Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain
the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in
Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles
and JavaScript.
We developed fatigue-resistant hydrogel optical fibers through the controlled growth of polymeric nanocrystalline domains to enable light delivery to peripheral nerves during locomotion. The hydrogel fibers withstand locomotion strain across more than 30,000 fiber stretch cycles and enable the optogenetic inhibition of pain hypersensitivity in naturally behaving mice.
Here we developed synthetic transactivation domains (TADs) built from human mechanosensitive transcription factors (MTFs). By linking MTF TAD segments together, we engineered compact and potent multipartite transcriptional activation modules. We then harnessed these modules to create a CRISPR activation system, which we termed the dCas9 recruited enhanced activation module (CRISPR-DREAM).
CryoREAD automatically builds DNA–RNA atomic structure from cryo-EM maps. Backbone accuracy is typically >85% and the method is applicable for maps with RNA-only, DNA-only and DNA–RNA–protein complex structures. CryoREAD uses deep learning to identify structure information and subsequently construct the 3D structure of nucleic acids.
We conducted a comprehensive long-read RNA sequencing (RNA-seq) benchmarking experiment by combining spike-ins and in silico mixtures to establish a ground-truth dataset. We used long- and short-read RNA-seq technology to deeply sequence samples and compared the performance of a range of analysis tools on these data.
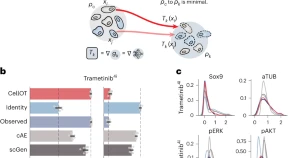
We developed CellOT, a tool that integrates optimal transport with input convex neural networks to predict molecular responses of individual cells to various perturbations. By learning a map between the unpaired distributions of unperturbed and perturbed cells, CellOT outperforms current methods and generalizes the inference of treatment outcomes in unobserved cell types and patients.
Modern high-throughput metagenomics is producing hundreds of thousands of metagenome-assembled genomes (MAGs), which is overwhelming traditional sequence-similarity search methods. We present a computational method, skani, that efficiently compares MAGs on a terabyte scale while being robust to the inherent noise in MAGs, enabling larger and more accurate analyses.
To capture expansive, seamless fields of view from frozen hydrated specimens by cryo-electron tomography, we developed methods for the collection and processing of montage data. This approach enables rapid acquisition of contiguous regions of specimens using a montaged tilt series collection scheme.
We introduce GelMap, a flexible workflow for reporting deformations and anisotropy in expansion microscopy. By intrinsically calibrating the expansion hydrogel using a fluorescent grid that scales with expansion and deforms with anisotropy, GelMap enables the reliable quantification of expansion factors and correction of deformations.
This Review discusses statistical and computational strategies for analyzing various spatial and temporal omics data types, with an emphasis on the common modeling principles.
Multiplexed spatial immunophenotyping has advanced our understanding of tissues in the context of homeostasis and disease. Two studies now provide additional tools to overcome challenges with multiplexed imaging: one procedure amplifies the detection of low-abundance antigens by integrating SABER and IMC technologies, and the other is an X-ray-based method that enables the non-destructive multiplexed detection of antigens in tissues at scalable resolution and speed.
A new chemically induced dimerization (CID) pair exhibits fluorescence upon dimerization for the first time. Moreover, the CID pair is small and offers easily reversible dimerization that can be repeated multiple times.
Leveraging nanopore long-read sequencing, scNanoHi-C identifies multiway interactions between enhancers and their target promoters within a single cell. Compared with short-read-based single-cell Hi-C or population-based multiway sequencing methods, scNanoHi-C offers new opportunities to investigate the heterogeneities of single-cell gene regulation networks mediated by high-order 3D chromatin structures.
The conversion of biological molecules into digital signals through sequencing is a complex process that often generates substantial systematic background noise. This noise can obscure important biological insights. However, by precisely identifying and removing this noise, we can bring the true signal into focus and eliminate misleading results from downstream analyses.
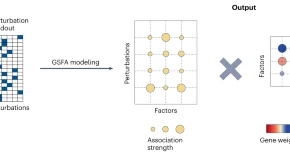
Single-cell perturbation screens are routinely conducted to study the effects of different perturbations on cellular state, yet such studies are easily confounded by nuisance sources of variation shared with control cells. We present a deep learning method that isolates perturbation-specific sources of variation, enabling a better understanding of the perturbation’s effects.
Recently proposed computational approaches explore casual links between chromatin and transcriptional changes that are provided by single-cell multimodal sequencing to bridge the knowledge gap in transcriptional regulatory control.
CheckM2 is a tool that applies machine learning to evaluate the quality of genomes from metagenomic data. CheckM2 is faster and more accurate than existing methods, and it outperforms them when applied to novel lineages and lineages with reduced genome sizes, such as Patescibacteria and the DPANN superphylum.
We developed, characterized and validated nLight sensors, a new family of genetically encoded green and red fluorescent norepinephrine indicators based on an alpha-1 adrenergic receptor. nLight probes can detect norepinephrine in living animals with superior sensitivity, ligand specificity and temporal resolution as compared with previous tools.