
Abstract
Motivated by recent experimental observations of opposite Chern numbers in R-type twisted MoTe2 and WSe2 homobilayers, we perform large-scale density-functional-theory calculations with machine learning force fields to investigate moiré band topology across a range of twist angles in both materials. We find that the Chern numbers of the moiré frontier bands change sign as a function of twist angle, and this change is driven by the competition between moiré ferroelectricity and piezoelectricity. Our large-scale calculations, enabled by machine learning methods, reveal crucial insights into interactions across different scales in twisted bilayer systems. The interplay between atomic-level relaxation effects and moiré-scale electrostatic potential variation opens new avenues for the design of intertwined topological and correlated states, including the possibility of mimicking higher Landau level physics in the absence of magnetic field.
Similar content being viewed by others


Introduction
The low-energy electronic structure of moiré superlattices can be described by Bloch electrons moving in a periodic potential that varies on the scale of the moiré period. The understanding of this moiré potential is pivotal to the realization of various topological states1,2,3,4,5,6, including the much coveted zero-field fractional Chern insulators7,8,9,10,11,12, recently discovered in twisted transition metal dichalcogenide (TMD) homobilayers13,14,15,16. Given the structural and chemical similarities among different TMDs, it is intuitive to expect that the moiré potentials of twisted TMD homobilayers, and thus the moiré band topology, would also be similar. However, recent experiments seem to suggest the contrary: at the integer hole filling of ν = −1, optical and transport measurements have found opposite Chern numbers in 3.7° twisted bilayer MoTe2 (tMoTe2)13,14,15,16 and 1.23° twisted bilayer WSe2 (tWSe2)6.
On the theory side, discrepancies in the Chern numbers were also found by two distinct approaches used to study the moiré electronic structures. The first approach involves deriving electronic structures from small unit cells containing local stacking arrangements17,18,19,20,21,22. The second approach relies on density-functional theory (DFT) calculations performed on reasonably sized moiré superlattices23,24,25,26. Curiously, for tMoTe2, the Chern number of the topmost spin-up (spin-down) moiré valence band is found to be −1 (+1) within the local stacking approximation20, whereas the DFT calculation conducted on a fully relaxed structure with a 3.89° twist yield opposite Chern numbers25. The latter is consistent with experimental observations. However, at smaller twist angles, the system size poses a substantial challenge to DFT calculations, and a direct comparison with experiments is currently unavailable.
In this letter, we perform large-scale DFT calculations for tMoTe2 and tWSe2 down to 1.25° twist angle. This is made possible by using a machine learning force field to obtain the relaxed structures, which enables a comprehensive exploration of the twist-angle dependence of the moiré lattice reconstruction. We show that the observed difference in Chern numbers is due to the twist-angle dependence of the moiré potential. Specifically, we find that as the twist angle varies, the location of the moiré potential maximum shifts from the MX stacking region to the XM stacking region (see Fig. 1 for the definition of MX and XM), causing a sign change of the Chern number. The shift of the moiré potential maximum is attributed to the competition between the in-plane piezoelectricity and the out-of-plane ferroelectricity, a mechanism associated with the broken inversion symmetry in TMDs and absent in the local stacking approximation. The large-scale calculations, enabled by machine learning methods, also reveal multiple flat bands with Chern numbers all equal to 1 in tMoTe2 at around 2° twist, indicating the possibility of mimicking higher Landau-level physics in the absence of magnetic field. The interplay between atomic-level relaxation effects and moiré-scale electrostatic potential variation opens new avenues for the design of intertwined topological and correlated states.

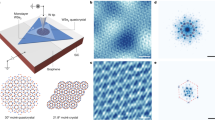
a The K-valley layer pseudospin Δ(r) skyrmion lattice, using parameters from the local stacking approximation20. The color denotes Δz(r) and the arrow denotes Δx,y(r). The black, green, and purple dots denote the three high-symmetry local stacking sites, MM, XM, and MX, respectively. b Similar to (a) but using parameters from the DFT calculation25.
Results
Following recent experiments6,13,14,15,16, we will focus on the valence bands of R-type twisted TMD homobilayers. In these systems, the emergence of nontrivial band topology can be understood as a consequence of the real-space layer pseudospin texture. Within the continuum model, the effective Hamiltonian for the K-valley electrons is given by20
where Δb/t(r) and ΔT(r) are the intra- and inter-layer moiré potential, respectively. Due to the spin-valley coupling, the band edge at each valley is spin split, and opposite valleys carry opposite spins as required by time-reversal symmetry27,28. The continuum Hamiltonian for the \({K}^{{\prime} }\)-valley spin-down electrons can be obtained by applying time-reversal symmetry to \({{{{{{{{\mathcal{H}}}}}}}}}_{K}^{\uparrow }\), resulting in moiré bands with opposite Chern numbers.
The moiré potential can be represented as an effective layer pseudospin magnetic field \({{{{{{{\boldsymbol{\Delta }}}}}}}}({{{{{{{\bf{r}}}}}}}})=(\,{{\mbox{Re}}}\,\,{\Delta }_{{{{{{{{\rm{T}}}}}}}}},-\,{{\mbox{Im}}}\,\,{\Delta }_{{{{{{{{\rm{T}}}}}}}}},\frac{{\Delta }_{{{{{{{{\rm{b}}}}}}}}}-{\Delta }_{{{{{{{{\rm{t}}}}}}}}}}{2})\). There are three high-symmetry local stackings in a moiré supercell, labeled as MM, XM, and MX (Fig. 1). It has been shown that Δ(r) forms a skyrmion lattice with its north/south poles located at the MX and XM points20. Curiously, for 3.89° tMoTe2, using parameters from the local stacking approximation20 and the DFT calculation25, we find a reversal in the positions of the north/south poles between the two cases as shown in Fig. 1, which results in opposite skyrmion numbers29. This contrast in skyrmion numbers, in turn, manifests as opposite Chern numbers for the topmost valence band, with only the DFT calculation matching the experiment.
Armed with the insight that the moiré potential landscape can affect the band topology, we now perform DFT calculations at even smaller twist angles. Because the system size at these angles (~13,000 atoms at 1.25°) is beyond the typical scale of DFT relaxations, we first trained a neural network (NN) inter-atomic potential to capture the moiré lattice reconstruction. The NN potentials are parameterized by using the deep potential molecular dynamics (DPMD) method30,31, where the training data are obtained from 5000 to 6000 ab initio molecular dynamics (AIMD) steps at 500 K for a 6° twisted homobilayer calculated using the VASP package32. We test the NN potential for a moiré bilayer at 5° and obtain a root mean square error of force <0.04 eV/Å. More details can be found in Supplementary Note 1. Figure 2a, b shows the calculated in-plane displacement field of the top-layer W atoms and interlayer distance in tWSe2 at 3.15° and 1.25°. The displacement field in the bottom layer shows the opposite pattern. It is clear that while the local stacking varies smoothly at 3.15°, at 1.25° large domains of the MX and XM regions form, with domain walls connecting the shrunken MM region. The difference between reconstruction patterns at the large and small twist angles also affects the strain tensor distributions, shown in Supplementary Fig. 6. We find that the shear strain (uxy) and uxx − uyy are much larger than the normal strain (uxx + uyy). As the twist angle decreases, the strains are mostly distributed near the domain boundaries due to the domain wall formation. These findings are consistent with previous calculations based on continuum model and parameterized inter-atomic potential26,33,34,35,36, as well as available experiments37,38,39,40,41,42,43.

a The in-plane displacement field of the top-layer W atoms at 3.15° and 1.25° for tWSe2. The color and arrow denote the amplitude and direction of in-plane displacement fields, respectively. b The interlayer distance (ILD) distribution at 3.15° and 1.25° for tWSe2. c, d Twist-angle dependence of the valence moiré bands of tWSe2 and tMoTe2, respectively. The labels indicate the spin orientations and the C3z eigenvalues at high-symmetry point with ξ = eiπ/3, ξ* = e−iπ/3, and \(\bar{1}=-1\). The C3z eigenvalue is the same at the κ and the \({\kappa }^{{\prime} }\) point.
We then calculate the moiré band structure for the relaxed atomic structures. To reduce the computational cost, we adopt the SIESTA package44 for band structure calculations. We first benchmark the accuracy of this local basis approach with the plane-wave basis approach by comparing the band structures at 6° obtained from SIESTA and VASP, and reach a qualitative agreement between the two (see details in Supplementary Note 4). Then we perform small twist-angle band calculations by using the SIESTA package. Figure 2c, d shows the twist-angle dependence of band structures for tWSe2 and tMoTe2, respectively. The top valence bands consist of folded K-valley and \({K}^{{\prime} }\)-valley minibands with opposite spins. A small band splitting can be seen, mostly between γ and m. Multiple factors, including trigonal warping and intervalley coupling, may contribute to the splitting. Nevertheless, we assume approximate spin z conservation, and separate moiré bands originating from the two valleys by adding a small Zeeman field in the calculation. In the following, we shall focus on the moiré bands from the spin-up K-valley.
To determine the Chern numbers for the moiré bands, we first calculate the eigenvalues of the DFT wave functions under three-fold rotational symmetry (C3z). The Chern number is then determined by the product of C3z eigenvalues at rotationally invariant momenta45: \(\exp (i\frac{2\pi }{3}C)=-{\xi }_{\gamma }{\xi }_{\kappa }{\xi }_{{\kappa }^{{\prime} }}\), where the ξ’s are the C3z eigenvalue at the high-symmetry point of the moiré Brillouin zone (mBZ). These eigenvalues are labeled in Fig. 2. Our assignment of the Chern numbers for tWSe2 at 3.15° and 1.70° agree with a recent calculation at 2.28° in which the Chern numbers were calculated by directly integrating the Berry phase over the entire mBZ26. The Chern numbers for tMoTe2 are further confirmed through the integration of the Berry curvature within the mBZ with the help of Wannier interpolations. The change of the Chern numbers with varying twist angles can be understood by tracking the evolution of the C3z eigenvalues, which signals band inversion. For example, in tWSe2 (Fig. 2c), when the twist angle changes from 1.70° to 1.54°, the first and second bands invert at the γ point, and the second and third bands invert at the κ and \({\kappa }^{{\prime} }\) points. As a result, the Chern numbers of the two topmost bands change from (+1, +1) to (0, 0).
The twist-angle dependence of the Chern numbers is in excellent agreement with experiments. First, it was reported that at ν = −1 the Chern numbers are opposite for tWSe2 at 1.23°6 and tMoTe2 at 3.7°13,14,15,16. Second, in 1.23° tWSe2, the Chern numbers are the same at filling factors ν = −1 and ν = −3, which indicates that the Chern numbers of the first two bands from the same valley must be the same6. Further increasing the twist-angle results in trivial insulators up to 1.6°6. Remarkably, all these observations are consistent with the trend in the twist-angle dependence of our calculations, confirming the validity of our machine learning-based approach. Our calculation also predicts that in tMoTe2, as the twist angle decreases, the Chern numbers of the two topmost bands change from (+1, −1) to (+1, +1), and finally to (0, 0) at the smallest angle of the calculation. In particular, our calculations reveal multiple flat bands with Chern numbers all equal to +1 at around 2°, indicating the possibility of mimicking higher Landau-level physics in the absence of magnetic field. The presence of multiple bands of Chern number +1 has been confirmed by a recent experimental measurement46.
Since we are interested in the sign change of the Chern number of the topmost band, in the following we will focus on tWSe2. As mentioned earlier, the evolution of band topology in momentum space is closely related to the change in the real-space moiré potential. In particular, the location of the north/south poles of Δ(r), which directly affects the skyrmion number, is given by the difference between the moiré potentials at the top and bottom layer.
The moiré potential can be inferred from the surface Hartree potential47, defined as the difference between the Hartree potential above and below the twisted bilayer surface in DFT calculations. Figure 3a shows the coarse-grained surface potential drop at 3.15° in tWSe2. More details can be found in Supplementary Note 4. The maximum is located at MX, zero at MM, and minimum at XM. Surprisingly, the surface potential drop shows a sign reversal at XM (and MX) as the twist angles decrease (see Fig. 3a–d). Going from 3.15°, to 1.70°, 1.47°, and eventually down to 1.25°, the potential at the high-symmetry point XM (and MX) changes sign, and the area of the flipped region grows in size. This sign switch suggests that the north pole of Δ(r) at ~3° becomes the south pole at ~1°. Additional features can be identified near the MM site, where the surface potential drop mimics the pattern of a six-petal flower with C3 symmetry. We find that the amplitude of potential inside the petal is comparable with that at XM (and MX) at 1.70°, suggesting unique quantum confinement effects which reshapes the electron wave function. The overall effects of these features can be clearly seen by a line cut along MM–XM–MX–MM, showing rich variations and multiple extremes in Fig. 3e. The intricate behavior of the surface potential goes beyond the continuum approximation of moiré potential based on the first-star expansion of the reciprocal lattice vectors alone, evident by their Fourier transform as shown in Fig. 3f.

a–d Twist-angle dependence of the difference of DFT Hartree potential, ΔvH, between the top-layer surface and the bottom-layer surface in tWSe2. The white dashed line denotes the path MM–XM–MX–MM. e The distribution of ΔvH along the dashed line in (a) for different twist angles. f The Fourier components of ΔvH for different twist angles. g1 is the length of the moiré reciprocal lattice vector. Inset in (f): schematics of the Hartree potential drop between the top-layer surface and bottom-layer surface.
The evolution of the surface potential implies that the layer polarization of the wave functions should also change with the twist angle. In Fig. 4, we plot the real-space wave function for the two topmost bands at the γ point of the mBZ at various twist angles for tWSe2. Since the time-reversal symmetry \({{{{{{{\mathcal{T}}}}}}}}\) and C2x symmetry are preserved at the γ point, only the wave function in the top layer is plotted, while the wave function in the bottom layer can be obtained by performing a \({{{{{{{\mathcal{T}}}}}}}}{C}_{2x}\) operation, under which MX/XM in the top layer is mapped to XM/MX in the bottom layer. At 3.15°, the wave function of the first band in the top layer is localized at MX. As the twist angle decreases, the localization region switches to MM and eventually to XM. The shift of the wave function location also coincides with the change of the C3z eigenvalue at the γ point (Fig. 2c). Similar changes have been found for the wave function of the second moiré band.

a–d Twist-angle dependence of the wave function of the first band (top row) and second band (bottom row) at the γ point for the top layer in tWSe2, respectively. Here, we map the weight of the projected wave function onto the W atomic orbitals. The value is normalized to its maximum in each plot. The dashed parallelogram denotes a moiré unit cell.
Two remarks are in order. First, the switch from MX to XM of the first band indicates the flip of the layer pseudospin, which gives rise to the sign change of the Chern number as shown in Fig. 2c. Second, aside from orbitals located at MX and XM, those at MM also play a significant role in deciding band topology, evident by the distribution of the wave functions (Fig. 4). Thus for a tight-binding model to properly describe the moiré band topology of tWSe2, one also needs orbitals from the MM site48. This goes beyond the real-space skyrmion picture discussed earlier.
What is the origin behind the change in surface potential drop as a function of twist angle? For a two-dimensional (2D) bilayer system in global charge neutrality, the electrostatic surface potential can be directly associated with the interlayer electric polarization. In twisted TMD homobilayers, two microscopic mechanisms contribute to this polarization: ferroelectricity and piezoelectricity. The ferroelectric effects arise from the inversion symmetry breaking in R-type TMD bilayers and have been termed “moiré ferroelectricity”49,50. In a moiré supercell, this leads to alternating out-of-plane ferroelectric polarization depending on the local stacking registry, with opposite dipoles in the XM/MX region50,51,52,53. On the other hand, since monolayer TMDs lack inversion symmetry, the strain field can produce piezoelectric polarization for each layer54. Because the two layers have opposite patterns of atomic displacement fields and the same piezoelectric coefficient, the polarization charge distributions are opposite between the two layers, which can produce a vertical potential drop35,36. As pointed out in ref. 36, these two types of polarization charges can be opposite in sign, and their competition will determine the potential drop.
Note that additional in-plane polarizations can also arise from the out-of-plane ferroelectricity and local symmetry breaking55. While these effects are present in our DFT calculations, they do not create additional Hartree potential that changes the Chern numbers.
Our machine learning-based first-principles simulations enable us to quantitatively study the polarization effects from the relaxed structure with atomic resolution. We start by separately calculating the piezoelectric charge and ferroelectric charge. For each layer, the piezoelectric charge density is directly proportional to the gradient of the strain field by \({\rho }_{{{{{{{{\rm{piezo}}}}}}}}}=-{\tilde{e}}_{11}[{\partial }_{x}({u}_{xx}-{u}_{yy})-2{\partial }_{y}{u}_{xy}]\), where \({\tilde{e}}_{11}\) is the independent non-zero component of the piezoelectric tensor35,54. The out-of-plane ferroelectric polarization is obtained directly from integrating the charge density along the z direction for each local stacking unit cell. More details about the polarization calculations can be found in Supplementary Note 3 and “Methods”. In Fig. 5, we plot the piezoelectric (further considering the dielectric screening effect35) and ferroelectric charge densities, as well as their sum at 3.15° and 1.25°, respectively. The total polarization charge matches qualitatively the pattern of the surface potential drop.

a The piezoelectric charge density, the ferroelectric charge density, and the total charge density, respectively, at 3.15° in tWSe2. b The same quantities at 1.25°.
Specifically, at 3.15°, the piezoelectric charges are mainly distributed in the XM and MX regions because of the larger gradient of shear strain and uxx − uyy in these regions (see Supplementary Fig. 6). The piezoelectric charge is negative at MX and positive at XM for the top layer. Because the bottom layer has the opposite atomic displacements, it has the opposite charge distributions. In contrast, the ferroelectric charge is positive (negative) at MX and negative (positive) at XM for the top (bottom) layer. Adding them together, we find the total charge density is negative (positive) at MX and positive (negative) at XM for the top (bottom) layer. As the twist angle decreases to 1.25°, the ferroelectric charge density at MX and XM remains virtually unchanged, but the total amount of ferroelectric charge within the MX and XM domains increases following the formation of the domain wall. In contrast, because the shear strain and uxx − uyy are mainly distributed along the domain wall and are uniformly small inside the XM and MX domains (see Supplementary Fig. 6), the piezoelectric charge density peaks near the domain wall but decreases at the interior of the domain. This explains the six-petal flower pattern that we discovered for the surface potential drop. As a consequence, the total charge density is now positive (negative) at MX and negative (positive) at XM for the top (bottom) layer. This trend from 3.15° to 1.25° is consistent with the variation of the surface moiré potentials and wave functions. In contrast, within the local stacking approximation, the sign of polarization charge is fixed and a sign reversal of the Chern number is not possible. Probing the predicted reversal of the polarization charges at MX and XM should be a clear experimental evidence of the change of band topology in momentum space.
Discussion
Up to this point, our discussion has focused on tWSe2. While the evolution of the moiré potential in tMoTe2 follows a similar trend, there are some quantitative differences in the band structures between tWSe2 and tMoTe2. This is mostly due to a couple of factors. WSe2 has a lighter effective mass (0.35) compared to MoTe2 (0.62), resulting in a larger bandwidth. In addition, differences in both elastic and piezoelectric coefficients also lead to quantitative changes in the moiré potential between these materials. More details can be found in Supplementary Information.
In summary, we have performed large-scale DFT calculations on R-type TMD homobilayers. Our results demonstrate machine learning as a powerful tool to study moiré systems, by revealing the importance of lattice relaxation that eventually leads to qualitative changes in band topology. This change is attributed to the competition between piezoelectricity and out-of-plane ferroelectricity, resulting in electrostatic potential variation that reshapes the potential landscape for any moiré electronic states. Our findings highlight the crucial long-range interactions arising from polarization charges, which change rapidly as the twist angle decreases. This behavior necessitates the explicit calculations of moiré electronic potential even at minimal twist angle.
Note added. We recently became aware of two related works in which machine learning force field is also used to calculate the moiré bands of twisted MoTe2 homobilayers56,57.
Methods
Machine learning
AIMD simulations are conducted to generate training datasets. These simulations utilize the VASP package32, employing the projector augmented wave pseudopotential58,59 and the Perdew–Burke–Ernzerhof (PBE) exchange-correlation functional60. Additionally, van der Waals corrections are incorporated using the D2 formalism61. 5000-step AIMD simulations using the canonical ensemble are performed at 500 K for 6° tMoTe2, and 6000-step for 6° tWSe2. The NN inter-atomic potential is generated using the DPMD method30,31. One thousand steps of the training data are used for validations. The embedding and fitting NNs include three hidden layers and the cutoff radius for each atom is 10.0 Å. One million steps (batches) with a batch size of 1 are used to minimize the loss function that includes energy and force contributions. One hundred new steps of MD trajectories at 500 K are used to test the NN potentials. The loss functions and comparisons between the NN inferences and DFT calculations can be found in Supplementary Note 1. The NN potentials are used to relax the superlattice within the LAMMPS package62 until the maximum atomic force is smaller than 10−4 eV/Å.
Band structure calculations
The SIESTA package is used to calculate band structures. Optimized norm-conserving Vanderbilt pseudopotentials63, PBE functional60, and double-zeta plus polarization basis are used. Spin–orbit coupling (SOC) is treated within the on-site approach64. We first perform self-consistent calculations without SOC. Then we include on-site SOC without iterating charge densities. The validations of the on-site SOC and the comparisons between the band structures from SIESTA and VASP can be found in Supplementary Note 4.
Electric polarization calculations
The proper piezoelectric coefficient is defined as65
which represents the response of the current with respect to the strain flow. Here Ji is the current component, ujk the strain component, E the macroscopic electric field, and T is the stress. Both E and T are zero in the DFT calculations. Since monolayer WSe2 has the symmetry of D3h, the only independent non-zero piezoelectric coefficient is \({\tilde{e}}_{11}\equiv\)\({\tilde{e}}_{111}\) and other non-zero coefficients are related to \({\tilde{e}}_{11}\) by54
\({\tilde{e}}_{11}\) is calculated from the change of electric polarization density with respect to the strain54. A rectangular unit cell of monolayer WSe2 and a 12 × 12 × 1 k-space grid are used. The in-plane polarization is calculated in SIESTA using the modern theory of polarization66.
The piezoelectric polarizations in the moiré superlattice are calculated from the strain,
and the piezoelectric charge density is calculated as
More details can be found in Supplementary Note 3. We use the method in ref. 35 to include the dielectric screening of piezoelectric charges.
The out-of-plane moiré ferroelectricity is calculated from the local stacking unit cell in the relaxed moiré superlattice. Within each unit cell, we calculate the out-of-plane dipole moment in SIESTA by integrating the charge density multiplied by z coordinates. Then the surface charge density due to ferroelectricity is obtained as ρferro = Pz/(Sdz), where Pz is the dipole moment, S is the area of the unit cell, and dz is the interlayer vertical distance between transition metal atoms.
Data availability
The files and datasets generated during this study, including typical input files for parameterizing NN potentials and DFT calculations, as well as the source data of band structures, Hartree potentials, wave functions, and polarizations, have been deposited in the Zenodo database67. Other data related to this paper are available from the corresponding authors upon request.
Code availability
The DFT calculations and parameterization of NN potentials are obtained from publicly available packages, following the procedure outlined in the paper. Automation and acceleration workflows are available from the corresponding authors on request.
References
Spanton, E. M. et al. Observation of fractional Chern insulators in a van der Waals heterostructure. Science 360, 62–66 (2018).
Serlin, M. et al. Intrinsic quantized anomalous Hall effect in a moiré heterostructure. Science 367, 900–903 (2020).
Chen, G. et al. Tunable correlated Chern insulator and ferromagnetism in a moiré superlattice. Nature 579, 56–61 (2020).
Xie, Y. et al. Fractional Chern insulators in magic-angle twisted bilayer graphene. Nature 600, 439–443 (2021).
Li, T. et al. Quantum anomalous Hall effect from intertwined moiré bands. Nature 600, 641–646 (2021).
Foutty, B. A. et al. Mapping twist-tuned multiband topology in bilayer WSe2. Science 384, 343–347 (2024).
Neupert, T., Santos, L., Chamon, C. & Mudry, C. Fractional quantum Hall states at zero magnetic field. Phys. Rev. Lett. 106, 236804 (2011).
Tang, E., Mei, J.-W. & Wen, X.-G. High-temperature fractional quantum Hall states. Phys. Rev. Lett. 106, 236802 (2011).
Sun, K., Gu, Z., Katsura, H. & Das Sarma, S. Nearly flatbands with nontrivial topology. Phys. Rev. Lett. 106, 236803 (2011).
Sheng, D., Gu, Z.-C., Sun, K. & Sheng, L. Fractional quantum Hall effect in the absence of Landau levels. Nat. Commun. 2, 389 (2011).
Regnault, N. & Bernevig, B. A. Fractional Chern Insulator. Phys. Rev. X 1, 021014 (2011).
Xiao, D., Zhu, W., Ran, Y., Nagaosa, N. & Okamoto, S. Interface engineering of quantum Hall effects in digital transition metal oxide heterostructures. Nat. Commun. 2, 596 (2011).
Cai, J. et al. Signatures of fractional quantum anomalous Hall states in twisted MoTe2. Nature 662, 63–68 (2023).
Park, H. et al. Observation of fractionally quantized anomalous Hall effect. Nature 622, 74 (2023).
Zeng, Y. et al. Thermodynamic evidence of fractional Chern insulator in moiré MoTe2. Nature 622, 69–73 (2023).
Xu, F. et al. Observation of integer and fractional quantum anomalous Hall effects in twisted bilayer MoTe2. Phys. Rev. X 13, 031037 (2023).
Bistritzer, R. & MacDonald, A. H. Moiré bands in twisted double-layer graphene. Proc. Natl Acad. Sci. USA 108, 12233–12237 (2011).
Jung, J., Raoux, A., Qiao, Z. & MacDonald, A. H. Ab initio theory of moiré superlattice bands in layered two-dimensional materials. Phys. Rev. B 89, 205414 (2014).
Wu, F., Lovorn, T. & MacDonald, A. H. Topological exciton bands in moiré heterojunctions. Phys. Rev. Lett. 118, 147401 (2017).
Wu, F., Lovorn, T., Tutuc, E., Martin, I. & MacDonald, A. Topological insulators in twisted transition metal dichalcogenide homobilayers. Phys. Rev. Lett. 122, 086402 (2019).
Yu, H., Chen, M. & Yao, W. Giant magnetic field from moiré induced Berry phase in homobilayer semiconductors. Natl. Sci. Rev. 7, 12–20 (2020).
Zhai, D. & Yao, W. Theory of tunable flux lattices in the homobilayer moiré of twisted and uniformly strained transition metal dichalcogenides. Phys. Rev. Mater. 4, 094002 (2020).
Naik, M. H. & Jain, M. Ultraflatbands and shear solitons in moiré patterns of twisted bilayer transition metal dichalcogenides. Phys. Rev. Lett. 121, 266401 (2018).
Devakul, T., Crépel, V., Zhang, Y. & Fu, L. Magic in twisted transition metal dichalcogenide bilayers. Nat. Commun. 12, 6730 (2021).
Wang, C. et al. Fractional Chern insulator in twisted bilayer MoTe2. Phys. Rev. Lett. 132, 036501 (2024).
Kundu, S., Naik, M. H., Krishnamurthy, H. & Jain, M. Moiré induced topology and flat bands in twisted bilayer WSe2: a first-principles study. Phys. Rev. B 105, L081108 (2022).
Xiao, D., Liu, G.-B., Feng, W., Xu, X. & Yao, W. Coupled spin and valley physics in monolayers of MoS2 and other group-VI dichalcogenides. Phys. Rev. Lett. 108, 196802 (2012).
Cao, T. et al. Valley-selective circular dichroism of monolayer molybdenum disulphide. Nat. Commun. 3, 887 (2012).
Pan, H., Wu, F. & Sarma, S. D. Band topology, Hubbard model, Heisenberg model, and Dzyaloshinskii-Moriya interaction in twisted bilayer WSe2. Phys. Rev. Res. 2, 033087 (2020).
Zhang, L., Han, J., Wang, H., Car, R. & Weinan, E. Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics. Phys. Rev. Lett. 120, 143001 (2018).
Wang, H., Zhang, L., Han, J. & Weinan, E. DeePMD-kit: a deep learning package for many-body potential energy representation and molecular dynamics. Comput. Phys. Commun. 228, 178–184 (2018).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Gargiulo, F. & Yazyev, O. V. Structural and electronic transformation in low-angle twisted bilayer graphene. 2D Mater. 5, 015019 (2017).
Carr, S. et al. Relaxation and domain formation in incommensurate two-dimensional heterostructures. Phys. Rev. B 98, 224102 (2018).
Enaldiev, V., Zolyomi, V., Yelgel, C., Magorrian, S. & Fal’Ko, V. Stacking domains and dislocation networks in marginally twisted bilayers of transition metal dichalcogenides. Phys. Rev. Lett. 124, 206101 (2020).
Magorrian, S. J. et al. Multifaceted moiré superlattice physics in twisted WSe2 bilayers. Phys. Rev. B 104, 125440 (2021).
Weston, A. et al. Atomic reconstruction in twisted bilayers of transition metal dichalcogenides. Nat. Nanotechnol. 15, 592–597 (2020).
McGilly, L. J. et al. Visualization of moiré superlattices. Nat. Nanotechnol. 15, 580–584 (2020).
Quan, J. et al. Phonon renormalization in reconstructed MoS2 moiré superlattices. Nat. Mater. 20, 1100–1105 (2021).
Weston, A. et al. Interfacial ferroelectricity in marginally twisted 2D semiconductors. Nat. Nanotechnol. 17, 390–395 (2022).
Van Winkle, M. et al. Rotational and dilational reconstruction in transition metal dichalcogenide moiré bilayers. Nat. Commun. 14, 2989 (2023).
Rupp, A. et al. Imaging lattice reconstruction in homobilayers and heterobilayers of transition metal dichalcogenides. 2D Mater. 10, 045028 (2023).
Tilak, N., Li, G., Taniguchi, T., Watanabe, K. & Andrei, E. Y. Moiré potential, lattice relaxation, and layer polarization in marginally twisted MoS2 bilayers. Nano Lett. 23, 73–81 (2023).
Soler, J. M. et al. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 14, 2745 (2002).
Fang, C., Gilbert, M. J. & Bernevig, B. A. Bulk topological invariants in noninteracting point group symmetric insulators. Phys. Rev. B 86, 115112 (2012).
Kang, K. et al. Evidence of the fractional quantum spin hall effect in moiré MoTe2. Nature 628, 522–526 (2024).
Liu, D., Zeng, J., Jiang, X., Tang, L. & Chen, K. Exact first-principles calculation reveals universal moiré potential in twisted two-dimensional materials. Phys. Rev. B 107, L081402 (2023).
Qiu, W.-X., Li, B., Luo, X.-J. & Wu, F. Interaction-driven topological phase diagram of twisted bilayer MoTe2. Phys. Rev. X 13, 041026 (2023).
Wu, M. & Li, J. Sliding ferroelectricity in 2D van der Waals materials: related physics and future opportunities. Proc. Natl Acad. Sci. USA 118, e2115703118 (2021).
Yasuda, K., Wang, X., Watanabe, K., Taniguchi, T. & Jarillo-Herrero, P. Stacking-engineered ferroelectricity in bilayer boron nitride. Science 372, 1458–1462 (2021).
Woods, C. et al. Charge-polarized interfacial superlattices in marginally twisted hexagonal boron nitride. Nat. Commun. 12, 347 (2021).
Vizner Stern, M. et al. Interfacial ferroelectricity by van der Waals sliding. Science 372, 1462–1466 (2021).
Wang, X. et al. Interfacial ferroelectricity in rhombohedral-stacked bilayer transition metal dichalcogenides. Nat. Nanotechnol. 17, 367–371 (2022).
Duerloo, K.-A. N., Ong, M. T. & Reed, E. J. Intrinsic piezoelectricity in two-dimensional materials. J. Phys. Chem. Lett. 3, 2871–2876 (2012).
Bennett, D., Chaudhary, G., Slager, R.-J., Bousquet, E. & Ghosez, P. Polar meron-antimeron networks in strained and twisted bilayers. Nat. Commun. 14, 1629 (2023).
Jia, Y. et al. Moiré fractional Chern insulators. I. First-principles calculations and continuum models of twisted bilayer MoTe2. Phys. Rev. B 109, 205121 (2024).
Mao, N. et al. Lattice relaxation, electronic structure and continuum model for twisted bilayer MoTe2. Preprint at https://arxiv.org/abs/2311.07533 (2023).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Thompson, A. P. et al. LAMMPS – a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 271, 108171 (2022).
Hamann, D. Optimized norm-conserving Vanderbilt pseudopotentials. Phys. Rev. B 88, 085117 (2013).
Fernández-Seivane, L., Oliveira, M. A., Sanvito, S. & Ferrer, J. On-site approximation for spin-orbit coupling in linear combination of atomic orbitals density functional methods. J. Phys. Condens. Matter 18, 7999 (2006).
Vanderbilt, D. Berry-phase theory of proper piezoelectric response. J. Phys. Chem. Solids 61, 147–151 (2000).
King-Smith, R. & Vanderbilt, D. Theory of polarization of crystalline solids. Phys. Rev. B 47, 1651 (1993).
Zhang, X.-W. et al. Dataset: polarization-driven band topology evolution in twisted MoTe2 and WSe2. https://doi.org/10.5281/zenodo.10888769 (Zenodo, 2024).
Acknowledgements
We acknowledge useful discussions with Jiaqi Cai, Ken Shih, and Xiaodong Xu. X.W.Z. is supported by DOE Award No. DE-SC0012509. C.W. is supported by ARO MURI with award number W911NF-21-1-0327. Y.F. is supported by the discovering AI@UW Initiative and DMR-2308979. This work was facilitated through the use of advanced computational, storage, and networking infrastructure provided by the Hyak supercomputer system and funded by the University of Washington Molecular Engineering Materials Center at the University of Washington (DMR-2308979). Access to GPUs is provided by Microsoft’s sponsorship of Azure credits to the Department of Materials Science and Engineering at UW.
Author information
Authors and Affiliations
Contributions
X.W.Z., T.C., and D.X. conceived the project. X.W.Z. performed the parameterization of NN potentials, DFT calculations, and polarization calculations with the help of C.W., T.C., and D.X. X.W.Z., T.C., and D.X. analyzed the results and wrote the paper with input from C.W., X.L., and Y.F.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks David Ruiz-Tijerina, Jiang Zeng, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, XW., Wang, C., Liu, X. et al. Polarization-driven band topology evolution in twisted MoTe2 and WSe2. Nat Commun 15, 4223 (2024). https://doi.org/10.1038/s41467-024-48511-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-48511-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
