regulatory response, promoting nutrient release into the blood and favouring substrate use over storage1. Well-recognized CNS pathways mediate crucial aspects of this counter-regulatory response. Recent data reveal a more general role for CNS pathways in the modulation of metabolism: various nutrient, energetic and hormonal cues (such as insulin and the adipose-derived hormone leptin) function in the hypothalamus to control glucose and lipid metabolism, in addition to overall energy balance2,3 (Fig. 1). Several hypothalamic areas, especially the ventromedial nucleus and arcuate nucleus (including the arcuate melanocortin system), as well as the hindbrain, make important contributions to these effects. Although the peripheral response to the hormones that control metabolism (for example, insulin and glucagon) tends to obscure these CNS contributions under many conditions, the brain controls overall metabolic tone and is crucial when the peripheral systems cannot compensate — especially when pancreatic hormone output is absent or cannot be modulated.

Recent findings have implicated medial basal hypothalamic nuclei (the arcuate and ventromedial nucleus) in the control of metabolism. Leptin, secreted from adipose tissue as an indicator of long-term energy stores, and insulin, reflective of recent food intake in addition to adipose stores, function in these nuclei to modulate the autonomic nervous system to increase hepatic glucose production and peripheral glucose disposal. Other signals of energy surfeit, including glucose and fatty acids, act in concert with these signals, whereas signals of energy deficit (such as hypoglycaemia and activation of AMP-dependent protein kinase (AMPK) and ghrelin) act in the opposite manner. Although the ventromedial nucleus neural pathways that mediate the responses to these stimuli have not been molecularly characterized, the arcuate nucleus melanocortin system (which is composed of pro-opiomelanocortin (POMC) neurons that act in part through melanocortin-3 receptor (MC3R) and melanocortin-4 receptor (MC4R), and is antagonized by Agouti-related protein (AgRP)-producing neurons) contributes.
Established autonomic and neuroendocrine roles
As anyone who has examined parameters of glucose homeostasis in mammals knows, stresses (such as pain or restraint) increase blood sugar. This reflects the activation of the SNS and the hypothalamic–pituitary–adrenal axis, which promote the breakdown of macromolecular forms of stored energy (such as glycogen, triglyceride and protein) for release into the bloodstream (as glucose, fatty acids and glycerol, and amino acids). Furthermore, SNS-mediated catecholamine action in the islets promotes increased glucagon release while suppressing insulin secretion; the parasympathetic nervous system (PNS) mediates opposing actions4. Therefore, stress signals, which are coordinated by the CNS, modulate autonomic and neuroendocrine function to increase blood glucose. Furthermore, this stress-induced hyperglycaemia complicates the treatment of diabetes by promoting poor glycaemic control5.
CNS-mediated modulation of glucose homeostasis is not unique to anxiogenic stresses, but it is also a crucial response to physiological stresses, such as exercise or infection, in which increased circulating nutrient availability is required to meet the needs of muscle contraction or the immune response, respectively6. Indeed, metabolic cues can also promote this response: hypoglycaemia — a common manifestation of iatrogenic insulin overdose — not only promotes glucagon secretion over insulin production at the level of the islets, but also acts directly in the brain to increase SNS activity and glucocorticoid production (as well as food intake), directing the return to normoglycaemia7,8,9. Within the brain, specialized glucose-sensing neurons that lie in several sites in the brainstem (including the nucleus tractus solitarius and other parts of the dorsal vagal complex) and hypothalamus (such as the ventromedial nucleus) respond to the decreased availability of glucose (the main metabolic fuel for the brain under most conditions) to promote this counter-regulatory response9. The importance of this system is underscored by the consequences of its failure to respond appropriately to hypoglycaemia in individuals, following repeated exposure to hypoglycaemia as a consequence of intensive insulin therapy. Such impairment increases the severity and frequency of hypoglycaemic events, and diminishes the patient's ability to effectively recognize their occurrence.
A larger role for the CNS in metabolic control
In addition to the well-established role for the brain in mediating important aspects of the counter-regulatory response to hypoglycaemia and other stresses, results over the past 15 years have demonstrated a more general role for the CNS in the sensing and control of whole-body metabolic homeostasis3,10. This theme has emerged from the search for signals that communicate the status of adiposity or energy stores to the brain to control food intake and energy expenditure. Insulin — the first hormone to be studied in this regard — exhibits elevated baseline blood concentrations with increasing adiposity, and increases rapidly in response to feeding. Acute administration of insulin directly into the CNS tends to suppress feeding10.
The cloning of the gene that encodes leptin, which is secreted by adipose tissue in approximate proportion to body fat stores, was a watershed event in our understanding of the role of the CNS in the control of energy balance and metabolism11. A lack of leptin or its predominantly CNS-expressed receptor, LRb, not only promotes massive hyperphagia and decreased energy expenditure (with subsequent obesity), but also results in early-onset insulin resistance, hyperglycaemia and metabolic dysfunction, which are, at least in rodents, disproportionately severe in relation to that expected with obesity alone. Indeed, whole-body or CNS-restricted leptin treatment of leptin-deficient ob/ob mice rapidly restores glycaemic control independently of changes in food intake or adiposity, suggesting that leptin acts in the brain to control blood glucose levels independently of energy balance12,13. These observations provoked a careful examination of the roles for CNS insulin and leptin action in the control of whole-body metabolism (mainly the control of glucose production and disposal)13,14. These studies reported that CNS insulin action contributes to the suppression of hepatic glucose output, and CNS leptin action promotes increased hepatic glucose flux by increasing gluconeogenesis while diminishing glycogenolysis. In diet-induced obese rodents, leptin mainly suppresses glycogenolysis to decrease net hepatic glucose production (HGP)15,16. In a similar vein, adiponectin and glucagon-like peptide 1 (GLP-1), like insulin, suppress gluconeogenesis and HGP following intracerebroventricular administration (although GLP-1-producing CNS neurons, rather than the gut, could represent the physiological source of CNS GLP-1 in this case)17,18. By contrast, intracerebroventricular ghrelin (which opposes leptin action in most cases) increases HGP, as does the adipose-derived hormone resistin (which attenuates insulin action)19,20.
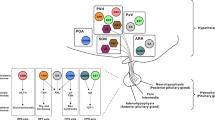
Subsequent genetic studies have demonstrated crucial roles for CNS leptin and insulin signals (including those mediated by elements of the insulin receptor substrate (IRS) 2 phosphatidylinositol (PI)-3-OH kinase–phosphoinositide-dependent kinase 1 (Pdk1) pathway that suppress the transcription factor FOXO1) in the control of glucose homeostasis21,22(Fig. 2). Furthermore, impairing leptin and insulin action in the same CNS circuits provokes substantially greater metabolic dysfunction than that observed with ablation of either one alone, suggesting a synergy between the insulin and leptin signalling pathways in the CNS control of metabolism23 (Fig. 2).

The insulin receptor (InsR) and the related insulin-like growth factor 1 receptor (IGF-1R) act through their intrinsic tyrosine kinases to promote the phosphorylation of receptor tyrosine residues (pY), leading to the recruitment and phosphorylation of the insulin receptor substrate (IRS) proteins-1 and -2. These recruit phosphatidylinositol 3-kinase (PI3K), which activates phosphoinositide-dependent kinase 1 (PDK1) and Akt to promote, among other things, the phosphorylation and nuclear exclusion of the FOXO1 transcription factor, inactivating FOXO1-mediated transcription. Phosphorylated IRS1 or IRS2 also recruit GRB2, which promotes extracellular-signal-regulated kinase (ERK) activation. Stimulation of leptin receptor (LRb) activates the associated Jak2 tyrosine kinase to promote the phosphorylation of intracellular tyrosine residues on LRb. One of these residues recruits the suppressor of cytokine signalling 3 (SOCS3) and the protein-tyrosine phosphatase SHP-2. SHP-2 recruits GRB2 to activate ERK signalling. Two additional phosphorylated LRb residues recruit latent signal transducers and activators of transcription (STAT3 and STAT5), which then translocate to the nucleus to modulate gene transcription. LRb also acts through undefined pathways to modestly promote PI3K pathway signalling.
In addition to hormones, nutrients and signals of cellular energy status also have a role in the CNS control of metabolism. As might be predicted from the response to hypoglycaemia, pathways that monitor cellular energy status in the hypothalamus also modulate food intake and whole-body metabolism. Activation of hypothalamic AMP-dependent protein kinase (AMPK) (which is stimulated by the depletion of cellular ATP levels) increases food intake and circulating glucose concentrations24,25. Roles in the CNS for amino acids, their sensing by the mammalian target of rapamycin pathway and mitochondria-derived reactive oxygen species in the control of peripheral metabolism have not yet been examined. However, these cellular signalling systems contribute to the control of food intake and energy homeostasis26,27, and could also participate in the CNS control of metabolism.
In addition to the response to hypoglycaemia, glucose sensing in the hypothalamus also controls peripheral glucose handling: intracerebroventricular glucose administration decreases HGP through a mechanism that requires cellular ATP generation and the subsequent closure of ATP-sensitive potassium (K+–ATP) channels3. Similarly, although fatty acids are not typically thought of as a major fuel for the CNS, in the hypothalamus fatty acids and the systems that mediate their mitochondrial import and oxidation control food intake and blood glucose levels28. Intracerebroventricular injection of oleic acid (and, to a lesser extent, other fatty acids) suppresses HGP, as do a variety of pharmacological and short-hairpin-RNA-mediated manipulations designed to alter hypothalamic fatty-acid oxidation and increase cellular concentrations of fatty-acid-coenzyme A (CoA). These studies suggest there is an important role for fatty-acid metabolism, and potentially for fatty-acid-CoA as an intracellular mediator, in nutrient sensing by hypothalamic neurons that modulate hepatic glucose handling.
Although most data that link the CNS to peripheral metabolism have examined the control of blood glucose concentrations as their surrogate for metabolism, a number of studies also suggest that these pathways may similarly modulate lipid handling throughout the body. Intracerebroventricular injection of leptin, for instance, suppresses lipogenesis in adipose tissue29. Moreover, genetic or pharmacological manipulation of CNS melanocortin action can alter systemic lipid metabolism30.
CNS circuits and cellular signals in metabolic control
Brain-wide and circuit-specific genetic manipulation of intracellular mediators of leptin or insulin action have demonstrated important roles for these signals in the CNS for the control of metabolism. Although genetic tools that manipulate many components of the cellular nutrient-sensing apparatus are not available or practical (because of factors such as redundant isoforms or embryonic lethality), direct injection of pharmacological inhibitors or short-hairpin RNA reagents to target these pathways has suggested that the hypothalamus, especially the medial basal hypothalamus (MBH) — which includes the ventromedial and arcuate nuclei — is an important site of action3. Other hypothalamic regions (such as the lateral hypothalamic area) and the brainstem contain neural populations that sense leptin and nutrients, including glucose and amino acids, but roles for these areas in the control of metabolism by direct leptin or insulin action have not been reported9,31. However, the dorsal vagal complex (including the nucleus tractus solitarius) participates in the sensing of hypoglycaemia and the counter-regulatory response, and represents a potential contributor to overall metabolic control. The dorsal motor nucleus of the vagus controls outputs to the periphery through the vagus nerve, and it is well-positioned to regulate metabolism.
The heterogeneity of brain tissue — in which even small and circumscribed brain areas (such as the arcuate nucleus) contain multiple types of neurons with distinct, or even opposing, functions — means that understanding the specific cell types and the circuits that are involved in metabolic control requires the use of cell-specific genetic tools32,33. Although it is difficult to analyse the cellular pathways that control fatty-acid-CoA levels and sensing of cellular energy using neuron-specific techniques, many aspects of insulin and leptin action can be investigated with the tools that are available. For instance, given the important and synergistic roles that insulin and leptin have in the CNS control of metabolism, insulin signalling in LRb neurons might be an important mechanism to analyse. The redundancy of signalling by insulin and insulin-like growth factor 1 (IGF-1) receptors renders it difficult to analyse through the simple deletion of insulin receptors from LRb neurons (indeed, the metabolic phenotype of insulin-receptor deletion throughout the brain is rather modest). However, deletion of IRS2 — which is the crucial shared second messenger protein for insulin and IGF-1 receptors — from the entire brain or specifically from LRb neurons results in early insulin resistance and glucose intolerance34,35.
Additionally, roles for direct LRb signalling in the control of energy homeostasis have been examined in mouse models, in which LRb mutants that are defective for specific signalling pathways replace the endogenous LRb. This analysis revealed crucial roles for LRb–signal transducers and activators of transcription 3 (STAT3) signalling in the control of energy balance, although the mice demonstrated improvements in glucose homeostasis relative to mice that were completely null for LRb36,37,38. However, the improvements in glycaemic control may be secondary to increased overall glucose disposal because HGP remains high in animals that carry mutations that block LRb–STAT3 signalling. Another pathway, potentially mediated by the LRb-associated tyrosine kinase Jak2, or other LRb signals that are distinct from phosphorylation sites on LRb, also seems to contribute to the control of glucose homeostasis by leptin39,40.
Thus, specific leptin- and insulin-mediated signals in LRb neurons contribute to glucose homeostasis. LRb neurons may be an important locus at which cellular nutrient and energy sensing could also have a notable metabolic role, although this has yet to be tested.
A role for the ventromedial nucleus
Within the MBH, the ventromedial nucleus is an obvious place to begin examining the hypothalamic circuits that control overall metabolism, owing to this region's important role in sensing glucose levels and in aspects of the counter-regulatory response41,42,43. Manipulation of AMPK activity in the ventromedial nucleus by stereotactic injection of inhibitors or activators suggests a role for AMPK signalling in this nucleus in sensing low glucose concentrations and mounting the counter-regulatory response to hypoglycaemia. A role for the ventromedial nucleus in glucose metabolism has also been implied by the manipulation of glucokinase and the K+–ATP channel by stereotactic injection of pharmacological agents or short-hairpin RNA reagents into this nucleus43,44.
Leptin activates at least some neurons in the ventromedial nucleus, which contains a substantial group of LRb neurons45. This region also seems to have a role in the control of glucose disposal by leptin. The injection of leptin into the ventromedial nucleus, but not the adjacent arcuate nucleus, increases sympathetic output and glucose uptake by muscle and brown adipose tissue (both of which have heavy SNS innervation)46. The deletion of LRb specifically from the ventromedial nucleus in mice has shown a role for ventromedial-nucleus leptin action in the control of food intake and overall energy expenditure47,48. Although glucose metabolism in these animals has not been completely characterized, they are less glucose tolerant at early ages than control mice — before the onset of frank obesity — consistent with the idea that leptin action in the ventromedial nucleus promotes glucose disposal independently of the control of energy balance.
Insulin action in the ventromedial nucleus also contributes to the control of metabolism. Injection of insulin into this area promotes peripheral glucose disposal, whereas knockdown of insulin receptors in the ventromedial nucleus of adult mice causes glucose intolerance49. Similarly, enhancing the action of insulin in the ventromedial nucleus by interference of the insulin-inhibited FOXO1 pathway promotes increased glucose disposal in muscle tissue50.
Although local hypoglycaemia (or AMPK activation) in the ventromedial-nucleus promotes a counter-regulatory response and glucose production, ventromedial-nucleus leptin and insulin signals promote glucose uptake by metabolically active tissues. Thus, whereas hypoglycaemia, and leptin or insulin in the ventromedial nucleus promote SNS-mediated processes, the specific outcome differs between stimuli. This discrepancy could reflect differences in the effects of each stimulus on the same cell type, or it might indicate that distinct (ventromedial nucleus) cell types respond to each stimulus. Unfortunately, systems to molecularly define and genetically perturb subtypes of ventromedial-nucleus-restricted neurons still need to be developed.
Arcuate nucleus and hypothalamic melanocortin system
Neural systems in the arcuate nucleus are thought to have a crucial role in the control of glucose homeostasis. Ablation of all arcuate-nucleus neurons with monosodium glutamate produces not only obesity but also frank hyperglycaemia, suggesting that the net tone of the arcuate nucleus tends to suppress hepatic glucose output51. By contrast, ventromedial hypothalamus lesions produce obesity, but also impair hypoglycaemia sensing. Of note, however, is that lesioning studies reveal nothing about the roles of specific populations of neurons within these nuclei, but only reveal the aggregate output for the entire area.
Arcuate-nucleus-directed LRb expression in rats and mice that are null for LRb alters body weight and food intake only modestly, but markedly ameliorates hyperglycaemia in these animals52,53. Similarly, adenoviral augmentation of insulin signalling pathways in the arcuate nucleus also improves glucose homeostasis in LRb-null animals54. Thus, leptin and insulin-like signalling in the arcuate nucleus are important for the suppression of glucose production.
Our understanding of the molecular make-up of neurons in the arcuate nucleus has advanced markedly over the past 15 years33,55. This advance has permitted the development of a substantial array of molecular genetic tools with which to probe the function of specific sets of arcuate-nucleus neurons. Although the arcuate nucleus contains other cell types, two main opposing types of output neurons have been defined: those that express pro-opiomelanocortin and those that contain Agouti-related protein (AgRP) and neuropeptide-Y. In the arcuate nucleus, pro-opiomelanocortin is processed to produce α-melanocyte stimulating factor (α-MSH) — an agonist for the melanocortin-3 receptor (MC3R) and melanocortin-4 receptor (MC4R) — which collectively promote activity, energy expenditure and suppress food intake. AgRP is an inverse agonist for MC3R and MC4R, opposing central melanocortin action, and neuropeptide-Y is an inhibitory neuropeptide that suppresses energy use and promotes food intake. AgRP neurons also contain and release the inhibitory neurotransmitter GABA (γ-aminobutyric acid) as do most of the other non-pro-opiomelanocortin neurons in the arcuate nucleus56. Insulin and leptin both inhibit AgRP neurons, and insulin action on these neurons suppresses HGP57,58.
Pro-opiomelanocortin neurons and their roles in controlling glucose homeostasis are more complicated. Recent data suggest there are multiple subtypes of pro-opiomelanocortin neurons, including those that respond to leptin or serotonin (each of which activates distinct subtypes of pro-opiomelanocortin neurons) and those that are inhibited by insulin23. Additional arcuate-nucleus neurons that transiently express pro-opiomelanocortin during development, but that later switch their fate to non-pro-opiomelanocortin-expressing cells have also been identified59. Pharmacological studies suggest that melanocortin action increases hepatic glucose output, as does insulin action on pro-opiomelanocortin neurons in a background null for insulin receptors outside the liver15,58. By contrast, expression of LRb in pro-opiomelanocortin neurons in an otherwise LRb-null background suppresses HGP, whereas simultaneous deletion of LRb and insulin receptors from pro-opiomelanocortin neurons increases HGP57,60,61. Impairing PI(3)K action in all pro-opiomelanocortin neurons also promotes insulin resistance62. Thus, although it is clear that the action of insulin and leptin on pro-opiomelanocortin neurons — along with the action of melanocortin in general — modulates HGP, the exact role of this pathway remains unclear. Presumably, the exact genetic, hormonal and physiological milieu of the animal affects the response to melanocortin activation. Although the relevant variables remain to be defined, the indirect regulation of pro-opiomelanocortin neurons by leptin action on other hypothalamic neurons could be a contributor56.
Genetic or pharmacological manipulation of central melanocortin action also alters systemic lipid metabolism in rodents30. Specifically, inhibition of melanocortin action promotes lipid uptake, triglyceride synthesis and accumulation of fat in white adipose tissue; central melanocortin blockade also increases circulating high-density lipoprotein cholesterol63. Importantly, these effects are independent of food intake and body weight. Central melanocortin stimulation results in fat-depot-specific increases in phosphorylated perilipin A and hormone-sensitive lipase, presumably through sympathetic outflow64. Whether these effects of melanocortin action will be recapitulated in humans remains to be determined, but these findings raise the possibility that central melanocortin receptors may be a therapeutic target for the lipid disorders associated with obesity and diabetes.
Melanocortin output
MC3R and MC4R are the predominant melanocortin receptors in the CNS. MC4R is expressed fairly widely in the CNS, whereas MC3R has more limited expression. Both receptors are expressed in the hypothalamus: MC3R is expressed heavily in the arcuate nucleus and ventromedial nucleus, whereas MC4R is expressed particularly strongly in the paraventricular hypothalamic nucleus.
Whole-body deletion of MC4R results in massive hyperphagic obesity, increased lean mass, and hyperinsulinaemia that is out of proportion to the obesity65. Much of the metabolic disturbance observed in Mc4r-knockout mice may be secondary to their obesity, although Mc4r-knockout mice display decreased energy expenditure and increased metabolic efficiency prior to the onset of hyperphagia66. Mc3r-knockout mice on the other hand do not display increased food intake and are more modestly obese, but they also have decreased lean mass and an abnormality in fuel partitioning that preferentially stores energy as fat67. When challenged with a high-fat diet, male Mc3r-knockout mice gain weight rapidly and develop hyperinsulinism similar to that seen with Mc4r-knockout mice67,68,69. This suggests a potentially important role for MC3R in mediating metabolic (rather than energy balance) responses to melanocortin action, although the extent to which this reflects altered nutrient partitioning relative to direct control of other metabolic parameters remains unclear. The function of these two melanocortin receptors in energy balance does not overlap because deletion of both receptors results in greater obesity and metabolic dysfunction than does the loss of either receptor alone69. This additive effect underscores the importance of delineating the identity and neural circuitry used by both MC3R and MC4R70.
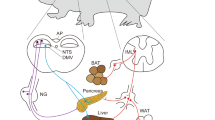
Unfortunately, the low expression of MC3R and MC4R in most melanocortin-responsive neurons has prevented the detailed mapping of the melanocortin circuitry with much cellular specificity. Nevertheless, pharmacological or viral manipulation of melanocortin action by site-specific injection in multiple areas of the brain has been shown to alter energy balance71,72. The effects of such manoeuvres on the metabolic actions of central melanocortins have not been fully explored. Genetic approaches have also been used to identify the specific neurons and circuits under melanocortin control that regulate metabolism, and these have suggested that central melanocortin action is dissociable in the CNS, with discrete brain regions mediating specific melanocortin effects (Box 1 and Fig. 3). Whether the metabolic effects of melanocortins are mediated mainly through neuroendocrine pathways or the autonomic nervous system or both has yet to be definitively determined.

The pro-opiomelanocortin (POMC) neurons of the arcuate nucleus produce melanocortin-receptor agonists, whereas Agouti-related protein (AgRP)-producing neurons antagonize melanocortin action. The two predominant CNS melanocortin receptors, MC3R and MC4R, mediate distinct effects. MC3R is expressed at high levels in the arcuate nucleus and ventromedial nucleus, and mainly controls the conversion of food to fat, nutrient partitioning and build-up of lean mass, whereas MC4R predominantly mediates effects on food intake. The specific neurons that express MC4R are unclear; however, genetic data suggest important roles for the paraventricular hypothalamic (PVH) nucleus and amygdala MC4R in these effects, whereas other data, including pharmacological data, suggest roles for MC4R at other sites such as the ventromedial (VMH) nucleus, lateral hypothalamus (LH), dorsal motor nucleus of the vagus (DMV) and nucleus tractus solitarius (NTS), as well as the lateral parabrachial nucleus. In addition to mediating metabolic effects attributable to body weight, MC4R mediates the control of insulin secretion through the dorsal motor nucleus of the vagus and energy expenditure through the intermediolateral cell column (IML) of the spinal cord. MC4R also contributes to lipid handling in the body, but the MC4R-expressing site or sites that mediate these effects remain undefined.
The brain and the physiological control of metabolism
Although the participation of the brain in the response to hypoglycaemia is well-established and has an impact on the therapy for patients with insulin-deficient diabetes, the more recently described role of sensing of hormones and nutrients in the brain for the control of whole-body metabolism was initially met with some scepticism73. Specifically, the roles of these brain systems have been defined largely at the limits of physiology — in many cases, under hyperinsulinaemic 'clamp' conditions that lock the contribution of the islets at a specific level, so that alterations in insulin or glucagon secretion cannot compensate for changes in glucose production or disposal. The study of these brain systems often requires such conditions because the reserve insulin and glucagon secretory capacity of the islets can otherwise compensate for alterations in glucose handling by the major metabolic organs, obscuring the effect of the brain on the control of glucose production and disposal. To what extent these brain systems are relevant is controversial. They do, however, have a role in leptin deficiency: not only does leptin ameliorate hyperglycaemia in animals3 that are genetically leptin deficient, but it also improves metabolic function — by decreasing blood sugar and lipids — in humans and mice with lipodystrophy syndromes74,75. In lipodystrophy (in which the lack of adipose tissue results in functional leptin deficiency), circulating insulin concentrations are elevated, and exogenous insulin administration fails to adequately suppress hyperglycaemia and hyperlipidaemia. Thus, in this situation, leptin action (presumably in the brain) improves metabolic parameters that cannot be ameliorated by very high insulin levels. More commonly, brain leptin and insulin make important contributions to metabolic homeostasis in β-cell failure (for example, in insulin-deficient diabetes)76,77. In rodents with insulin insufficiency due to chemical or autoimmune β-cell destruction, leptin (including intracerebroventricular leptin) normalizes blood glucose in the absence of exogenous insulin, and improves glycaemic control as an adjunct to insulin therapy. Leptin action is also likely to have an important role in glucoprivation, in which insulin is either fixed and artificially high (in insulin overdose) or fixed near zero (with inhibitors of glucose metabolism, such as 2-deoxyglucose).
Thus, the need to eliminate the islet contribution to metabolic control in order to observe the effects of the brain-hormone and nutrient-sensing pathways does not diminish the importance of these brain systems in the control of peripheral metabolic homeostasis. Instead, it suggests that the CNS serves to raise and lower the overall tone of the peripheral response, and dominates the control of metabolic homeostasis when pancreatic islets are unable to compensate.
Questions for the future
Although we now have a much clearer picture about how the brain contributes to metabolic homeostasis than we did a decade ago, many questions still remain. CNS perturbations have been seen to modulate processes such as glucose production and lipid metabolism in the periphery, but the circuits and mechanisms that intervene between the brain and systemic nutrient handling are poorly defined. Certainly, the SNS contributes to some of the outcomes, such as in the promotion of glucose disposal into skeletal muscle. Similarly, vagotomy — including sub-diaphragmatic vagotomy — prevents many of the changes in hepatic-glucose metabolism that occur in response to intracerebroventricular or MBH-directed injections, suggesting that the parasympathetic nervous system has a role3. Additionally, the control of glucagon secretion may contribute to the suppression of blood glucose by CNS leptin76. Determining how each of these systems contributes to the various responses to brain nutrient and hormone action, and the specific brain regions and circuits that control the response, will be important for our understanding of the physiology of metabolism.
Within the brain itself, we have an enormous amount left to learn. In addition to resolving the mechanisms through which the leptin and melanocortin systems contribute to various metabolic conditions, perhaps the most important issue to resolve is the cellular specificity of the system. Clearly, some progress has been made in this regard, especially in the arcuate nucleus, for which some genetic reagents are available for use in analysis; however, we desperately need other methods of genetic analysis to examine defined subpopulations of ventromedial-nucleus and arcuate-nucleus neurons, and to clarify the roles of other hypothalamic and brainstem circuits that have been implicated in metabolic control.
References
Levin, B. E. Neuronal glucose sensing: still a physiological orphan? Cell Metab. 6, 252–254 (2007).
Levin, B. E. & Sherwin, R. S. Peripheral glucose homeostasis: does brain insulin matter? J. Clin. Invest. 121, 3392–3395 (2011).
Pocai, A., Obici, S., Schwartz, G. J. & Rossetti, L. A brain–liver circuit regulates glucose homeostasis. Cell Metab. 1, 53–61 (2005).
Taborsky, G. J. Jr. Islets have a lot of nerve! Or do they? Cell Metab. 14, 5–6 (2011).
Wiesli, P. et al. Acute psychological stress affects glucose concentrations in patients with type 1 diabetes following food intake but not in the fasting state. Diabetes Care 28, 1910–1915 (2005).
McCowen, K. C., Malhotra, A. & Bistrian, B. R. Stress-induced hyperglycemia. Crit. Care Clin. 17, 107–124 (2001).
Levin, B. E., Routh, V. H., Kang, L., Sanders, N. M. & Dunn-Meynell, A. A. Neuronal glucosensing: what do we know after 50 years? Diabetes 53, 2521–2528 (2004)
Evans, M. L. & Sherwin, R. S. Blood glucose and the brain in diabetes: between a rock and a hard place? Curr. Diab. Rep. 2, 101–102 (2002).
Ritter, S., Li, A. J., Wang, Q. & Dinh, T. T. The value of looking backward: the essential role of the hindbrain in counter regulatory responses to glucose deficit. Endocrinology 152, 4019–4032 (2011).
Schwartz, M. W. & Porte, D. Jr. Diabetes, obesity, and the brain. Science 307, 375–379 (2005).
Zhang, Y. et al. Positional cloning of the mouse obese gene and its human homologue. Nature 372, 425–432 (1994).
Rossetti, L. et al. Short term effects of leptin on hepatic gluconeogenesis and in vivo insulin action. J. Biol. Chem. 272, 27758–27763 (1997).
Liu, L. et al. Intracerebroventricular leptin regulates hepatic but not peripheral glucose fluxes. J. Biol. Chem. 273, 31160–31167 (1998).
Obici, S., Zhang, B. B., Karkanias, G. & Rossetti, L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nature Med. 8, 1376–1382 (2002).
Gutierrez-Juarez, R., Obici, S. & Rossetti, L. Melanocortin-independent effects of leptin on hepatic glucose fluxes. J. Biol. Chem. 279, 49704–49715 (2004).
Pocai, A. et al. Central leptin acutely reverses diet-induced hepatic insulin resistance. Diabetes 54, 3182–3189 (2005).
Ahima, R. S. Central actions of adipocyte hormones. Trends Endocrinol. Metab. 16, 307–313 (2005).
D'Alessio, D. A., Sandoval, D. A. & Seeley, R. J. New ways in which GLP-1 can regulate glucose homeostasis. J. Clin. Invest. 115, 3406–3408 (2005).
Briggs, D. I. & Andrews, Z. B. Metabolic status regulates ghrelin function on energy homeostasis. Neuroendocrinology 93, 48–57 (2011).
Heppner, K. M., Tong, J., Kirchner, H., Nass, R. & Tschop, M. H. The ghrelin O-acyltransferase-ghrelin system: a novel regulator of glucose metabolism. Curr. Opin. Endocrinol. Diabetes Obes. 18, 50–55 (2011).
Kleinridders, A., Konner, A. C. & Bruning, J. C. CNS-targets in control of energy and glucose homeostasis. Curr. Opin. Pharmacol. 9, 794–804 (2009).
Hribal, M. L., Oriente, F. & Accili, D. Mouse models of insulin resistance. Am. J. Physiol. Endocrinol. Metab. 282, E977–E981 (2002).
Williams, K. W., Scott, M. M. & Elmquist, J. K. Modulation of the central melanocortin system by leptin, insulin, and serotonin: co-ordinated actions in a dispersed neuronal network. Eur. J. Pharmacol. 660, 2–12 (2011).
Minokoshi, Y. et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428, 569–574 (2004).
Pocai, A., Muse, E. D. & Rossetti, L. Did a muscle fuel gauge conquer the brain? Nature Med. 12, 50–51 (2006).
Cota, D. et al. Hypothalamic mTOR signaling regulates food intake. Science 312, 927–930 (2006).
Diano, S. & Horvath, T. L. Mitochondrial uncoupling protein 2 (UCP2) in glucose and lipid metabolism. Trends Mol. Med. 18, 52–58 (2012).
Lam, T. K. et al. Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nature Med. 11, 320–327 (2005).
Vanpatten, S., Karkanias, G. B., Rossetti, L. & Cohen, D. E. Intracerebroventricular leptin regulates hepatic cholesterol metabolism. Biochem. J. 379, 229–233 (2004).
Nogueiras, R. et al. The central melanocortin system directly controls peripheral lipid metabolism. J. Clin. Invest. 117, 3475–3488 (2007). Using pharmacological and genetic approaches in rodents, this article demonstrates a role for endogenous CNS melanocortin action in the control of whole-body lipid metabolism.
Burdakov, D. & Alexopoulos, H. Metabolic state signalling through central hypocretin/orexin neurons. J. Cell. Mol. Med. 9, 795–803 (2005).
Myers, M. G. Jr, Munzberg, H., Leinninger, G. M. & Leshan, R. L. The geometry of leptin action in the brain: more complicated than a simple ARC. Cell Metab. 9, 117–123 (2009). This review highlights the idea that most LRb neurons in the brain are distinct from the canonical pro-opiomelanocortin and AgRP neurons and lie outside the arcuate nucleus.
Xu, Y., Elmquist, J. K. & Fukuda, M. Central nervous control of energy and glucose balance: focus on the central melanocortin system. Ann. NY Acad. Sci. 1243, 1–14 (2011).
Sadagurski, M. et al. IRS2 signaling in LepR-b neurons suppresses Foxo1 to control energy balance independently of leptin action. Cell Metab. 15, 703–712 (2012).
Taguchi, A., Wartschow, L. M. & White, M. F. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science 317, 369–372 (2007).
Patterson, C. M. et al. Leptin action via LepR-b Tyr1077 contributes to the control of energy balance and female reproduction. Mol. Metab. http://dx.doi.org/10.1016/j.molmet.2012.05.001 (25 July 2012).
Bates, S. H. et al. STAT3 signaling is required for leptin regulation of energy balance but not reproduction. Nature 421, 856–859 (2003).
Buettner, C. et al. Critical role of STAT3 in leptin's metabolic actions. Cell Metab. 4, 49–60 (2006).
Robertson, S. et al. Insufficiency of Janus kinase 2-autonomous leptin receptor signals for most physiologic leptin actions. Diabetes 59, 782–790 (2010).
Jiang, L. et al. Tyrosine-dependent and -independent actions of leptin receptor in control of energy balance and glucose homeostasis. Proc. Natl Acad. Sci. USA 105, 18619–18624 (2008).
Barnes, M. B., Lawson, M. A. & Beverly, J. L. Rate of fall in blood glucose and recurrent hypoglycemia affect glucose dynamics and noradrenergic activation in the ventromedial hypothalamus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R1815–R1820 (2011).
Levin, B. E., Magnan, C., Dunn-Meynell, A. & Le Foll, C. Metabolic sensing and the brain: who, what, where, and how? Endocrinology 152, 2552–2557 (2011).
Chan, O., Lawson, M., Zhu, W., Beverly, J. L. & Sherwin, R. S. ATP-sensitive K+ channels regulate the release of GABA in the ventromedial hypothalamus during hypoglycemia. Diabetes 56, 1120–1126 (2007).
Kang, L. et al. Glucokinase is a critical regulator of ventromedial hypothalamic neuronal glucosensing. Diabetes 55, 412–420 (2006).
Elias, C. F. et al. Chemical characterization of leptin-activated neurons in the rat brain. J. Comp. Neurol. 423, 261–281 (2000).
Haque, M. S. et al. Role of the sympathetic nervous system and insulin in enhancing glucose uptake in peripheral tissues after intrahypothalamic injection of leptin in rats. Diabetes 48, 1706–1712 (1999).
Dhillon, H. et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 49, 191–203 (2006).
Bingham, N. C., Anderson, K. K., Reuter, A. L., Stallings, N. R. & Parker, K. L. Selective loss of leptin receptors in the ventromedial hypothalamic nucleus results in increased adiposity and a metabolic syndrome. Endocrinology 149, 2138–2148 (2008).
Klockener, T. et al. High-fat feeding promotes obesity via insulin receptor/PI3K-dependent inhibition of SF-1 VMH neurons. Nature Neurosci. 14, 911–918 (2011).
Kim, K. W. et al. FOXO1 in the ventromedial hypothalamus regulates energy balance. J. Clin. Invest. 122, 2578–2589 (2012).
Bergen, H. T., Mizuno, T. M., Taylor, J. & Mobbs, C. V. Hyperphagia and weight gain after gold-thioglucose: relation to hypothalamic neuropeptide Y and proopiomelanocortin. Endocrinology 139, 4483–4488 (1998).
Coppari, R. et al. The hypothalamic arcuate nucleus: a key site for mediating leptin's effects on glucose homeostasis and locomotor activity. Cell Metab. 1, 63–72 (2005). This article reports the reactivation of LRb specifically in the arcuate nucleus; although this only slightly altered food intake and adiposity, glucose homeostasis was markedly improved, suggesting an important role for arcuate-nucleus leptin action in the control of glucose homeostasis independently of energy balance.
Morton, G. J. et al. Arcuate nucleus-specific leptin receptor gene therapy attenuates the obesity phenotype of Koletsky (fak/fak) rats. Endocrinology 144, 2016–2024 (2003).
Morton, G. J. et al. Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab. 2, 411–420 (2005). Using molecular–viral and pharmacological approaches, the authors show that insulin-dependent signalling pathways in the arcuate nucleus are crucial for the control of glucose homeostasis by leptin.
Schwartz, M. W. Central nervous system regulation of food intake. Obesity (Silver Spring) 14, 1S–8S (2006).
Vong, L. et al. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71, 142–154 (2011).
Hill, J. W. et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 11, 286–297 (2010).
Lin, H. V. et al. Divergent regulation of energy expenditure and hepatic glucose production by insulin receptor in agouti-related protein and POMC neurons. Diabetes 59, 337–346 (2010).
Padilla, S. L., Carmody, J. S. & Zeltser, L. M. Pomc-expressing progenitors give rise to antagonistic neuronal populations in hypothalamic feeding circuits. Nature Med. 16, 403–405 (2010).
Berglund, E. D. et al. Direct leptin action on POMC neurons regulates glucose homeostasis and hepatic insulin sensitivity in mice. J. Clin. Invest. 122, 1000–1009 (2012).
Huo, L. et al. Leptin-dependent control of glucose balance and locomotor activity by POMC neurons. Cell Metab. 9, 537–547 (2009).
Hill, J. W. et al. Phosphatidyl inositol 3-kinase signaling in hypothalamic proopiomelanocortin neurons contributes to the regulation of glucose homeostasis. Endocrinology 150, 4874–4882 (2009).
Perez-Tilve, D. et al. Melanocortin signaling in the CNS directly regulates circulating cholesterol. Nature Neurosci. 13, 877–882 (2010).
Shrestha, Y. B. et al. Central melanocortin stimulation increases phosphorylated perilipin A and hormone-sensitive lipase in adipose tissues. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299, R140–R149 (2010).
Huszar, D. et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88, 131–141 (1997).
Ste, M. L., Miura, G. I., Marsh, D. J., Yagaloff, K. & Palmiter, R. D. A metabolic defect promotes obesity in mice lacking melanocortin-4 receptors. Proc. Natl Acad. Sci. USA 97, 12339–12344 (2000).
Butler, A. A. et al. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology 141, 3518–3521 (2000).
Sutton, G. M. et al. Diet–genotype interactions in the development of the obese, insulin-resistant phenotype of C57BL/6J mice lacking melanocortin-3 or -4 receptors. Endocrinology 147, 2183–2196 (2006).
Chen, A. S. et al. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat Genet. 26, 97–102 (2000).
Butler, A. A. The melanocortin system and energy balance. Peptides 27, 281–290 (2006).
Grill, H. J. Distributed neural control of energy balance: contributions from hindbrain and hypothalamus. Obesity (Silver Spring) 14, 216S–221S (2006).
de Backer, M. W. et al. Melanocortin receptor-mediated effects on obesity are distributed over specific hypothalamic regions. Int. J. Obes. (Lond.) 35, 629–641 (2011).
Cherrington, A. D. The role of hepatic insulin receptors in the regulation of glucose production. J. Clin. Invest. 115, 1136–1139 (2005).
Shimomura, I., Hammer, R. E., Ikemoto, S., Brown, M. S. & Goldstein, J. L. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature 401, 73–76 (1999).
Oral, E. A. et al. Leptin-replacement therapy for lipodystrophy. N. Engl. J. Med. 346, 570–578 (2002).
Wang, M. Y. et al. Leptin therapy in insulin-deficient type I diabetes. Proc. Natl Acad. Sci. USA 107, 4813–4819 (2010).
German, J. P. et al. Leptin deficiency causes insulin resistance induced by uncontrolled diabetes. Diabetes 59, 1626–1634 (2010). In this article, the authors show that brain leptin injection — in the absence of endogenous or exogenous insulin — suffices to normalize blood glucose.
Grill, H. J., Ginsberg, A. B., Seeley, R. J. & Kaplan, J. M. Brainstem application of melanocortin receptor ligands produces long-lasting effects on feeding and body weight. J. Neurosci. 18, 10128–10135 (1998).
Williams, D. L., Kaplan, J. M. & Grill, H. J. The role of the dorsal vagal complex and the vagus nerve in feeding effects of melanocortin-3/4 receptor stimulation. Endocrinology 141, 1332–1337 (2000).
Boghossian, S., Park, M. & York, D. A. Melanocortin activity in the amygdala controls appetite for dietary fat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R385–R393 (2010).
Balthasar, N. et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 123, 493–505 (2005).
Rossi, J. et al. Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab. 13, 195–204 (2011).
Begriche, K. et al. Melanocortin-3 receptors are involved in adaptation to restricted feeding. Genes Brain Behav. 11, 291–302 (2012).
Acknowledgements
The authors thank members of the Myers and Olson laboratories for discussions and scientific insight. M.G.M. is supported by the Marilyn H. Vincent Foundation and by grants from the National Institutes of Health and the American Heart Association.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reprints and permissions information is available at www.nature.com/reprints.
Rights and permissions
About this article
Cite this article
Myers, M., Olson, D. Central nervous system control of metabolism. Nature 491, 357–363 (2012). https://doi.org/10.1038/nature11705
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature11705
This article is cited by
-
Hypothalamic control of energy expenditure and thermogenesis
Experimental & Molecular Medicine (2022)
-
Brain fractalkine-CX3CR1 signalling is anti-obesity system as anorexigenic and anti-inflammatory actions in diet-induced obese mice
Scientific Reports (2022)
-
PI3K signalling at the intersection of cardio-oncology networks: cardiac safety in the era of AI
Cellular and Molecular Life Sciences (2022)
-
Stimulation of the hepatoportal nerve plexus with focused ultrasound restores glucose homoeostasis in diabetic mice, rats and swine
Nature Biomedical Engineering (2022)
-
Prostaglandin in the ventromedial hypothalamus regulates peripheral glucose metabolism
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
