
Abstract
The light-induced ultrafast switching between molecular isomers norbornadiene and quadricyclane can reversibly store and release a substantial amount of chemical energy. Prior work observed signatures of ultrafast molecular dynamics in both isomers upon ultraviolet excitation but could not follow the electronic relaxation all the way back to the ground state experimentally. Here we study the electronic relaxation of quadricyclane after exciting in the ultraviolet (201 nanometres) using time-resolved gas-phase extreme ultraviolet photoelectron spectroscopy combined with non-adiabatic molecular dynamics simulations. We identify two competing pathways by which electronically excited quadricyclane molecules relax to the electronic ground state. The fast pathway (<100 femtoseconds) is distinguished by effective coupling to valence electronic states, while the slow pathway involves initial motions across Rydberg states and takes several hundred femtoseconds. Both pathways facilitate interconversion between the two isomers, albeit on different timescales, and we predict that the branching ratio of norbornadiene/quadricyclane products immediately after returning to the electronic ground state is approximately 3:2.

Similar content being viewed by others
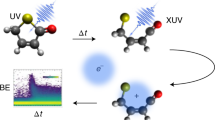
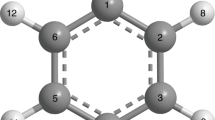
Main
Molecular photoswitches are an exciting area of research with emerging applications in catalysis, photochromic materials, molecular machines, logic devices and energy storage1. The last example includes molecular solar thermal (MOST) energy storage solutions, whereby molecular isomers are exploited to absorb, store and later release solar energy as heat2,3. An important model system for such applications is the photoswitchable pair of isomers quadricyclane (QC), a highly strained multicyclic hydrocarbon, and its lower-energy isomer norbornadiene (NBD)4,5,6,7,8,9, whose International Union of Pure and Applied Chemistry (IUPAC) names are tetracyclo[3.2.0.02,7.04,6]heptane (QC) and bicyclo[2.2.1]hepta-2,5-diene (NBD). The isomers, shown in Fig. 1, interconvert upon photoabsorption in the ultraviolet (UV) range10,11,12,13. More broadly, the photoisomerization of NBD → QC is an example of a [2 + 2] cycloaddition reaction, and the behaviour of the molecule is reminiscent of the prototypical ethylene dimerization, with the bridging framework acting to hold the ethylene moieties together14,15. We investigate the mechanism of the reverse interconversion, QC → NBD, which is of both fundamental photochemical interest and practical importance since it represents the undesired UV-induced photoreversion process in QC/NBD-derived MOST systems.

The isomerization reaction breaks (or forms) the 1–2 and 3–4 carbon–carbon bonds in QC, with the energy of the QC ground state located ~1 eV above the ground state of NBD. The interconversion can be driven by light absorption, allowing the system to act as a molecular photoswitch.
Early time-resolved ion mass spectrometry experiments on gas-phase NBD noted signatures of ultrafast molecular dynamics upon UV excitation at 200 nm (6.20 eV), which were speculatively attributed to transitions from a superposition of Rydberg and valence states to the electronic ground state via a series of conical intersections on a sub-100 femtosecond timescale15. A subsequent study on QC excited at 208 nm (5.96 eV) employed time-resolved Rydberg fingerprint photoelectron spectroscopy16. The latter work suggested that QC isomerizes to NBD on the 3s Rydberg state within 140 fs and that relaxation to the electronic ground state occurs within 400 fs. Notably, that study could not observe the dynamics all the way to the ground state since the probe photons had insufficient energy to ionize ground-state molecules. Theoretical work has focused on the photorelaxation of NBD, examining the electronic structure4,9,17 and the dynamics18,19. In both cases, the key role of interactions between the Rydberg manifold and valence states in NBD was highlighted.
In the present study, we use time-resolved photoelectron spectroscopy (TRPES) with extreme ultraviolet (XUV) probe pulses, capable of tracking the entire reaction path from the initial UV-excited states through to the electronic ground states of both isomers. TRPES with XUV pulses is a powerful method for mapping complex chemical dynamics20,21,22,23,24. Our experimental data, supported by high-level simulations, provide a much more complete description of the deactivation mechanisms in QC, thus providing mechanistic insights that can be used to design more-efficient MOST systems.
Results
We initiate the photochemical reaction in gas-phase QC by a UV excitation pump pulse at 200.6 nm (6.18 eV), followed by an XUV ionization probe pulse at 18.97 eV (65.35 nm) from a seeded free-electron laser (FEL). The TRPES spectra obtained as the difference between the spectra acquired with and without the pump pulse (Methods and Supplementary Section 1.4 for details) are shown as a two-dimensional false-colour map in Fig. 2a. One-dimensional spectra obtained by averaging over selected time-delay ranges are displayed in Fig. 2b,c (additional spectra are shown in Extended Data Fig. 2). Several prominent features are apparent. A narrow and long-lived feature at ~2.3 eV binding energy (BE), visible as a faint horizontal line in Fig. 2a and as a clear peak in Fig. 2b, is assigned to two of the three 3p Rydberg states of QC in accordance with the literature16. The 3s Rydberg state can be seen at a BE of ~2.9 eV in Fig. 2b but has a weaker signature than in earlier studies16, suggesting less excitation to the 3s state at 200.6 nm compared to 208 nm. Figure 2a also shows a spectrally broad and short-lived vertical feature in the BE range of ~1.5–7 eV, which merges into a long-lived horizontal band at ~7–8 eV BE, both of which were not observed in the earlier work that did not have a sufficient photon energy to ionize this BE region. These observations suggest the existence of a rapid (<100 fs) deexcitation pathway leading to the formation of vibrationally hot photoproducts, while the longer timescale of the Rydberg-associated features indicates the existence of a second, slower decay mechanism, which corresponds to the pathway identified in earlier work16. Finally, there is a pronounced negative signal in the difference spectrum in the BE range 8–9 eV, assigned to the depletion of the QC ground state, followed by a partial recovery.

a, Photoelectron spectra as a function of BE (with respect to the first ionization potential, D0) and pump–probe time delay between UV and XUV pulses, plotted as the electron yield difference between spectra taken with and without the UV-excitation pulse (Extended Data Fig. 1). The respective electron yield at each delay is normalized to the FEL pulse energy before subtraction, and the difference signal is normalized to the integrated electron yield without UV excitation, both summed over all shots at the respective delay. Negative delays correspond to the FEL pulse preceding the UV excitation pulse. b,c, Photoelectron BE spectra in the excited-state region (b) and at large BE values that sample the electronic ground state (c) at specific time delays. d,e, Delay-dependent electron yield in the denoted BE regions, normalized to have the same peak amplitudes. The data and error bands (shading) in b and c, and the error bars in d and e, represent the mean value of approximately 40,000 single-shot digitizer traces and the 68% confidence interval obtained from a bootstrapping analysis (Methods).
Delay-dependent photoelectron yields in selected spectral regions are shown in Fig. 2d,e. They display a nearly Gaussian shape at low BE values with a temporal width that is mostly given by the instrument response function (Methods), while clear exponential ‘tails’ are visible in the range of the Rydberg excitations and in the high-BE yields. Least-square fits of the delay-dependent yields are provided in Supplementary Figs. 9–11.
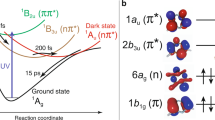
The nature of the previously unreported broad, short-lived feature with BE values in the range ~1.5–7 eV is investigated further using high-level electronic structure calculations and non-adiabatic dynamics simulations. Figure 3 shows calculated potential energy curves for the ground (S0) and first five singlet excited (S1–S5) electronic states identified as being active in the dynamics probed by the present experiment, plotted along a linear interpolation in internal coordinates (LIIC) from the equilibrium geometry of QC(S0) to the S1/S0 minimum energy conical intersection (MECI) and then continued along a second LIIC from this MECI to the NBD(S0) equilibrium geometry. The dominant electronic characters and adiabatic energies at the equilibrium geometries and the MECI are given in Table 1. In the Franck–Condon (FC) region of QC, the S1 adiabatic state has 3s Rydberg character; S3 and S4 are 3p Rydberg states; and S2 and S5 are a pair of mixed 3px/valence (V) states. As the QC molecule approaches the MECI geometry, the S1 state acquires valence character, which persists into the FC region of NBD. A complete discussion of the vertical electronic spectra of both isomers, complementing the work by Palmer et al.17,25, is reserved for a separate publication26.

Potential energy curves for the ground state S0 and excited states S1–S5, as well as the cation ground state D0 and first excited state D1 (dashed lines), calculated along the LIICs, first from QC to the S1/S0 MECI and then from the MECI to NBD, using RMS-CASPT2(2,6)/6-31G* + D (Methods and Supplementary Section 2.1). The two proposed reaction pathways are indicated schematically by black arrows, with the slow Rydberg pathway supporting vibrational motions on the Rydberg states before relaxing via the S1 state, while the fast valence pathway descends on S1, bypassing the MECI on the initial descent, before crossing onto the S0 ground state to form both products. The molecular geometry at the MECI has a distinct rhombic distortion (Extended Data Fig. 3). The Estorage indicates the energy stored in QC relative to NBD (~1 eV). Corresponding vertical excitation energies are listed in Table 1, and a plot including higher excited states and the approximate state characters is provided in Extended Data Figure 4.
Discussion
The potential energy curves in Fig. 3 suggest the possible mechanisms that underpin the features observed in the TRPES data, with the two pathways indicated schematically by black arrows. From the QC ground state, the bandwidth of the pump pulse (0.6 nm) allows excitation to a manifold of closely spaced states with 3p Rydberg character (Extended Data Fig. 7). Population in the 3py/z Rydberg states (S3 and S4 in the FC region) then evolves via a (comparatively) slow Rydberg pathway on the rather flat Rydberg manifold at excitation energies of ~6 eV; we anticipate that this would correspond to the pathway observed previously16. Due to the similar topographies of the Rydberg and ground-state ion (D0) adiabatic potential energy surfaces, the BE values for the slow Rydberg pathway can be expected to be relatively constant—as observed experimentally. Conversely, in the fast valence pathway, the population in the strongly mixed Rydberg–valence 3px/V state (S2 in the FC region) is subject to efficient non-adiabatic coupling to S1, funnelling down towards a conical intersection with the S0 state. In the proposed model, the BE values for the fast valence pathway would increase rapidly, consistent with the short-lived structure observed for experimental BE values in the range of ~1.5–7 eV.
We confirm these hypothesized mechanisms using non-adiabatic mixed quantum–classical trajectory simulations (Methods). The simulations divulge that the photodynamics of excited QC are strongly dependent on the initial excited state. Figure 4 shows the results separated into two sets. Dynamics initiated on the S2 (3px/V) state are labelled fast valence, shown in the top row, while dynamics initiated on the S3 and S4 (3py and 3pz) states are labelled slow Rydberg and shown in the bottom row. The plots in Fig. 4a show false-colour maps of the nuclear dynamics along the coordinate associated with the making and breaking of the 1–2 and 3–4 carbon–carbon bonds (r12 and r34, respectively) during the interconversion between QC and NBD (Methods). The left and right sides show the trajectories on the excited states and ground state, respectively. Figure 4b shows the time-dependent electronic-state populations and the fraction of population best defined as NBD-like, while the false-colour maps in Fig. 4c finally show the contributions to the simulated TRPES spectrum to aid comparison with the experimental data in Fig. 2a.

The simulations are separated into the fast valence mechanism (top row, corresponding to trajectories starting on the S2 state) and the slow Rydberg mechanism (bottom row, trajectories originating on the S3/4 Rydberg states). A zoomed-in depiction of the first 200 fs is shown in Extended Data Fig. 6. a, False-colour maps for average carbon–carbon distance rCC = (r12 + r34)/2 versus time (Methods). The left and right sides show the nuclear dynamics in the excited states and the ground state (S0), respectively. b, Populations of the electronic states as a function of time, where Sn ≥ 2 shows the cumulative population of the excited states other than S1. These plots also show the fraction of molecules with NBD-like geometry (defined as rCC > 2.0 Å). c, Simulated TRPES data, convoluted by Gaussian functions with full-width at half-maximum of 0.1 eV and 94 fs. Signals with BE > 3 eV (below the horizontal dashed line) have been multiplied by a factor of three prior to display.
We first examine the simulation results for the fast valence mechanism shown in the top row of Fig. 4, corresponding to trajectories initiated on the S2 (3px/V) state. One-photon excitation to the 3px/V state from the S0 state is symmetry forbidden at the exact equilibrium geometry, but the transition gains strength from the Herzberg–Teller effect (45% excitation fraction; Methods). The nuclear dynamics in the electronic excited states (Fig. 4a, left side) break the carbon–carbon bonds along r12 and r34, allowing the molecule to deform towards NBD-like geometries (rCC =(r12 + r34)/2 > 2.0 Å). The wave packet then returns towards QC-like geometries (smaller rCC) and crosses to the S0 ground state via a conical intersection, appearing on the right side. It immediately partitions into QC-like and NBD-like geometries. The recurrences in the upper map reflect the persistence of the ‘butterfly’-like vibrational motion of the NBD products when they first reach the S0 state (with wave number \(\widetilde{\upsilon }\) ≈ 428 cm–1)27. The corresponding motion in QC has a higher wave number (\(\widetilde{\upsilon }\) ≈ 721 cm–1)28 but can also be discerned in the lower maps.
This picture is confirmed in the upper plot of Fig. 4b, which shows almost immediate population transfer from S2 to S1 via internal conversion (zoomed-in view in Extended Data Fig. 6), followed by another rapid internal conversion to S0, with only a small fraction (<10%) of the population remaining in the Rydberg manifold after 1 ps. The time-dependent fraction of molecules with NBD-like geometries (that is, with rCC > 2.0 Å) in the same graph shows a rapid early increase and then decline (mirroring the S1 state population), finally converging to a branching ratio of ~3:2 for NBD-like to QC-like geometries in the S0 state. This ratio is set by the decay dynamics rather than energetics, as the timescales are far too short for thermalization on the ground state.
The theoretically predicted TRPES signal in Fig. 4c shows a curved band of intensity evolving from the BE values of the Rydberg state (~2.1 eV) down to that of the S0 state (~7 eV), in good accord with the ‘hockey-stick’-shaped feature in the experimental data (Fig. 2a). In part, this broad feature reflects the very fast motion of the wave packet down the 3px/V surface, with a corresponding rapid increase in the BE with respect to D0. In addition, there is a smaller contribution from the D1 ionization channel when the wave packet approaches the MECI. Figure 4a (upper left map) shows that the bond breaks and reforms in under 100 fs, faster than the instrument response function of the experiment, making these features appear almost simultaneously at time t = 0. A more detailed analysis of the components of the photoelectron signal is provided in the Supplementary Information.
Next, we consider the slow Rydberg pathway with trajectories initiated on the S3/4 (3py/z) states, shown in the bottom row of Fig. 4. Already on the left side of Fig. 4a, it is apparent that the excited-state dynamics persist for substantially longer than in the fast valence mechanism, and that the dynamics are more centred at QC-like molecular geometries (rCC < 2.0 Å), with only a small fraction of the trajectories exploring NBD-like geometries in the 3p excited states, discernable as a faint signal at rCC > 2.0 Å. Examining the populations in Fig. 4b, we see a comparatively slow but steady deactivation of the Rydberg manifold states (S2 and higher) with a matching increase in S0 population. The decay proceeds via the same 3px/V state as the fast valence mechanism, evidenced by the build-up of population in the S1 intermediate state, which acts as a funnel towards the S0 state via the conical intersection. It appears that this conical intersection has a strong influence on the NBD/QC branching ratio in the S0 state, since the fraction of NBD (Fig. 4b, bottom) approaches the same value as the fast valence pathway.
The simulated TRPES signal in Fig. 4c shows a long-lived feature with BE ≈ 2.1 eV, corresponding to the 3p Rydberg states (with potentials along the LIIC of interest that closely track that of the D0 ionic state), in good accord with the experimental data, which show the corresponding feature attributable to ionization from these Rydberg states at BE ≈ 2.3 eV (Fig. 2a). At long times, the TRPES signal seen in the slow Rydberg and fast valence dynamics are essentially identical. Both pathways involve similar ultimate decays to the S0 state, yielding similar branching ratios of hot QC and NBD products. Note that the vertical ionization potentials of QC and NBD are similar, differing by ~0.3 eV (Methods). Thus, the TRPES signals of hot ground-state QC and NBD species overlap, and the experimentally observed bleach recovery in Fig. 2e includes components from both isomers.
Notwithstanding the very good agreement between experiment and theory, some discrepancies are evident. The simulations return longer Rydberg state lifetimes than observed experimentally, which could reflect shortcomings in the electronic structure theory, for example, regarding the strength and location of the couplings with the 3px/V state, additional decay paths not accounted for by the active space used or the accuracy of the predicted ionization cross-sections.
In summary, TRPES studies using XUV photons from a seeded FEL have provided new and detailed insights into the relaxation pathways of electronically excited QC molecules formed by photoexcitation at 200.6 nm, from Rydberg states through to vibrationally excited hot ground-state QC and NBD molecules. We present observations of the dominant relaxation pathway in QC, involving strong Rydberg–valence coupling, alongside the previously observed Rydberg-state-mediated isomerization16. The present combined experimental and theoretical study shows that all three 3p Rydberg states of QC are excited by the 200.6 nm pump pulse. The lowest-energy (3px) state is strongly coupled to a valence state, giving rise to the fast valence pathway characterized by ultrafast electronic decay and nuclear motion. The other two support initial nuclear dynamics in the Rydberg manifold, prior to comparatively slower decay to the ground state (identified here as the slow Rydberg pathway). The simulations suggest that ~40% of the vibrationally excited ground-state products sampled at pump–probe delays of ~1 ps correspond to QC-like structures, irrespective of the decay pathway followed, and that this early-time partitioning is determined by the molecular dynamics rather than thermodynamics.
The insights provided by the present study point to key features that affect the photodynamics: the partial absorption cross-sections, the strength of the non-adiabatic couplings to the valence excited state and the relative stabilities and geometries of the regions of maximal interstate coupling, that is, the conical intersection between the S1 and S0 states. For potential applications, these properties could be manipulated using substituent groups, using spatial confinement and/or by the choice of excitation wavelength5,8,19,29. For instance, the detailed nature of valence/Rydberg mixing in the valence state controls its energy and accessibility, which could be used to affect the system’s function as a molecular photoswitch. The manner in which the wave packet approaches the S1/S0 conical intersection region is also important, and this could be manipulated either in the preparation of the wave packet or by altering the position and shape of the conical intersection.
From a broader photochemical perspective, one might conjecture that the bipartite dynamics found here will be a common feature whenever a manifold of Rydberg states intersects valence states in the FC region30,31,32. The excitation energies to Rydberg states typically fall within ~3 eV of the associated ionization limit, EIP (IP, ionization potential), and, in most small to medium-sized molecules, will overlap with those of one or more excited valence states. Rydberg excitations will manifest as a longer-lived trapping component, whereas excitations to valence states (or, as here, to a mixed Rydberg–valence state) will lead to more-rapid relaxation that gives rise to broad features in the photoelectron spectrum. Broadband excitation can excite both types of states, leading to both types of mechanism. Bifurcation of wave packets has been reported previously, for example, in the photoisomerization of stilbene33 and in photoexcited inverse dithienylethene derivatives34, but in most such cases studied hitherto, the bifurcation arises as a result of sampling a ‘transition-state’-like region of a common excited state (for example, a ππ* state), the potential energy surface of which supports multiple minima.
The current results demonstrate the utility of TRPES with XUV probes for characterizing complex photochemical mechanisms. Comparison with previous experiments16 indicates that a systematic examination of the effect of excitation wavelength would be useful. Ideally, this should be coupled to complementary experimental work more sensitive to molecular geometry. For instance, ultrafast electron diffraction and ultrafast X-ray scattering experiments35,36,37,38 might offer further insights into the dominant conical intersection and, at longer times, reveal fragmentation products from the hot ground-state molecules. We anticipate that future experiments should provide an even deeper understanding of the intriguing ultrafast ring reconfigurations in this photoswitch molecule.
Methods
Experiment
The experiment was performed at the Free Electron laser Radiation for Multidisciplinary Investigations (FERMI) FEL facility39 at the Low Density Matter (LDM) beamline40,41. XUV probe pulses with a photon energy of 18.97 eV were generated at 50 Hz as the fourth harmonic of the seed laser wavelength at 261.4 nm. The UV pump pulses with a central wavelength of 200.6 nm and a bandwidth of 0.6 nm (6.18 ± 0.02 eV) were generated as the fourth harmonic of a Ti:sapphire laser with a sequence of three barium borate crystals. For the data presented here, the pump–probe delay was scanned between −500 and 1,000 fs with a step size of 25 and 50 fs near and far from the pump–probe overlap, respectively. At each delay point, 3,000 shots were acquired. For every second shot, the UV pulse was blocked by inhibiting the trigger of the Ti:sapphire amplifier. Shots without UV excitation pulses present were used to subtract static features in the signal and for additional normalization (Supplementary Section 1.4). The pump–probe instrument response function was obtained by monitoring the UV-induced ionization of helium atoms following resonant 1s → 4p excitation at 23.72 eV (fifth harmonic of the seed laser), which yielded an instrument response function of 186 ± 28 fs (full-width at half-maximum), as shown in Supplementary Fig. 3 and Supplementary Table 1.
QC was synthesized by adding recrystallized Michler’s ketone (0.35 wt%; 98%, Sigma-Aldrich) to NBD (97%, Alfa Aesar) and irradiating the mixture for 48 hours (Thorlabs, M365LP1; 365 nm). The product was distilled, and the irradiation and distillation process repeated. The final product was checked by 1H NMR (400 MHz) and showed a 99% photoconversion of NBD to QC, without significant contamination from other products (Supplementary Section 1.3 for further details). The molecules were delivered into an ultrahigh vacuum chamber as a supersonic molecular beam using a pulsed Even–Lavie valve without further heating and with helium at a backing pressure of 6 bar as a carrier gas.
Photoelectron spectra were measured with a magnetic bottle electron spectrometer described previously23. For the data shown here, an effective retardation voltage of 9.7 V was used to increase the resolution in the BE region of interest containing the excited states as well as the lowest-BE peak of ground-state QC. The conversion from time of flight to kinetic energy was calibrated by measuring the helium 1s photoline at 28.46 eV (sixth harmonic of the seed laser) and 33.20 eV (seventh harmonic) photon energy. Weaker photoelectron signals corresponding to ionization with the third harmonic of the FEL tuned to the fourth, fifth, sixth and seventh harmonic were also included in the calibration. For the conversion to BE values, the photoelectron kinetic energy was subtracted from the central FEL photon energy. By comparing the XUV photoelectron spectrum of QC measured here with a high-resolution spectrum reported in the literature16 (Supplementary Fig. 4), we estimate the kinetic energy resolution to be δE/E ≈ 0.03 (that is, ±0.3–0.5 eV) in the kinetic energy range shown in Fig. 2.
The statistical uncertainty of the experimental signal was estimated using a standard bootstrapping analysis. The ~40,000 single-shot digitizer traces that were analysed to obtain Fig. 2 were randomly resampled with replacement 150 times. The conversion from time of flight to electron kinetic energy (including Jacobian correction) was performed on each bootstrap dataset separately. The mean and standard deviation of these 150 datasets define the values and the corresponding error bars shown in Fig. 2b–e. They were checked for convergence by repeating the bootstrapping analysis a second time and comparing the results of both procedures.
Calculations
Electronic structure calculations for the neutral species were performed using RMS-CASPT2 (ref. 42) based on SA(9)-CASSCF(2,6) with the 6-31G(d) basis augmented by additional diffuse functions (8s8p contracted to 1s1p) centred on the bridging CH2 fragment. The active space contained two valence, one 3s Rydberg and three 3p Rydberg orbitals (shown in Extended Data Fig. 5). No 3d orbitals were included in this study, as their presence was not seen in the experimental data. The lowest six (S0–S5) states were deemed sufficient to describe the dynamics observed, though state averaging over the first nine roots returned by the calculations was required for convergence. All nine states were active in the simulations; however, only a very small fraction of population ever appeared in the states S6 and above during the dynamics. We note that this approach fails to capture well the second valence excited state of NBD4, but as discussed in the Supplementary Information, this omission is judged to have little impact on the dynamics initiated by photoexciting QC. Electronic structure calculations for the molecular ion were performed with the same method and basis, although with one electron removed, that is, RMS-CASPT2/SA(6)-CASSCF(1,6), using the neutral orbitals as an initial guess. This accounts for all ionizations from the active electron, including the D0 and D1 ionic states of interest (further details in Supplementary Information). The absence of Rydberg states in the ion wavefunction leads to lower relative energies in the ion than the neutral, leading to a bias towards lower EIP values (Supplementary Information). All calculations included an imaginary shift of 0.5Eh, in units of hartree Eh=27.2114 eV, to remove intruder states and were performed in a development branch of OpenMolcas (ref. 43) implementing gradients for RMS-CASPT2 (ref. 44).
The potential energy curves in Fig. 3 are calculated along a LIIC that connects the ground-state equilibrium geometry of QC to the S1/S0 MECI, and then along a LIIC from this MECI geometry to the ground-state equilibrium geometry of NBD. The C4-ring geometry at the MECI is rhombic4,18 and does not lie directly between the equilibrium geometries of the two isomers, as shown in Extended Data Fig. 3. Since this is an essentially barrierless process, there is no obvious conceptual advantage in considering a minimum energy pathway over the LIICs.
Non-adiabatic trajectories were run using the surface-hopping method with a locally modified version of SHARC v.2.1 (ref. 45). Couplings were calculated using the local-diabatic approach. Limited numerical issues with the stability of the gradients were mitigated by a variable time step for the integration (Supplementary Section 2.3 for details). The surface-hopping simulations were performed using an electronic time step 1/25 times smaller than the nuclear time step, and used the energy-based decoherence correction with the standard C = 0.1Eh.
The initial conditions were selected from a Wigner-sampled ground-state QC geometry using the delta-pulse approximation46 inside a 5.75 ± 0.07 eV window, chosen to overlap well with the calculated partial absorptions to the 3p Rydberg states of QC (Extended Data Fig. 7). Trajectories were categorized into those excited into S2 (121 trajectories, 45%) and those excited into S3 and S4 (146 trajectories, 55%). Strictly, one-photon excitation S0 →S2 is forbidden at the equilibrium geometry, and the excitation is therefore attributable to the Herzberg–Teller effect with the transition gaining strength from the mixing of valence character into the S2 state. Inspection showed that within the envelope of the Wigner sampling, the character of the states remained separated, with the S2 state corresponding to the 3px/V configuration, whereas the S3 and S4 states each involved a mixture of excitations to the 3py and 3pz orbitals. This separation is confirmed by the distinctly different, excitation-character-dependent dynamics displayed by the respective sets of trajectories. A very small sample (5 trajectories) was excited to the S1 (3s) state at this energy. These are not considered further in the current analysis.
For the analysis, the trajectories were sorted according to initial state, allowing resolution of the dynamics into state-specific components. A small portion (7% of the total) of trajectories undergo a retro-Diels–Alder reaction from NBD-like geometries after passing through the MECI geometry, yielding C5H6 and C2H2 fragments, but these were excluded from the analysis as the selected active space cannot describe the fragmentation pathway accurately. Molecular geometries were differentiated by the average carbon–carbon distance rCC = (r12 + r34)/2 (atom numbers shown in Fig. 1), with QC-like geometries defined to have rCC < 2.0 Å and NBD-like geometries defined to have 2.0 Å < rCC < 3.5 Å.
Photoionization cross-sections and energies were calculated by selecting a random time point in each 5 fs period within each trajectory. The squared Dyson norm47 was calculated between the active state in the surface hopping and all ionic states, and used as an approximation to the photoionization cross-section. The method yields qualitatively lower cross-sections for valence states than for Rydberg states when compared to experiment, so for display purposes, the intensities in Fig. 4c with BE > 3 eV have been multiplied by a factor of three (further discussion in Supplementary Section 2.3). The difference in vertical ionization potential between QC and NBD is roughly 0.3 eV with EIPQC = 7.67/8.4 eV and EIPNBD = 8.02/8.67 eV (first value from present vertical calculations in Table 1 and second value from experiment (ref. 16), respectively).
Data availability
Data generated or analysed during this study are included in this Article (and its Supplementary Information). Source data are provided with this paper.
Code availability
The analysis codes used to generate the data presented in this study are available from the corresponding authors upon reasonable request.
References
Pianowski, Z. L. Molecular Photoswitches: Chemistry, Properties, and Applications Vols 1 and 2 (Wiley, 2022).
Franz, E. et al. Tunable energy release in a reversible molecular solar thermal system. ACS Catal. 12, 13418–13425 (2022).
Wang, Z., Hölzel, H. & Moth-Poulsen, K. Status and challenges for molecular solar thermal energy storage system based devices. Chem. Soc. Rev. 51, 7313–7326 (2022).
Antol, I. Photodeactivation paths in norbornadiene. J. Comput. Chem. 34, 1439–1445 (2013).
Jorner, K. et al. Unraveling factors leading to efficient norbornadiene–quadricyclane molecular solar-thermal energy storage systems. J. Mater. Chem. A 5, 12369–12378 (2017).
Dreos, A. et al. Liquid norbornadiene photoswitches for solar energy storage. Adv. Energy Mater. 8, 1703401 (2018).
Orrego-Hernández, J., Dreos, A. & Moth-Poulsen, K. Engineering of norbornadiene/quadricyclane photoswitches for molecular solar thermal energy storage applications. Acc. Chem. Res. 53, 1478–1487 (2020).
Alex, W. et al. Solar energy storage: competition between delocalized charge transfer and localized excited states in the norbornadiene to quadricyclane photoisomerization. J. Am. Chem. Soc. 144, 153–162 (2022).
Coppola, F., Nucci, M., Marazzi, M., Rocca, D. & Pastore, M. Norbornadiene/quadricyclane system in the spotlight: the role of Rydberg states and dynamic electronic correlation in a solar-thermal building block. ChemPhotoChem 7, e202200214 (2023).
Van Ingen, J. W. F., Van Tieghem, C. H. C. & Cramer, W. A. Radiation-induced isomerization of [2,2,1] bicycloheptadiene (norbornadiene) to [2,2,1,02,6,03,5] quadricycloheptane (quadricyclene) in cyclohexane solutions. J. Chem. Phys. 53, 3665–3674 (1970).
Jones, G., Chiang, S.-H., Becker, W. G. & Greenberg, D. P. Structure-reactivity factors for exciplex isomerization of quadricyclene and related compounds. J. Chem. Soc., Chem. Commun. 15, 681–683 (1980).
Jones, G., Chiang, S. H., Becker, W. G. & Welch, J. A. Photosensitization of quadricyclene isomerization by electron acceptors. A short-circuit nonradiative decay mechanism for electron donor-acceptor quenching in polar media. J. Phys. Chem. 86, 2805–2808 (1982).
Hillers-Bendtsen, A. E., Iuel Lunøe Dünweber, P. G., Olsen, L. H. & Mikkelsen, K. V. Prospects of improving molecular solar energy storage of the norbornadiene/quadricyclane system through bridgehead modifications. J. Phys. Chem. A 126, 2670–2676 (2022).
Hoffmann, R. Interaction of orbitals through space and through bonds. Acc. Chem. Res. 4, 1–9 (1971).
Fuß, W., Kuttan Pushpa, K., Schmid, W. E. & Trushin, S. A. Ultrafast [2 + 2]-cycloaddition in norbornadiene. Photochem. Photobiol. Sci. 1, 60–66 (2002).
Rudakov, F. & Weber, P. M. Ultrafast structural and isomerization dynamics in the Rydberg-exited quadricyclane:norbornadiene system. J. Chem. Phys. 136, 134303 (2012).
Palmer, M. H. et al. High-level studies of the ionic states of norbornadiene and quadricyclane, including analysis of new experimental photoelectron spectra by configuration interaction and coupled cluster calculations. J. Chem. Phys. 153, 204303 (2020).
Valentini, A., van den Wildenberg, S. & Remacle, F. Selective bond formation triggered by short optical pulses: quantum dynamics of a four-center ring closure. Phys. Chem. Chem. Phys. 22, 22302–22313 (2020).
Hernández, F. J., Cox, J. M., Li, J., Crespo-Otero, R. & Lopez, S. A. Multiconfigurational calculations and photodynamics describe norbornadiene photochemistry. J. Org. Chem. 88, 5311–5320 (2023).
Adachi, S., Sato, M. & Suzuki, T. Direct observation of ground-state product formation in a 1,3-cyclohexadiene ring-opening reaction. J. Phys. Chem. Lett. 6, 343–346 (2015).
von Conta, A. et al. Conical-intersection dynamics and ground-state chemistry probed by extreme-ultraviolet time-resolved photoelectron spectroscopy. Nat. Commun. 9, 3162 (2018).
Smith, A. D. et al. Mapping the complete reaction path of a complex photochemical reaction. Phys. Rev. Lett. 120, 183003 (2018).
Squibb, R. J. et al. Acetylacetone photodynamics at a seeded free-electron laser. Nat. Commun. 9, 63 (2018).
Pathak, S. et al. Tracking the ultraviolet-induced photochemistry of thiophenone during and after ultrafast ring opening. Nat. Chem. 12, 795–800 (2020).
Palmer, M. H. et al. High-level studies of the singlet states of quadricyclane, including analysis of a new experimental vacuum ultraviolet absorption spectrum by configuration interaction and density functional calculations. J. Chem. Phys. 158, 234303 (2023).
Cooper, J. C. et al. Valence-shell electronically excited states of norbornadiene and quadricyclane, J. Chem. Phys., https://doi.org/10.1063/5.0187707 (2024).
Jensen, J. O. Vibrational frequencies and structural determination of cyanogen isocyanate. J. Mol. Struct. THEOCHEM 715, 1–5 (2005).
Zhou, X. & Liu, R. Density functional theory study of vibrational spectra. 3. Assignment of fundamental vibrational modes of quadricyclane. Vib. Spectrosc. 12, 65–71 (1996).
Jacovella, U. et al. Photo- and collision-induced isomerization of a charge-tagged norbornadiene–quadricyclane system. J. Phys. Chem. Lett. 11, 6045–6050 (2020).
Robin, M. B. Higher Excited States of Polyatomic Molecules Vol. 1 (Academic Press, 1974).
Robin, M. B. Higher Excited States of Polyatomic Molecules Vol. 3 (Academic Press, 1985).
Forbes, R. et al. Vacuum ultraviolet excited state dynamics of the smallest ketone: acetone. J. Phys. Chem. Lett. 12, 8541–8547 (2021).
Weir, H., Williams, M., Parrish, R. M., Hohenstein, E. G. & Martínez, T. J. Nonadiabatic dynamics of photoexcited cis-stilbene using ab initio multiple spawning. J. Phys. Chem. B 124, 5476–5487 (2020).
Lietard, A. et al. Competitive direct vs. indirect photochromism dynamics of constrained inverse dithienylethene molecules. Phys. Chem. Chem. Phys. 16, 22262–22272 (2014).
Minitti, M. P. et al. Imaging molecular motion: femtosecond X-ray scattering of an electrocyclic chemical reaction. Phys. Rev. Lett. 114, 255501 (2015).
Stankus, B. et al. Ultrafast X-ray scattering reveals vibrational coherence following Rydberg excitation. Nat. Chem. 11, 716–721 (2019).
Wolf, T. J. A. et al. The photochemical ring-opening of 1,3-cyclohexadiene imaged by ultrafast electron diffraction. Nat. Chem. 11, 504–509 (2019).
Yang, J. et al. Simultaneous observation of nuclear and electronic dynamics by ultrafast electron diffraction. Science 368, 885–889 (2020).
Allaria, E. et al. Highly coherent and stable pulses from the FERMI seeded free-electron laser in the extreme ultraviolet. Nat. Photon. 6, 699–704 (2012).
Lyamayev, V. et al. A modular end-station for atomic, molecular, and cluster science at the low density matter beamline of FERMI@Elettra. J. Phys. B At. Mol. Opt. Phys. 46, 164007 (2013).
Svetina, C. et al. The Low Density Matter (LDM) beamline at FERMI: optical layout and first commissioning. J. Synchrotron Rad. 22, 538–543 (2015).
Battaglia, S. & Lindh, R. On the role of symmetry in XDW-CASPT2. J. Chem. Phys. 154, 034102 (2021).
Aquilante, F. et al. Modern quantum chemistry with [Open]Molcas. J. Chem. Phys. 152, 214117 (2020).
Nishimoto, Y., Battaglia, S. & Lindh, R. Analytic first-order derivatives of (X)MS, XDW, and RMS variants of the CASPT2 and RASPT2 methods. J. Chem. Theory Comput. 18, 4269–4281 (2022).
Mai, S., Marquetand, P. & González, L. Nonadiabatic dynamics: the SHARC approach. WIREs Comput. Mol. Sci. 8, e1370 (2018).
Barbatti, M. & Sen, K. Effects of different initial condition samplings on photodynamics and spectrum of pyrrole. Int. J. Quantum Chem. 116, 762–771 (2016).
Ruckenbauer, M., Mai, S., Marquetand, P. & González, L. Revealing deactivation pathways hidden in time-resolved photoelectron spectra. Sci. Rep. 6, 35522 (2016).
Acknowledgements
We acknowledge Elettra Sincrotrone Trieste for providing access to the FERMI facility and gratefully acknowledge the staff at FERMI and Elettra for their outstanding support during the beamtime, which was performed as a remote-user experiment during the COVID pandemic, and for the excellent operation of the FEL. We also happily acknowledge the insightful comments provided by the reviewers of the original submission. This work was primarily supported by the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, Office of Science, US Department of Energy, grant no. DE-SC0020276 (M. Centurion), which funded S.B., S.B.M., M. Centurion, J.P.F.N., A.O., A. Rudenko, P.M.W., A.K. and D.R.; and grant no. DE-FG02-86ER13491 (A. Rudenko), which funded K.D.B. and S.P. The theoretical and computational part of this work used facilities at the University of Oxford. A.K. acknowledges funding from the Engineering and Physical Sciences Research Council (EPSRC), grants EP/V006819 and EP/V049240 (A.K.), and from the Leverhulme Trust, grant RPG-2020-208 (A.K.); and J.C.C. acknowledges a doctoral studentship from the University of Oxford. A.S.V. is funded through the National Science Foundation (NSF) grant no. PHYS-1753324 (D.R.); C.S.H., through the Australian Research Council grant no. DE200100549 (C.S.H.); D.M.P.H., through the Science and Technology Facilities Council; and R.L., through the Swedish Research Council (VR) grant no. 2020-03182 (R.L.). B.F.E.C. acknowledges the European Union Horizon 2020 research and innovation programme (SINDAM), grant no. 803718 (B.F.E.C.) and the EPSRC grant EP/V026690/1 (B.F.E.C.). R.F. acknowledges support from the Swedish Research Council and the Knut and Alice Wallenberg Foundation, Sweden. R.J.G.F. gratefully acknowledges support from the Linac Coherent Light Source, SLAC National Accelerator Laboratory, which is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE-AC02-76SF00515. R.A.I. acknowledges funding from the Royal Society grant RGS/R2/202009 (R.A.I.), and H.G.M. is supported by EPSRC Doctoral Training Grant nos EP/N509577/1 and EP/T517793/1. J.B. and M. Bosch acknowledge A. Zahl (Friedrich-Alexander-Universität Erlangen-Nürnberg) for NMR measurements and funding by the Deutsche Forschungsgemeinschaft, grant 392607742 (J.B.). We are grateful to N. Ekanayake at Deutsches Elektronen-Synchrotron (DESY) for providing some optical elements for the fourth harmonic generation. D.R. thanks Linac Coherent Light Source and the Stanford PULSE Institute for their hospitality and financial support during a sabbatical.
Author information
Authors and Affiliations
Contributions
P.M.W. and D.R. conceived the experiment with further input from M. Centurion and A. Rudenko. The experiment at the FERMI FEL facility was conducted by M. Bonanomi, C.C., M.B.D., A.D., M.D.F., O.P. and K.C.P. with remote participation by K.D.B., M.N.R.A., S.B., R.B., M. Centurion, B.E., R.F., R.J.G.F., C.S.H., D.M.P.H., R.A.I., L.M., H.G.M., S.B.M., J.P.F.N., A.O., S.P., A. Rouzée, A. Rudenko, R.J.S., A.S.V., P.M.W. and D.R.; R.F. and R.J.S. provided the magnetic bottle spectrometer. M. Bonanomi, C.C., M.D.F., O.P. and A.S. prepared and operated the beamline and the LDM instrument. A.D. and M.B.D. prepared the optical laser set-up, and P.R. operated the FEL. M.Bosch synthesized and characterized the sample with input from J.B.; J.C.C. and A.K. performed the ab initio simulations, with contributions from B.F.E.C. and R.L. Experimental data were analysed by K.D.B. and D.R.; K.D.B, J.C.C., M.N.R.A., D.M.P.H., P.M.W., A.K. and D.R. interpreted the results with input from all the authors, and K.D.B., J.C.C., A.K. and D.R. wrote the initial manuscript with further input from all the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemistry thanks Mariachiara Pastore, Lionel Poisson and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Time-dependent photoelectron spectra of QC with and without pump pulse.
During the pump-probe scans, the UV laser was present only for every second FEL shot, such that data with and without pump pulse were recorded in parallel. All data presented in Fig. 2 of the main text are shown as a difference signal of all the accumulated ‘pump on’ (a) minus ‘pump off’ (b) spectra at each delay point. Note the log scale for the color map in the above spectra to show the weak pump-probe signal in the non-subtracted spectra.
Extended Data Fig. 2 Time-dependent photoelectron difference spectra of QC in the excited-state region at different pump-probe delays.
Same data as shown in Fig. 2b but presented in finer delay steps and over a larger delay range.
Extended Data Fig. 3 Optimized geometries.
The molecular geometries for QC, NBD, and the MECI, optimized using RMS(9)-CASPT2(2,6)/6-31 G* + D level theory. The geometries are shown looking down the z-axis (as shown in Fig. 1 in the main text). Note the rhombic nature of the MECI geometry, which does not lie on the totally symmetric displacement between the two equilibrium geometries of QC and NBD.
Extended Data Fig. 4 State characters.
LIIC for all electronic states considered in the RMS(9)-CASPT2(2,6)/6-31 G* + D model, with approximate character labelled (color coded). Each state is labelled by its principal character (for example, 3py indicates a state of mostly HOMO → 3py character). DE indicates a doubly excited state, with the character of the orbital(s) excited into in parentheses.
Extended Data Fig. 5 Active space orbitals calculated at QC.
The active space consists of 2 electrons in a space of 2 valence and 4 Rydberg orbitals. Extending the active space to include one additional pair of virtual and occupied orbitals creates the (4,8) active space of Valentini et al. (Valentini et al., 2020). The two orbitals that extend the (2,6) active space are identified by a (4,8) in their caption. The isosurface cutoff value has been adjusted such that 80% of the total density is shown for each orbital, and Rydberg orbitals are shown zoomed out to fully render their spatial extent. A rendering of the molecule is included in each frame.
Extended Data Fig. 6 Early-time dynamics from simulations.
The same data as shown in Fig. 4 in the main text but zoomed in on the first 200 fs. a, False-colour maps for average carbon–carbon distance rCC = (r12 + r34)/2 versus time. b, Populations of the electronic states and fraction of molecules with NBD-like geometry (defined as rCC > 2.0 A) as a function of time. c, Simulated TRPES data, convoluted by Gaussian functions with full-width at half-maximum of 0.1 eV and 94 fs. Signals with BE > 3 eV (below the horizontal dashed line) have been multiplied by a factor of three prior to display.
Extended Data Fig. 7 Calculated absorption spectrum.
Wigner sampled absorption spectrum of QC using 10000 initial conditions broadened by a Gaussian (FWHM = 0.1 eV). Calculated using geometries and energies at the RMS(9)-CASPT2(2,6)/6-31 G* + D level of theory.
Supplementary information
Supplementary Information
Supplementary Information with further details on the experiment and calculations is available.
Source data
Source Data Fig. 1
QC, MECI and NBD coordinates in .xyz format (in angstroms).
Source Data Fig. 2
Experimental photoelectron difference spectra.
Source Data Fig. 3
Absolute energies in hartrees; the first six columns are S0 to S5, and the final two columns are D0 and D1, from left to right respectively.
Source Data Fig. 4
ASCII data.
Source Data Extended Data Fig. 1
Photoelectron spectra with and without pump pulse.
Source Data Extended Data Fig. 2
Time-dependent photoelectron difference spectra.
Source Data Extended Data Fig. 3
QC, MECI and NBD coordinates in .xyz format (in angstroms).
Source Data Extended Data Fig. 4
Absolute energies in hartrees.
Source Data Extended Data Fig. 6
ASCII data.
Source Data Extended Data Fig. 7
Cross-sections. Headers of columns indicate data in the column. The 1, 2, 3 and so on indicate the excitation cross-section into states S0, S1, S2 and so on, respectively. Total is the total excitation cross-section.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Borne, K.D., Cooper, J.C., Ashfold, M.N.R. et al. Ultrafast electronic relaxation pathways of the molecular photoswitch quadricyclane. Nat. Chem. 16, 499–505 (2024). https://doi.org/10.1038/s41557-023-01420-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41557-023-01420-w
