
Abstract
The traditional Haber-Bosch process for ammonia production is energy-intensive and relies on harsh conditions. Plasmonic materials are considered a sustainable alternative to this process. In plasmonics, the ability to localize and enhance light fields beyond the diffraction limit offers a promising avenue for harnessing visible light for photocatalysis. Moreover, the integration of plasmonic metals with various catalysts presents a promising avenue to improve efficiency and selectivity. However, cost and stability challenges hinder large-scale applications. Researchers are actively exploring new materials, optimizing catalyst design, and improving stability to overcome these hurdles. This Review delves into the intricacies of plasmonic catalysis, including material design, co-catalyst selection, and the use of nanotechnologies for precise control of metal properties in plasmonic catalysis. It emphasizes understanding energy flow at material interfaces. Ultimately, the Review aims to establish a foundation for efficient nitrogen fixation through plasmonic photocatalysis, paving the way for a more sustainable future.
Similar content being viewed by others
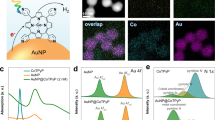
Introduction
Nitrogen fixation is a fundamental chemical process crucial for the ecosystem. Ammonia synthesis, relying on nitrogen (constituting 78% of the atmosphere), produces a versatile, non-carbon-based compound with broad industrial applications, including fertilizer production, pesticides, dyes, wastewater treatment, fermentation, and explosives1,2,3,4. Given its significance, nitrogen fixation procedures warrant thorough examination. Since the 1930s invention of the Haber-Bosch process, which utilizes pure hydrogen (H2) and atmospheric nitrogen (N2) to produce ammonia (NH3) (N2 + 3H2→2NH3, Eq. 1), global ammonia production has primarily relied on Haber’s method due to its higher yield. In 2016, statistics indicated the mass production of 175 million metric tons of ammonia annually for agricultural and industrial needs5. The process involves high temperature (300–500 °C) and pressure (150-200 atm) along with Iron or Ruthenium catalysts. However, a persistent challenge is finding ways to utilize nitrogen under ambient and milder conditions.
The formidable strength (941 kJ mol−1) and non-polar nature of triple-bonded nitrogen molecules contribute to a substantial activation barrier. This characteristic highlights the energy-intensive nature of industrial sites employing the Haber-Bosch process, leading to significant CO2 emissions, surpassing 300 million metric tons annually from fossil fuels6,7. Notably, 1–3% of the world’s electrical energy and 2–5% of the world’s natural gas are consumed exclusively for mass ammonia synthesis. Furthermore, carbon dioxide generated as a byproduct during hydrogen production in the Haber-Bosch process is a major contributor to global carbon emissions. The continuous reliance on fossil fuels also raises concerns about their diminishing availability, prompting research efforts to develop sustainable and environmentally friendly methods for nitrogen fixation4.
Although highly effective nitrogen fixation occurs in biological systems, particularly through nitrogenase in bacteria (Azotobacter, Bacillus, Clostridium, etc.), mimicking enzymatic reduction has proven insufficient to meet the increasing demands for ammonia production6,8. Nitrogenase, featuring a catalytic site in a heterotetrameric MoFe protein, facilitates nitrogen fixation activities through metal hydride intermediates, leading to a several-fold increase in turnovers9,10,11,12. However, this approach lacks the robustness required for large-scale ammonia production. To address the growing demand for ammonia, researchers are exploring the use of renewable energy sources as inputs.
This review focuses on the utilization of plasmonic nanomaterials for green nitrogen fixation, leveraging advances in nanotechnology. Implementing metals as catalysts at the nanoscale has demonstrated significant enhancements in optical properties, plasmonic effects, surface-area-to-size ratio, magnetic properties, and more13. According to the free-electron theory, nanosized metals support localized surface plasmon resonance (LSPR). LSPR arises from the collective oscillation of free electrons induced by incident light, creating strong light-matter interactions at the metal surface. The recognition that LSPR excitation facilitates field localization beyond the diffraction limit and yields exceptionally high field enhancement opens vast opportunities for light harvesting in photocatalysis. The intense field energy near the nanoparticle (NP) surface dissipates either through the radiative scattering of photons or via nonradiative decay, leading to the generation of a nonequilibrium distribution of hot charge carriers14,15. Depending on the electronic band structure of the nanomaterial, the potential energy of these charge carriers dictates their contribution to the overall free energy of chemical reactions occurring on the nanoparticle surface16,17. Consequently, these hot charge carriers show significant potential in reshaping the potential landscape of both thermodynamically and kinetically challenging chemical reactions18. Nevertheless, the extremely brief lifetime and short mean free paths remain fundamental hurdles to overcome for the efficient extraction and utilization of these hot charge carriers19. Various methods for employing plasmonic nanomaterials, such as electrocatalytic nitrogen reduction, single plasmonic nanosheets, metal-organic frameworks, hybrid nanostructures, and surface defect-induced reduction, are explored (Fig. 1).

Nitrogen fixation via plasmonic catalysis can proceed through two primary pathways: direct involvement of metal nanoparticles (NPs) or indirect involvement utilizing semiconductor substrates. The former entails intricate mechanistic processes, while the latter necessitates additional substrate materials. In the direct approach, a metal facilitates both light harvesting and the chemisorption of nitrogen molecules. Conversely, the indirect method employs a semiconductor in intimate contact with the plasmonic metal for nitrogen chemisorption. This indirect approach was developed to address limitations associated with plasmonic metals in nitrogen chemisorption. Commonly used semiconductors include titania (TiO2), (BiO)2CO3, CeO2, SrTiO3, among others.
Two widely accepted mechanisms can induce activation of the reactant and consequent product formation. The first mechanism relies on the transient exchange of hot charge carriers between the NP and adsorbate/reactant, leading to vibrational excitation of the reactant. This mechanism pertains to transient charge transfer20 (the charge stays in the hybrid state for a few femtoseconds ~10−15 s)21, where the energy is transferred in the form of vibrational excitation of the N2 molecule. It is followed by the immediate decay of the hot electrons back to the metal surface following one of the many dissipative pathways (vide infra). It is to be noted that transient charge transfer is sufficient to initiate chemical transformations on the adsorbates, but to make the process more competent for light-harvesting applications, permanent charge extraction is needed. Thus, it is crucial to develop interfaces that permanently extract the energetic charge carriers once they are generated so they do not relax back into the metal. This process requires a hole scavenger that compensates for the hole (h+) left behind after permanent electron (e−) injection into the adsorbate from the metal center, thereby prolonging the electron–hole (e-h) separation lifetime20,22. Permanent extraction of the charge carriers from the metals and their transfer either to the antibonding orbital of nitrogen or the conduction band of a semiconductor, depending on the system used is catalytically more desired than transient charge extraction. This method of extracting charge carriers from photoexcited plasmonic metal NPs resembles the classical photocatalytic reactions on semiconductors or molecular photocatalysts. The electric field induced by LSPR also facilitates direct charge transfer within the molecule-adsorbate system, a topic that has garnered significant attention in recent years23. Extensive research efforts have been dedicated to achieving optimal charge extraction and separation at interfaces by exploring various plasmonic nanostructures to drive selective and efficient N2 reduction24,25,26,27,28,29,30. Traditional catalysis methods for N2 dissociation require elevated temperatures and pressures, leading to substantial energy consumption. In contrast, plasmonic catalysis utilizes the unique properties of metallic nanostructures, particularly their ability to generate hot electrons through surface plasmon resonance decay. These hot electrons can selectively transfer energy to N2 molecules, potentially breaking the robust N≡N bond under milder reaction conditions, offering a promising avenue for more efficient and selective nitrogen fixation.
This review aims to provide a comprehensive understanding of N2 reduction, emphasizing its thermodynamic and kinetic attributes and discussing various challenging mechanisms involved. It delves into the intricacies of plasmonic catalysis, offering insights into material design optimization and the role of co-catalysts for enhanced efficiency and selectivity. Advances in nanotechnologies are explored, showcasing diverse metal structures in plasmonic photocatalysis. Additionally, the review highlights the processes associated with plasmonic photocatalysis, emphasizing how plasmonic chemistry aids in this energy-intensive process. Furthermore, the latest developments in devising plasmonic nanostructures for N2 reduction and the fundamental physical principles governing the nitrogen reduction reaction (NRR) are thoroughly examined. Finally, the review addresses bottlenecks to efficient N2 fixation, proposes an ideal catalyst scenario, and discusses strategies for refining catalytic activity to overcome current limitations. By offering insights into these aspects, the review contributes to enhancing nitrogen fixation efficiency and provides a comprehensive overview of the field’s current state and future prospects.
Key considerations in choosing plasmonic and nonplasmonic materials
Plasmonic metal nanoparticles manipulate electromagnetic energy at the nanoscale by exciting LSPR upon light illumination. This resonance concentrates energy into amplified electromagnetic fields at the nanoparticle surface, which dissipates either through radiative scattering or nonradiative excitation of energetic charge carriers31,32,33. Utilizing these hot carriers offers control over reaction selectivity in catalysis, altering the potential landscape of challenging chemical reactions based on the nanomaterial’s electronic band structure34,35. The dynamics of plasmon decay and the characteristics of absorption processes vary depending on the type of plasmonic metal36. This is because the generation of charge carriers through nonradiative decay can occur via intraband s-to-s excitation or interband d-to-s excitation, with interband transitions dominating when energetically feasible. Noble metals such as Ag have d-bands situated well below the Fermi level, making interband excitations forbidden with visible light22,37. However, metals like Au and Cu have d-bands closer to the Fermi level, allowing for interband transitions with visible light above a specific threshold. In non-noble metals, the d-states intersect the Fermi level, enabling interband transitions across the entire visible range38.
Understanding the dynamics of plasmon decay is crucial for charge extraction before relaxation through various pathways39,40. Following plasmon excitation and charge carrier generation within a short timeframe of 1–100 fs, the charge carriers undergo electron–electron relaxation, electron–phonon relaxation, and phonon–phonon relaxation steps. Hot electron–hole pairs primarily dissipate their energy through electron–electron scattering on a 100 fs–1 ps timescale, followed by electron–phonon scattering on a 1–10 ps timescale, and phonon–phonon scattering on a 100 ps–10 ns timescale, leading to heating of the nanoparticle and its surroundings21,41.
The energetics, lifetime, and transport of these generated charge carriers determine the overall efficiency of the targeted catalytic reaction42. The relative position of the s and d-bands of the plasmonic metal can influence the energy distribution of hot charge carriers, while electron–phonon interactions regulate hot-carrier energy loss and transport43. Energy distribution, relaxation time, and mean free path of hot charge carriers significantly differ between intraband and interband excitation regimes17. Extracting hot holes poses challenges due to their ultrashort lifetime, shorter mean free path, and faster relaxation dynamics, necessitating considerable efforts to overcome these constraints44. Charge carrier transport is affected by the energetics of hot electrons and holes, particularly across various interfaces, and must be considered when engineering interfaces for charge localization19,45.
Moreover, plasmonic metals have the capability to create complexes with adsorbates, facilitating the direct excitation of charge carriers from the metal-adsorbate interface. This capability enhances the catalytic efficiency of nitrogen fixation processes by enabling a higher rate of direct excitation of charge carriers into the adsorbate surface states compared to the indirect injection of carriers into the adsorbate states26. Such investigations have paved the way for novel approaches to mitigate energy losses.
In addition, careful consideration of co-catalysts becomes crucial in tailoring the catalytic activity and selectivity of these processes while mitigating potential issues like adsorbate poisoning. Overall, plasmonic catalysis offers an innovative and sustainable route to achieve nitrogen fixation into ammonia under more benign conditions, presenting a viable alternative to the energy-intensive Haber-Bosch process. Thanks to the advancements in nanotechnologies, the surface energies, numbers of adsorption sites, and optical properties of metals can be finely tuned with several techniques. The size and shape of the metal NPs govern the surface energies, which direct the adsorption strategies46,47,48. Better adsorption of the adsorbate is detected at sites with higher surface energies. Several metal structures, including mesoporous (honeycomb), spheres, rods, wires, cages, cubical, helical frameworks, ultrathin sheets, and many others, were nanoengineered to tweak the plasmonic characteristics according to the materials used. For instance, ruthenium-embedded Au composite1, plasmonic metal governed metal-organic framework49, hybrid hollow Au-Ag2O cages50, ultrathin titania sheets decorated with gold particles51, and surface imperfections52 are few of the widely investigated areas in plasmonic photocatalysis.
Recently, the plasmonic local heating effect has also been used to promote solar ammonia synthesis beyond the limits of thermal catalysis53. Understanding the complex synergy between plasmonic/nonplasmonic heterostructures, which dictates the flow of energy at the interface, is crucial to take this field forward. Efficiently extracting and utilizing charge carriers in various applications depends on designing plasmonic nanostructures with highly localized charge generation. Coating these structures with nonplasmonic metals can preferentially dissipate energy in the nonplasmonic metal, influenced by the constituent metals’ electronic structure. The energy flow in multimetallic plasmonic nanostructures relies on two critical factors: the electric field intensity at LSPR frequencies and the availability of direct transitions in the nonplasmonic metal relative to the plasmonic metal. Nanoparticles with higher field intensities under LSPR conditions more effectively dissipate energy through the nonplasmonic metal shell. Moreover, the extent of energy transfer to the nonplasmonic shell depends on the ratio of the imaginary part of the material’s dielectric function (ε2) of the core and shell materials at the LSPR wavelength. A higher shell-to-core ε2 ratio results in more energy transfer to the shell. This framework enables the design of hybrid nanostructures that localize charge carriers and potentially extract charge carriers with different energy distributions than those generated in plasmonic metals54.
Nitrogen fixation
The conversion of nitrogen and water into ammonia under ambient conditions is a highly attractive yet challenging reaction pathway. Despite the abundance of nitrogen in the atmosphere and the exothermic nature of the ammonia synthesis reaction according to thermodynamics, the process encounters significant obstacles due to the chemical inertness and stability of N2. In the following section, we delve into the intricate thermodynamic aspects of nitrogen fixation. It provides insights into the energetics associated with breaking the strong triple bond between nitrogen atoms in atmospheric nitrogen. This bond requires a substantial amount of energy to be overcome, thereby we shed light on how realizing each step of the reduction process can aid in optimizing the catalytic processes for NRR. N2 reduction reactions primarily occur on surfaces, and N2 adsorption can take place via associative or dissociative methods. Therefore, this section aims to enhance the mechanistic understanding of these two different mechanisms and highlight their contributions in NRR.
Thermodynamic and kinetic standpoint
The chemical inertness of nitrogen molecules attributes to their strong π bonding. This results in a significantly high energy gap (10.82 eV) between the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) as shown in Fig. 2a, which thereby makes the ionization potential relatively high (~15.5–15.8 eV). Besides this, the non-polar nature of this diatomic molecule also accounts for its’ negative electron affinity (−1.9 eV)55,56,57. These parameters contribute to its high activation barrier, curtailing the chemical transformations under mild conditions5.

a Simplified molecular orbital diagram of N2, the highest occupied molecular orbitals of N2 (σ2p and π2px, π2py) are strongly bonding: the energy of the σ2p orbital is –15.6 eV, and of the π2px, π2py orbital is –17.1 eV. The lowest unoccupied orbital energy (π2p*) is strongly antibonding: its energy is +7.3 eV. b For the reduction of N2 to NH3 with the subsequent intermediate generation (transient species indicated with a star (*) indicates adsorbed species), the plot of state free energy (eV) versus Reaction coordinate is shown. The maximum state free energy is found during the injection of the first electron into the LUMO of the N2 molecule, suggesting that this is the rate-limiting step59 (b reprinted with permission from ref. 59. Copyright. 2018, American Chemical Society).
Owing to the high threshold values of the intermediates, the stepwise reduction of nitrogen to ammonia is energetically cumbersome despite having favorable thermodynamics (\(\Delta {G}_{f}^{0}\) = −16.7 kJ mol−1; \(\Delta {H}_{f}^{0}\) = −92.2 kJ mol−1 at room temperature)5,58.
The above data suggests that the initial nitrogen reduction demands a significant amount of energy, hinting at the potential rate-determining nature of the first step (Fig. 2b)59. This inference aligns with previous studies highlighting the substantial energy barrier associated with the initial stages of nitrogen reduction processes. Figure 2b illustrates the state free energy (eV) versus the reaction coordinate for the reduction of N2 to NH3, with the generation of subsequent intermediate species (adsorbed species indicated with a star *).
The rate-limiting behavior can be understood by considering nitrogen’s initial bond dissociation energy, 410.0 kJ mol−1, which is over half the energy necessary for its complete dissociation (941.0 kJ mol−1). This is in direct contrast to the energy required for acetylene’s initial bond cleavage (222.0 kJ mol−1)60. The nitrogen activation begins with the first electron insertion into the LUMO, partially stretching the bond length (Eq. 2). In an aqueous medium, the reduction is preferable because the successive intermediates have larger proton affinities (N2H2: 803.0 kJ mol−1; N2H4: 853.2 kJ mol−1) than nitrogen (493.8 kJ mol−1)57. Furthermore, a smaller HOMO-LUMO gap for the intermediates (N2H4: 7.4 eV) enhances the kinetic accessibility of nitrogen (10.8 eV). Even though the reaction is exothermic overall, mediators are used to generate the first intermediate. One such influential and well-known biologically mediated reaction occurs via the nitrogenase enzyme. The enzyme functions efficiently at room temperature and fixates about 108 T/y of N2 to NH3. Other approaches, namely, photocatalytic and electrocatalytic methods, have been investigated to execute this reduction reaction.
Surface mechanisms for nitrogen reduction to ammonia
It is important to understand the mechanistic pathway of a catalytic N2 reduction process to design and tune the catalyst. Most N2 reduction reactions are surface reactions, where N2 adsorption may occur in one of two ways: associative or dissociative (Fig. 3)61.

Adapted with permission from ref. 61. Copyright. 2016 Elsevier B.V. All rights reserved.
In the dissociative method, the N2 molecule is dissociated before any hydrogenation and then chemically adsorbed onto the surface, like conventional thermal catalysis such as Haber’s process. On the other hand, the molecule adsorbs in an associative manner. There are two methods for this to happen: distal and alternating mechanisms. In the distal mechanism, the N2 molecule is assumed to be coupled in an end-on coordination mode. The nitrogen farthest from the surface undergoes successive hydrogenation, resulting in the M≡N unit, which can also be further hydrogenated. A single molecule of N2 creates one ammonia molecule at a time. In the alternating mechanism, both nitrogen atoms are protonated simultaneously, forming two equivalents of ammonia per N2.
Compared to the distal, the alternating mechanism is not thermodynamically inclined considering the end-on mode. This is because the proximity of one nitrogen is more than the other. Nonetheless, it is preferred over the distal mechanism during side-on bridging. Throughout this review, the primary aim is to emphasize the benefits of plasmon-driven nitrogen fixation employing energy in the form of photoexcitation, not simply stoichiometrically but catalytically, ensuring the mechanistic understanding of the overall process.
Plasmonic catalysis
Solar energy as a light source can be used as a broadband light source or can be accommodated by selecting a single-colored light depending on the need and catalyst design. Metals whose LSPR peaks lie outside the solar spectrum are seldom used for photocatalysis62. The metals reported for their plasmon effect are noble metals (Au, Ag, Ru, Pt, Pd) and non-noble metals (Bi, Fe, Cu, Al, Mg, Co, Ti, Ni, In, and Ga)63. Amongst plasmonic metals, Au remains extensively researched. It is to be comprehended that surface plasmonic excitations are not limited to metals but can also be induced in other 2D materials with sufficient charge carrier densities on the surface, such as MXenes64,65, chalcogenides66,67,68, graphene69,70,71, black phosphorous72 and, hexagonal boron nitrides73.
Surface-confined electrons on metals at the nanoscale can resonate with the visible light of a specific wavelength. Unique optoelectronic properties are observed when the frequency of the incident electromagnetic radiation and the free electrons on the metal surface are in phase. It leads to the generation of an oscillating dipole constituting hot charge carriers in the same direction as the electric field of light, thereby enabling the LSPR. This dramatically amplifies the collision frequency between the oscillating electron density and the lattice atoms, causing Joule heating to induce additional vibrational energy74. Sole focus on employing this heat, however, shifts the paradigm to another field of plasmonics, namely thermoplasmonics. This entire enhancement of inherent characteristics of plasmonic metals can be used effectively in catalytic processes at the surface, also referred to as plasmonic catalysis.
Plasmonic excitation induced by the illumination of visible light plays a crucial role in influencing the activation of adsorbates on nanoparticle surfaces, potentially facilitating chemical transformations75,76. Essentially, the generation of hot charge carriers subsequent to plasmonic excitation enables the transfer of energy to adsorbates, thereby initiating oxidation/reduction half-reactions42,77,78,79,80. The collective resonance of hot electrons on nanoparticles with the incident visible light frequency results in excess energy, necessitating equilibration. Prior to plasmonic excitation, adsorbates reside at the ground state of the potential energy surface, requiring an energy of Ea to initiate chemical transformations (see Fig. 4a). Following excitation, two possibilities arise. Firstly, transient electronic excitation may guide the evolution of adsorbates along an excited potential energy surface, with excess energy inducing atom reconfiguration, ultimately perturbing bond length or angle and initiating chemical transformation at the excited potential energy surface. Secondly, if the reactant does not remain in the excited state long enough to undergo a chemical reaction, it returns to the ground state with excess vibrational energy, facilitating the overcoming of the high activation energy barrier with greater ease (see Fig. 4b)75,76,81,82. It is important to emphasize that the activation of adsorbates in this context does not entail the permanent extraction of charges from the metal, leaving it in a charged state. Rather, it involves a transient electronic exchange between the metal and the reactant, resulting in the formation of transient adsorbates that persist for tens of femtoseconds before relaxation. This transient state is adequate to initiate chemical reactions or impart excess vibrational energy with a longer lifetime, such as picoseconds. In fact, for weakly bonded molecules on plasmonic metals (e.g., Au or Ag), energy dissipation routes are highly restricted, thus it is reasonable to assume that excited vibrational states will have a longer lifetime. This implies that a molecule on the metal surface can participate in a chemical reaction while in an excited vibrational state76,82,83.

a The adsorbate is initially in an equilibrium position on its ground-state potential energy surface, demanding activation energy Ea to dissociate. b The plasmonic NP’s photoexcitation deposits plasmon energy in the adsorbate and raises it to an excited potential energy surface. The adsorbate gains kinetic energy and may react in the excited state as it travels along the excited potential energy surface. If the adsorbate does not react in the excited state, it falls back to the ground-state potential energy surface in a vibrationally excited state. This lowers the barrier to dissociation. (a, b Adapted with permission from ref. 76. Copyright. 2018, Nature catalysis).
For a plasmonic metal to exhibit LSPR properties, the wavelength of the incident light must be greater than the size of the metal. The total radiant flux, known as the extinction cross-section (\({\sigma }_{{ext}}\)), is significantly higher than the geometric cross-section of the metal. The \({\sigma }_{{ext}}\) is determined by the dielectric constants of the metals, expressed as:
This relationship, known as the Mie approximation (Eq. 3), is applicable primarily to spherical nanoparticles84. Here, \({\sigma }_{{abs}}\) represents the absorption cross-section, \({\sigma }_{{scat}}\) denotes the scattering cross-section, and \({\varepsilon }_{2}\) (the imaginary dielectric function) signifies dielectric loss, which indicates the amount of energy dissipated or lost from the electric field in the form of heat. The decay of energy in LSPR modes via the generation of energetic carriers is favored for large \({\varepsilon }_{2}\) values. It is apparent from the aforementioned relation that \({\sigma }_{{ext}}\) achieves its maximum value when \({\varepsilon }_{1}\) (the real part of the dielectric function of the metal) is approximately equal to – \({2\varepsilon }_{m}\) (where \({\varepsilon }_{m}\) represents the dielectric constant of the medium). Experiments have shown that such conditions are predominantly met in the ultraviolet-visible range for plasmonic metals.
The selection of plasmon metals for surface chemical reactions primarily hinges on the prominence of specific transitions (Fig. 5)85. These transitions leading to the formation of electron–hole pairs entail two possibilities: indirect s-to-s intraband excitation from filled s states below the Fermi level to empty s states above it, and direct interband d-to-s excitation from d band below the Fermi level to s band above it. The proximity of d states to the Fermi level dictates the accessibility of these transitions. While the filled d band of Ag, situated well below the Fermi level, does not undergo interband transitions under visible light, the fully filled d-bands of Au and Cu lie closer to the Fermi level. In noble metals like Pt and Pd, interband transitions can occur across the visible light spectrum due to the overlap between the Fermi level and the d band (Fig. 5a). Noble plasmon metals are chosen to have a filled d orbital with the Fermi level positioned above them. A lower Fermi level facilitates electron capture by the metal, significantly curtailing intraband transitions (Fig. 5a)76. Consequently, interband transitions prove more advantageous in initiating chemical transformations, with a rate constant as high as \({10}^{15}{{{{{{\rm{s}}}}}}}^{-1}\)86,87,88.

a Depiction of the density of states in a plasmonic metal. The accessibility of interband d-to-s transitions depends on the position of the Fermi level concerning the d band. However, the intraband s-to-s transitions can be attained via visible light photons. In the case of non-noble metals and for Pd and Pt noble metals, interband excitation occurs throughout the visible range due to the intersection of the d band and Fermi level76. (a Adapted with permission from ref. 76. Copyright 2018, Nature Catalysis) b The difference in relative contributions of resistive, geometry-assisted, phonon-assisted, and direct transition to absorption in Gold as a function of frequency for a semi-infinite surface, 40 nm, 20 nm, and 10 nm diameter sphere17 (b reprinted with permission from ref. 17. Copyright 2016, American Chemical Society). c Representation of the multiple steps involved in the internal relaxation of a collective plasmonic mode in a nanocrystal following a pulsed excitation on the ultrafast and fast time scales89 (c adapted with permission from ref. 89. Copyright 2020, American Chemical Society).
Upon excitation of a plasmonic mode in a conductive nanostructure, nonradiative energy is dissipated either through a cascade of charge transfer events or via near-field interaction, contributing to the heating of the nanostructure and dispersing heat into its surroundings89. The plasmonic energy generated arises from various mechanisms including phonon-assisted, surface, geometry-assisted, and direct transitions, encompassing resistive losses or Landau damping. These contributions vary with incident light frequency and are influenced by the geometries and sizes of the nanocrystals. The Atwater group corroborated similar findings for both bulk gold surfaces and spherical gold nanoparticles of varying sizes (Fig. 5b)17.
Similarly, Clavero emphasized the importance of plasmonic nanostructure size and shape in excitation and charge carrier generation90. Not only do they govern the LSPR excitation wavelength, but they also influence the charge separation process. Small nanostructures of Au and Ag (20–40 nm) tend to exhibit nonradiative decay in experiments, such as electron–electron scattering (<100 fs), phonon–phonon (~100 ps) coupling, and electron–phonon scattering (1–10 ps). In contrast, larger nanostructures (>50 nm) favor radiative decay via photon emission (see Fig. 5c)90. The optical characteristics of the nanostructures determine the dimension at which radiative decay becomes the predominant relaxation mechanism. Only electrons decaying through nonradiative pathways facilitate the chemical transformation.
N2 fixation using plasmonic nanocomposites with enhanced charge separation
When passed over a plasmonic metal surface under specific conditions, atmospheric nitrogen chemisorbs onto the metal surface. Instead of directly photoexciting the nitrogen molecule, highly energetic charge carriers (electron–hole pairs) activate the nitrogen molecule91. These energetic electrons transiently occupy the nitrogen’s empty antibonding orbital, allowing sufficient time to energize the molecule vibrationally. The generation of hot electrons and their insertion into the nitrogen molecule occurs within femtoseconds, as experimentally proven using ultrafast femtosecond visible and infrared pump-probe transient absorption spectroscopy 92,93. The temporary hole formed during electron injection initiates charge separation through the electron–hole pair, inducing instability in the system, compelling the electron to return to its source metal. If the transient hole remains uneliminated within nanoseconds to picoseconds of electron injection, the charge carrier recombines20,90. This transient process facilitates the charge carrier’s residence in the adsorbate’s empty orbital (Fig. 6a, b). The insertion of hot electrons, whether transiently or permanently, results in bond elongation. Permanent electron transfer leads to a more significant decrease in bond order than transient transfer1,21,34,41.

a SPR-induced mechanism for electron transfer. (i) electron excited from the metal Fermi level to surface plasmon (SP) states, (ii) Electron transfer to the nearest conduction band of the semiconductor, (iii) transferred electron utilized for reduction such as hydrogen evolution here, the same premise could be implemented for nitrogen reduction as well. b Energy transfer from photoexcited SP state to unpopulated adsorbed states wherein SP states are seen to interact with the adsorbed states via electron scattering processes.
LSPR plays a crucial role here, as highlighted by the inhibition of nitrogen reduction in dark reactions, underscoring the significance of plasmonic assistance94. The enhanced electric field resulting from the localization effect and larger surface area lays the groundwork for sustained reaction pathways under benign conditions94. Electron transfer from the plasmonic metal reduces the high thermodynamic activation energy barrier essential for nitrogen fixation. Semiconductors offer additional active sites to assist in the passage of accumulated hot electrons to adsorbed molecules, resulting in the formation of active intermediate species95. When plasmonic metals interact with semiconductors, two main events occur at the semiconductor interfaces: Schottky barrier formation and surface plasmon resonance-induced changes in interfacial charge transfer dynamics.
The band alignment and possible charge transfer mechanism in a metal and metal/semiconductor assembly are shown in Fig. 7a–c. The parameter φM is the metal’s working function, defined as the energy needed to bring an electron from the metallic Fermi energy to the vacuum. XS is the electron affinity, the energy difference between the lowest-lying conduction band (CB) and the vacuum (Vac) energy. The description of the semiconductor dictates the shape of the CB after combining the semiconductor with the metal. Metals with lower Fermi energy than semiconductors are more likely to trigger electron flow from semiconductor to metal, and vice versa when the metal’s Fermi energy is higher than the semiconductor. This process continues until their Fermi levels align in equilibrium. A Schottky barrier caused by these deformations at the interface must be crossed for effective charge separation. The work function of the metal is greater than the work function of the semiconductor in n-type electronics, and the electrostatic attraction between the positively charged semiconductor surface and the negatively charged metal surface ends up bending the valence band (VB) and CB upwards (Fig. 7b) at the boundary of the metal/semiconductor. Additionally, only the energetic electrons from the plasmonic metal can transcend the Schottky barrier to travel across the semiconductor’s conduction band, leaving the hot holes in the metal96,97. The lower the Fermi level, the easier it is for the metal to capture electrons, enable efficient charge extraction, and thus, they serve as potential electron reservoirs98. Likewise, for p-type semiconductors, the opposite flow of electrons (from metal to semiconductor) is linked to the lower work function of the plasmonic metal relative to the semiconductor. Consequently, in contrast to the n-type semiconductor, the Schottky barrier is generated with the downward bending of the VB and CB at the interface (Fig. 7c), and only the hot holes having adequate energy are carried to the VB of the semiconductor, resulting in an effective separation of charges99. These pathways extend the lifespan of the hot electron–hole pair requisite for the photoreaction in both n-type and p-type semiconductors. Such isolation of the charge carriers becomes valuable and relevant for accelerating chemical reactions.

Plasmon-induced energetic charge carrier generation in the a clean metal system and relaxation, b, c metal/n type, and metal/p-type semiconductor, respectively. Localized surface plasmons can decay radiatively through reemitted photons or non-radiatively through the excitation of hot electrons. Schottky barrier forms at the interface; electrons with sufficient energy to overcome the Schottky barrier (φSB = φM - χS) are injected into the conduction band Ec of the neighboring semiconductor. Where φM is the work function of the metal, φSB is the work function of the Schottky barrier, χS is the electron affinity of the semiconductor. d–f Hot electron transfer or back transfer in metal/adsorbate assemblies. The shaded area represents the electron state density90,95. (Adapted with permission95. Copyright 2018, American Chemical Society).
Electron transfer is facilitated in a nearby electron-trapping orbital of a semiconductor or an adsorbate. The photoexcited electrons transfer to the vacant LUMO of the adsorbed molecules on the metal surface. Numerous surface-enhanced Raman spectroscopic (SERS) and photocurrent studies have corroborated this hot electron flow100. Fig. 7 (d–f) highlight the two probable charge transfer mechanisms at the interface of metal/adsorbate. In such an assembly, it is noteworthy that orbital overlap is a prerequisite for direct and indirect electron transfer from the metal to the adsorbate.
Various transitions elucidate the mechanism of nitrogen fixation. Water serves both as a hydrogen source and a hole scavenger101. The general outline is shown in Eq. 4, where it is used as a hole scavenger.
Equation 5 shows the successive protonation to the surface-adsorbed dinitrogen molecule.
The size and shape of NPs predominantly determine the efficacy of the light absorption and adsorption of the nitrogen molecule. Especially the surface energies are primarily dictated by the shapes of the metal nanostructures. Density functional theory (DFT) calculations can assist in pre-determining the surface sites at which maximum absorption can occur as per the sites having high surface energies. While size can ascertain the predominant decay, shape governs the surface energies and the wavelength at which LSPR peaks are observed102. The LSPR peak obtained for an Au rod differs from that of spheres.
Surface imperfections, metal, and semiconductor doping, and hetero interface fabrication can all overcome the electron–hole pair’s ultrashort lifespan and poor chemisorption on the metal surface. When a small quantity of another material is attached to the surface of a plasmonic NP to create a hybrid plasmonic material, the NP’s optical, physical, and chemical properties vary 38. Heterogeneous catalysts have been frequently employed to drive chemical processes. Numerous Au-based hybrid nanostructures have been actively used to dissociate nitrogen among plasmonic metals. This review analyzes Au and a few other noble and non-noble metal-based hybrid structures that have already performed admirably in reducing N2. Many of these techniques impose a competition between hydrogen evolution reaction (HER) and nitrogen fixation. We have also covered ways to circumvent this hurdle by modifying specific reaction conditions.
Advancements and challenges in plasmonic catalysis and engineering of plasmonic photocatalysts
In the realm of plasmonic photocatalyst engineering, a deep understanding of the catalytic process is crucial. Nanoplasmonics, with their ability to manipulate light and heat at the thermodynamic limit, offer exciting prospects. One promising avenue involves utilizing ‘hot’ carriers generated through surface plasmon decay. Theoretical insights into plasmon decay processes, supported by first-principles calculations, have unveiled microscopic mechanisms governing this decay and enhanced our grasp of excited carrier distributions. These calculations, involving phonon-assisted optical excitations and energy-dependent lifetimes, expand the potential of nonequilibrium plasmonics across various domains, including photodetection, photovoltaics, chemical transformations, and spectroscopy.
Plasmonic catalysis and nonequilibrium plasmonics
Nonequilibrium plasmonics has advanced to quantify carrier energy distributions impacted by electron−phonon and electron−electron scattering, as well as optical responses linked to direct and phonon-assisted transitions. To collect hot carriers efficiently for plasmon-driven chemistry and photodetection, understanding momentum conservation, surface scattering, and charge carrier transfer to adsorbates is vital. Small plasmonic nanoparticles near metal-environment interfaces are promising for charge injection, despite challenges like rapid relaxation times, low excitation rates for high-energy carriers, and momentum constraints. Innovative approaches and precise system design invigorate research. However, a comprehensive theoretical framework for efficient nanoengineering of hot-carrier injection systems remains a challenge. Further research is needed, addressing momentum distribution in plasmonic excited electrons, energy profiles of initial carriers, and interfaces’ roles in hot-carrier dynamics. Quantifying energy distribution and collection efficiency remains active research. Theoretical models and experiments clarify the effects of charge separation across metal/semiconductor interfaces on available hot carriers for photocatalysis. Resolving discrepancies between ultrafast spectroscopy and steady-state measurement, understanding interfacial states, and quantization effects are essential for plasmonic hot-carrier catalysis. Investigating nonlinearities in high-field plasmonic systems and modulating plasmonic absorption through thermal injection hold promise. In summary, plasmonic catalysis aims to harness sunlight for external manipulation of chemical reactivity through theory and experimentation.
Engineering plasmonic photocatalysts
The field of engineering plasmonic photocatalysts has shown promise in recent years, where notable achievements include the precise control of plasmonic nanostructures, enabling tailored light absorption and catalytic enhancement. Several promising strategies and emerging techniques are being explored that account for excited states involved in photocatalytic reactions on plasmonic surfaces are essential. Robust multiscale methods are needed to understand mechanism interactions and guide nanoengineering efforts for efficient plasmonic reactors. Modeling catalytic reactions using well-established mechanisms and quantifiable parameters can deepen our understanding of processes and exclude side reactions. Small-scale studies at the single-particle level provide valuable insights into localized temperature effects and their role in accelerating thermo-chemical reactions. Optimizing plasmonic metasurfaces for efficient light-to-heat conversion can enable large-scale thermal reactors with potential applications in various fields. Designing high-temperature plasmonic systems should consider the mutual influence between optical and thermal responses, leveraging nonlinear photothermal phenomena for optimization. Sustainable plasmonic materials and understanding excited states in molecular targets contribute to sustainable plasmonic photocatalysis. Exploring atomic-scale catalysis and controlling selectivity within picocavities represents a promising frontier in plasmonic research. In conclusion, the field of engineering plasmonic photocatalysts faces challenges but is evolving towards comprehensive theoretical models, experimental advancements, and diversified applications. Progress in scalability, control, and sustainability is expected through the exploration of emerging strategies in the coming years. The following section examines the evolution of plasmonic photocatalysis seeking inspiration from earlier endeavors. For instance, it could be clubbing semiconductors with noble metals and introducing vacancies onto the surface to augment the lifetime of electron–hole separation. It could also be by incorporating Au NPs in the porous metal-organic framework (MOF) or by utilizing electrochemical reduction in synergy with plasmon resonance to improve the surface adsorption of N2 molecules. Furthermore, we have also highlighted how leveraging the properties of non-noble metals and the development of 2D plasmonic nanosheets seem to be promising for overcoming the barriers associated with conventional noble plasmonic metals. We have presented a detailed outline for the readers to establish a comprehensive picture and offer new insights into the fabrication of prospective plasmon-based photocatalysts (Table 1).
Noble metal-mediated plasmonic catalysts for NRR
For a surface reaction to occur proficiently, the plasmonic properties must cooperate with the surface adsorption sites with close reference to the gaps between two nano-plasmonic metals or the overlapping region of the electric field between adjacent plasmonic metals termed hotspots81. The inability of the plasmonic metal on its own to effectively chemisorb nitrogen molecules on its surface has led to the formation of composite structures. Anchoring the semiconductor to the plasmon metal overcomes the inefficacies of the plasmon metal, keeping the crux of the mechanism as plasmon resonance intact. Plasmon/Semiconductor composites improve charge separation between hot electrons and hot holes across the composite interface, minimizing their recombination rate103. Additionally, photocurrent increases significantly when an appropriate semiconductor is anchored to the plasmon metal104,105. The mechanistic path to couple the plasmonic metal with a semiconductor is generally through an interfacial hybridization between them, depending upon the choice of the semiconductor. A spike in semiconductor photoluminescence for nanocomposites confirmed the radiative energy transfer from an excited plasmonic metal to the semiconductor, supporting the metal SPR34. The choice of semiconductors depends on several factors, the pivotal of which is bandgap106. Bandgap energy keeps a hold on numerous elements of the reaction mechanism, including impacting the resonance wavelength and altering the probability of the electron–hole pair recombination.
LSPR coupled with semiconductors majorly initiates surface chemical reactions through three different mechanisms, namely DET—Direct electron transfer, where the hot electrons from the Fermi level of the plasmon metal inject themselves into the conduction band of the semiconductor, overcoming the Schottky barrier (vide supra). Oscillating dipole propagating on the metal surface allows for another mechanism—resonance energy transfer (RET), where the excitation energy is decayed into the electron–hole pairs in semiconductors. Another possible mechanism for plasmon energy flow into the semiconductor is local electromagnetic field enhancement, where the charge separation of hole and electron is amplified81,107,108. However, these mechanistic details are subject to improvement and further research. Several semiconductors have been used and researched as nonstoichiometric photocatalyst to fix nitrogen. Some used quite widely are Ru, TiO2, ZnO, AgO, CeO2, SrTiO3, CdS, In2S3, MoS2, InVO4, Bi2MO6, Bi2WO6, Na2Ti3O7, BiOBr, and many others are still in progression101,109,110,111,112,113,114,115,116.
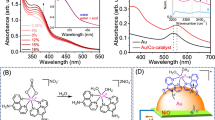
Among these, (BiO)2CO3 (BSC) has attracted research attention due to its diverse uses in photocatalysis, supercapacitors, sensing, and anti-bacterial agents117,118,119,120. Xiao et al. studied the hybrid Au/(BiO)2CO3 to fix nitrogen where Au NPs have homogeneously been distributed over the (BSC) nanodisks (NDs) (Fig. 8)117.

a Schematic depiction of the charge transfer in Au/BSC hybrids where the synergetic effect of Au NPs and Bi2O2CO3 facilitates the triple bond cleavage of the N2 molecule. b Comparison of ammonia yield between BSC and Au/BSC over different periods. c Rate of ammonia production under different conditions. A considerable amount of NH3 was detected with Au/BSC illuminated in the N2 atmosphere, while no NH3 was detected in the argon atmosphere or the dark. A higher yield of NH3 with Au/BSC in N2 compared to only BSC in N2 justifies the role of plasmonic metal in enhancing the catalytic process117 (a, b, c reprinted with permission from ref. 117. Copyright, 2017, American Chemical Society). d Performance of KNbO3 and with 0.5% deposition of Ag under different light irradiation122. (d reprinted with permission from ref. 122. Copyright, 2019, American Chemical Society).
Au NPs are highly attracted to the surface of the BSC nanodisks; together, they operate synergistically to improve charge separation and transfer. In addition to augmenting the visible light absorption by LSPR, Au NPs function as an electron sink by trapping and transferring electrons to adsorbed nitrogen molecules accountable to its high work function (Fig. 8a). It was observed that with the exact reaction conditions, BSC resulted in a far smaller quantity of ammonia output than the Au/BSC hybrid (Fig. 8b), and this further substantiated the function of plasmon metal in the chemical process121. Rate of ammonia production with Au/BSC illuminated in the N2 atmosphere, while no NH3 was detected in the Argon atmosphere or the dark further highlights the role of N2 and light (Fig. 8c).
In the recent past, ABO3-type metal-oxide perovskite has been used to extend the lifetime of the photogenerated carrier when decorated with a plasmonic metal. Extrapolating this, KNbO3 has been considered with Ag to construct composite such as Ag/KNbO3122. While hot electrons generated in Ag NPs due to LSPR could travel to the CB of KNbO3 under visible light exposure, the higher work function of Ag (4.3 eV) stimulates electron transport from KNbO3 to the metal center (vide supra). Therefore, the loaded Ag NPs act as the electron trapper and photosensitizer, similar to the aforementioned Au. Both electron transfers are opposite; hence excessive Ag loading can inhibit NH3 production. The highest ammonia yield is obtained at 0.5% Ag loading with a value of 385.0 μmol L−1 g−1, four times that of KNbO3, which showed almost no catalytic activity under visible light illumination (Fig. 8d).
Large-scale solar energy harvesting could be impeded by the Au NPs’ narrow and sharp LSPR. Jiang et al. used an intriguing strategy to develop a novel Schottky barrier-free catalyst to overcome the inadequacy associated with limited LSPR. Recently discovered plasmonic semiconductors (vide infra) with carefully tailored morphology and surface treatment can display tunable LSPR over the entire solar spectrum (visible to NIR), rendering them suitable alternatives for Au. Accordingly, they constructed hierarchical nanoarrays comprising Au/KxMoO3/Mo/KxMoO3/Au to leverage the LSPR of MoO3 and the active sites of Au123. Besides, the HER was attenuated by conducting the NRR in a KClO4 than in an aqueous solution. They reported the highest NH3 production at 808 nm, which perfectly aligned with the Au/KxMoO3 LSPR. We argue that inserting defects in such nanoarrays to produce a composite such as Au/KxMoO3-x/Mo/KxMoO3-x/Au might provide an attractive alternative. However, this would need a careful study of hot electron flow (vide supra).
Plasmonic metals, notably gold, have been at the forefront recently and are the subject of in-depth research by experts. Numerous hybrid catalysts containing Cu, Ru, Fe, Sr, Mo, Rh, and other metals have lately been investigated and display promising outcomes in heterogeneous catalysis. Systems comprising plasmonic NPs (the antenna) decorated with reactor particles such as islands, clusters, or single atoms of platinum groups metals (PGMs) are typically known as antenna reactor complexes15. Herein, the catalytic activity and charge separation efficiency is governed mainly by the spatial distribution of the two components124.
Hu et al. used Ru as a photocatalyst that adsorbs nitrogen via an end-on configuration1. Through the alignment of energy levels and interfacial hybridization, bimetallic noble hybrids such as Ru-coated Au generated the proper channel for energy and charge carrier transport, resulting in nitrogen fixation via a dissociative mechanism the same as the one involved in the Haber-Bosch process (Fig. 9a). However, instead of elevated temperature and pressure, AuRu composite allows a novel technique to accomplish the same reaction at low temperature and pressure through an identical route. The plasmon-enhanced local electric field activates the chemisorbed nitrogen molecules on the Ru surface1. This justification seems quite reasonable from Fig. 9b, which shows a characteristic peak around 500 nm. Figure 9c highlights the relevance of light intensity in stimulating NRR under ambient conditions. The precise catalytic process at the boundary of the hybrid nanostructures remains a matter of contention, and scientists are designing experiments to unravel the feasible charge transfer pathways.

a Schematic illustration of N2 photo fixation of AuRux via a dissociative mechanism, showing Ru dispersed over the gold core. b Calculated apparent quantum efficiency (AQE) for N2 photo fixation over AuRu0.31 in pure water under 20 mW cm−2 monochromatic light irradiation about UV-vis spectrum which indicates the high light utilization by the catalyst. c Linear dependence of the NH3 fixation rate with AuRu0.31 on the light intensity. This suggests that a single charge carrier drives the reaction. d Catalytic ammonia formation rates by bare Au and different compositions of AuRu catalyst in the first two hours (full spectrum, 400 mW cm−2, pure water, and 2 atm N2). Maximum efficiency of NH3 was found for AuRu core antenna structure with 31 wt% of Ru (molar ratio)1 (a, b, c, d reprinted with permission from ref. 1. Copyright 2019, American Chemical Society).
Structures with plasmonic metal cores may gather light and are known to channel energy flow selectively to the desired metal site with adsorbed nitrogen. They can increase overall extinction and electric field intensities125. Besides that, the ammonia evolution rate parallels the apparent quantum efficiency (AQE), which is expressed as follows (Eq. 6)126
Under the irradiation of a 300 W Xe lamp, an ammonia production rate of 101.4 μmol g−1 h−1 has been recorded without any sacrificial donor and with a Ru loading of 31% (Fig. 9d). Analogous to the Au-Ru hybrid, a bimetallic Au-Mo composite has also been reported to perform nitrogen reduction26. As per the emb-NEVPT2 (A density functional embedding theory in combination with n-electron valence second-order perturbation is used to estimate the corrected ground and electronic state energies) computed barrier for N2 dissociation, the Au-Mo catalyst outperforms any existing commercial catalyst, including Iron126. The LSPR reaction shown here demonstrates how one may change the surface chemistry of Mo-doped Au NPs, eventually overcoming the constraints of ground-state catalysts.
Au NP’s catalytic performance was upgraded with black silicon (bSi) and Cr. Ali et al. achieved improved results with Au NPs by exploiting the properties of bSi and Cr (Fig. 10a). The bSi’s ability to operate as a light absorber is due to its low reflectance, large and chemically active surface area, high hydrophobicity, and high luminescence efficiency. On the other hand, Cr functions as a sacrificial hole sink, limiting Si oxidation and promoting charge separation (Fig. 10b)127. Sodium sulfite was retained as an alternative photogenerated hole scavenger to preserve the Cr anode from oxidizing. After 24 h at ambient temperature and pressure, the final ammonia production was reported to be \(13.3\, {{{{{\rm{mg}}}}}}{{{{{{\rm{m}}}}}}}^{2}{{{{{{\rm{h}}}}}}}^{-1}\) under two suns illumination. Although the precise mechanism has yet to be discovered, it is crucial to note that a thorough knowledge of the reaction’s kinetics may allow it to be scaled up several times.

a A schematic depiction of a photoelectrochemical cell. The photo reduction cell was illuminated with 300 W light (artificial solar light) with steady N2 bubbling over the surface of the GNP/bSi/Cr composite. b NH3 output recorded over 24 h on various composites. The improved NH3 generation rate when Cr was employed confirmed the role of Cr as a hole sink127 (a, b reprinted with permission from ref. 127. Copyright 2016, Springer Nature).
Besides conventional semiconductors, MXenes have been marginally explored for their plasmonic properties. These are 2D materials with the general formula Mn+1XnTx where M-early transition metal, X-C/N, T-surface terminating moieties such as -O, -OH, -F, x-population of such surface terminated entities. Ti3C2 is the most studied MXene by far. Chang et al. reported a sandwich-like r-Ti3C2/Au composite and demonstrated N2 photo fixation in aqueous media128. r-Ti3C2 (partially reduced Ti3C2 with -O, -F, and -OH functionalization on the surface) effectively exposed low-coordinated Ti sites for N2 chemisorption. Upon adsorption, a substantial lengthening of the N2 bond is seen (Free N2 −1.114 Å to 1.345-1.351 Å depending on the Ti sites). It is essential to notice that they were able to avoid any competitive HER.
Besides the experimental proof, the ongoing study has backed the use of several different semiconductors to modulate their energy levels with the plasmon noble metal. Semiconductors, metal, or metal oxide coupled with plasmonic metals, offer intriguing signs of accelerated reduction-oxidation reactions under atmospheric temperature and pressure.
Vacancy-assisted composites
Vacancies are induced as the result of imperfections or low coordination sites that alter the electrical and optical properties of the solid structures. DFT studies have revealed that oxygen vacancies (OVs) on oxide surfaces have low formation energy, which has encouraged several researchers to explore the possibilities of using OVs to increase the rate of catalytic processes at the surface129. Imitating a biological phenomenon that uses a Mo-dependent enzyme nitrogenase where two metalloprotein clusters (electron-donating Fe and MoFe protein) work cooperatively to produce NH3 in a placid condition has always been an ultimate goal. In this process, however, the high energy barrier of N2 makes the route more laborious than that of CO2 reduction and H2 evolution. It turned out that such inadequacy could be incapacitated by introducing several types of imperfections, such as OVs, Sulfur vacancies (SVs), and Nitrogen vacancies (NVs) on the semiconductor surface. Upon literature survey we have observed two types of vacancy-based photocatalyst into multi and single components; single nanosheets will be discussed in “2D-Nanosheets: defects induced plasmonic catalysis” section.
The number of applications for plasmon absorption and scattering in Au NPs is remarkable. The unique characteristic of gold to exhibit LSPR has been broadly used to improve product formation. The selective growth of Ceria (n-type semiconductor) onto Au NPs anchored by a bilayer of cetyltrimethylammonium bromide (CTAB) bears promise toward initiating N2 activation (Fig. 11a; vide supra). The surface of the grown ceria, abundant in OVs, not only expedites the N2 adsorption but also ensures the activation. The Au/end-CeO2 catalyst’s spatial separation design provides reaction sites for oxidation and reduction (Fig. 11b), producing 114.3 μmol g−1 h−1 of NH3, which is almost six times faster than the core-shell Au-CeO2 catalyst (Fig. 11c)111.

a Schematic illustration of an Au/end-CeO2 nanostructure synthesis process. b Comparison of NH3 production on Au NRs, Au core-shell, and Au/end-CeO2. The core-shell nanostructures fully encapsulate the Au nanocrystal in the oxide shell; this inhibits the energetic heated electrons from reaching the reactant molecules, inhibiting catalytic activity. On the other hand, the spatially separated end-on modification permits the reactant to interact with the hot electrons and holes, boosting photocatalytic activity. c Hot-carrier separation behaviors of the Au/end-CeO2 nanostructure with that of the core-shell nanostructure111 (a, b, c reprinted with permission from ref. 111. Copyright, 2019, American Chemical Society).
Moreover, Li et al. reported a new plasmonic metal catalyst of Au supported on BiOCl, which contains OVs that utilize the synergistic behavior of hot electrons and holes to oxidize benzyl alcohol130. Generally, OVs act as catalytic sites for N2 molecules and enhance the lifetime of charge carriers, leading to the weakening of the triple bond of N2 (Fig. 12a)51. Although the hot holes keep the substrate from being over-oxidized, low group velocities and short mean free paths cause a rapid decay rate22 of the energetic charge carriers in the range of femtoseconds, which demands careful attention. The lifetime of the charge carriers can also be fostered by introducing oxygen vacancy-induced low-lying localized electronic states in the adsorbate (Fig. 12b), which typically act as very efficient trapping sites.

a Illustration of oxygen vacancy mediated catalytic process. OVs act as catalytic sites for N2 molecules and boost the lifetime of charge carriers, thereby weakening the triple bond of N2 (a reprinted with permission from ref. 51. Copyright 2017, American Chemical Society). b Schematic depiction of hot electron dynamics within Au-BiOCl-OV130 (b reprinted with permission from ref. 130. Copyright 2017, American Chemical Society).
A UV-Vis study of Au-BiOCl-OV with defect-free BiOCl, BiOCl-OV, and Au-BiOCl revealed that Au-BiOCl-OV has the combined effect of both BiOCl-OV and Au-BiOCl. The absorption of Au-BiOCl-OV is in perfect accordance with the solar emission range (450–650 nm), which verifies the high uptake of light energy. The structure of the OVs has proven to be a crucial parameter in ammonia production. BiOCl is utilized as a model catalyst to investigate the effect of OVs on the thermodynamic and kinetic activation of N2 (Fig. 12a).
Compared to BOC (BiOCl Single-Crystalline nanosheets) 001, controlled tests have indicated that the N2 binding mode in BOC 010 is more effective in activating nitrogen (4.6 μmol h−1 ammonia production rate). This accounts for the excellent stability of the side-on bridging mode of N2 on BOC 010 than the terminal end-on binding on BOC 001131. End-on coordination involves an asymmetric distal pathway in which consecutive protonation is performed on one remote nitrogen atom to generate the first NH3, and the coordinating nitrogen is hydrogenated to yield the second NH3. The side-on bridging involves alternate hydrogenation on both nitrogen atoms of the coordinated N2 moiety to generate N2H2 and N2H4 intermediates successively132. The formation of intermediates is a high-energy demanding process; yet, due to the low-energy reaction steps, an OV-mediated reduction on BOC surfaces has proven to be thermodynamically advantageous. Theoretical analysis suggests that as long as N2 is bound to the OVs of BiOCl, the first electron addition to the π* antibonding orbital, which usually is an endothermic step, changes into an exothermic step, with a free energy change of 41.7 kcal mol−1 for the terminal end-on mode and 53.3 kcal mol−1 for the side-on binding mode. Similarly, a BiOBr surface with surface defects has been claimed to trigger N2 activation and generate NH3 in pure water without requiring a sacrificial agent116. Researchers were tempted to manufacture composite structures of morphologically modified δ-Bi2O3 for nitrogen photofixation since the p-type bismuth oxide is substantially rich in anionic OVs. Gao et al. devised a simple method for producing 2D ultrathin Ag/AgI- δ-Bi2O3 and recorded an NH3 output of 420.0 μmol.L−1 g−1 h−1, which is much higher than that of δ-Bi2O3 (Fig. 13a)133. The bigger surface area increased the visible light absorption by the plasmonic Ag metal, oxygen vacancy-driven charge separation, adsorption, and activation of N2 molecules on the surface, and they are all involved in the elevated photochemical activity98.

a Illustration of proposed nitrogen reduction pathway on Ag/AgI-δ-Bi2O3. Effective adsorption occurs at higher concentrations of oxygen vacancies. Upon illumination, Ag NPs create energetic electrons, which are transferred to the δ-Bi2O3 and captured by OVs, while hot holes move from the δ-Bi2O3 to AgI. The energetic electrons participate in the reduction of N2, and the photogenerated holes oxidize the H2O to O2133 (a reprinted with permission from ref. 133. Copyright 2019, Nanomaterials). b Photocatalytic nitrogen fixation performance control to determine the role of CNNVs. c Comparison of performance of CNNVs v/s AuCNNVs under visible and full spectrum radiation under 300 W Xe lamp. d CN with different hydrogen reduction temperatures (b, c, d reprinted with permission from ref. 135. © 2020 Wiley‐VCH Verlag GmbH & Co. KGaA, Weinheim).
In addition to OVs, nitrogen vacancies (NVs) and sulfur vacancies (SVs) have been the scientists’ focus for quite some time. SVs have assumed a prominent role when combined with a noble plasmonic metal, in line with the catalysts stated in the preceding sections. Yuan et al. demonstrated sulfur vacancy-induced plasmonic catalysis to degrade N2 to NH3 with a yield of 438.0 μM g−1 h−1. They synthesized a bimetallic structure of Ru/CoSx containing sulfur vacancies on g-C3N4 nanosheets. The increased electric field produced by the LSPR effect of the plasmonic Ru NP triggered a six-fold increase in the vis-NIR absorption of the Ru-SV-CoS/CN composite, which in turn stimulated the generation of highly energetic charge carriers, and thereby, the N2 fixation rate in pure water was amplified. Due to their low coordination, Ru and Co atoms near the interface are excellent active sites for the side-on chemisorption of nitrogen molecules134. The Ru/CoSx interface was shown to have a ninefold increase in electron-oscillation absorption, making it a catalytic hotspot. The asymmetrical transfer of electrons by Ru and Co to the N atoms at both ends of N2 leads to a highly polarized and significantly weakened N–N bond. On the downside, the LSPR of Ru is not that effective without the support of localized electron states and is exhibited mainly in the UV region.
Apart from SVs, NVs operate as excellent electron-capturing sites, and the associated transient defect states can act to promote the lifetime of the energetic electron–hole pair. They promote the chemisorption and activation of N2 during the reduction process. The effect of Au-loaded g-C3N4 (n-type semiconductor) with numerous NVs in the N2 reduction process was addressed by Wu et al. 135. Au NPs were loaded on the CNNVs (Carbon nitride nitrogen vacancies) using an impregnation-calcination procedure. The increased yield of NH3 on the vacancy-incorporated surface of CN compared to bulk CN outlined the value of NVs in driving chemical reactions (Fig. 13b). The reaction is further stimulated by the LSPR effect of the plasmonic Au NPs, yielding a 93.0 μmol g−1 and 184.0 μmol g−1 NH3 without any sacrificial agent under visible and full light irradiation, respectively (Fig. 13c, d).
The Schottky barrier between Au NPs and g-C3N4 permits energetic electrons to travel over the surface while preventing them from being transferred from g-C3N4 to Au. The energetic hot electrons populate the g-C3N4 conduction band, which subsequently diffuses and becomes confined in the NVs with the chemisorbed N2 molecules (vide supra). The hot electron degraded the activated nitrogen molecules on NVs sites to ammonia. Au embellished over hollow carbon nitride spheres (Au@HCNSs-NV) has also been reported with an NH3 production rate of 783.4 μmol h−1 gcat−1 under visible light136. It is to be recognized that NVs outperform OVs in N2 bond elongation (Free N2−1.114 Å; NVs adsorbed- 1.236 Å; OVs adsorbed- 1.173 Å).
This study presents a novel methodology for engineering highly efficient vacancy-based photocatalysts in solar-driven ammonia synthesis. It is fair to say that combining plasmon noble metals with vacancy-incorporated semiconductors might significantly enhance catalytic processes. We are optimistic that the fabrication of such hybrid structures will introduce more refined ways of heterogeneous catalysis in the near future.
Fabricating ensembles and plasmon-embedded frameworks
When inorganic clusters or metal ions combine with organic molecules, they form a highly organized porous framework known as a metal-organic framework (MOF). Variable pore sizes and high surface area enable these frameworks to suit the demands of the applications, making them versatile. MOFs work on the molecular sieving effect, which utilizes the porous interior area to encapsulate molecules of various sizes137,138. Research has been conducted employing MOFs to improve the efficiency of plasmon-mediated processes such as water splitting and carbon dioxide reduction137,139,140. This prompted whether the same process could fixate nitrogen.
Chen et al. reported using a metal-organic framework (UiO-66) for direct plasmonic NRR (PNRR) with gold inserted within the porous region (Fig. 14)49. On the surface of Gold embedded metal-organic framework (Au@MOF), they synthesized ammonia in a gas membrane solution. Besides the improved localized electric field generated by the Au NPs, the intimate interaction of the Au NPs with the adsorbed nitrogen molecules aids activation by improving the energy transfer (ET) process and electric field (EF) polarization (Fig. 14a). These three mechanisms work in tandem to promote more vibrational transitions in the N2 molecule, which lengthens the N–N bond and promotes fixation. Other mechanisms stimulating the N2 molecule to higher energetic levels include energy transfer, photothermal effect, and a strong localized electric field. This contributes to the production of intermediates via the lowest energy pathway. A lifetime of vibrational relaxation is a few picoseconds that further support the gas-phase chemical reactions. It paves the way for a reactant molecule to reach the transition state quickly. Nitrogen was purged via the PTFE (polytetrafluoroethylene) polymer linked to the matrix’s other interface (Fig. 14b). This polymer has a low friction coefficient and an optimum pore size, allowing nitrogen flow to permeate quickly to the Au@MOF surface. Considering the adaptability of inorganic NPs, plasmonic metals have been integrated into the porous matrix (Fig. 14c). Their design allows gold to harvest solar energy to generate hot electrons via surface plasmon resonance and boosts its dispersibility and stability. The confinement of gold NPs in this matrix assures nitrogen availability in the vicinity of the Au NPs and accelerates the molecule’s mass transfer (Fig. 14d)141. It also overcomes nitrogen’s poor solubility in water (20 mg L−1). Using this setup, their group attained an ammonia evolution rate of 18.9 mmol gAu−1 h−1 (in the visible light range of >400 nm) with a quantum efficiency of 1.5 percent at 520 nm.

a Schematic depiction of direct PNRR on Au NPs encapsulated in UiO-66 matrix. a The activation of adsorbed N2 molecules (*N2) and other critical chemical intermediates (e.g., *NNH) is aided by synergistic LSPR effects such as hot electron formation, electron transfer (eT), energy transfer (ET), and localized-electric-field polarization (EF-polarization). When hot electrons (e−) and protons (H+) approach, N2 is reduced and protonated to NH3 (NH4+). b Gas membrane solution (GMS) reaction interface model. Nitrogen gas can be diffused in high flux through the gas-permeable Au@UiO-66 membranes supported by porous polytetrafluoroethylene (PTFE) films. c, d Schematic diagram showing the dispersion of plasmonic Au NPs within the cavities of MOFs matrix. The cage-like structure of the Au @UiO-66 matrix promotes N2 gas diffusion. As an outcome, the proximity orientation is far more effective, allowing for a higher radiant flux absorbed by N249 (a, b, c, d reprinted with permission from ref. 49. Copyright 2021, American Chemical Society).
Multiple metal-based frameworks have been investigated during the past few decades, for instance—Zr (UiO-66, UiO-67, ZIF-8)142,143,144, Cr (MIL- 100), Zn (MOF-5), etc. Amongst these, Zr-based UiO-66 has been demonstrated to reduce nitrogen145. UiO-66 has the edge over other common MOFs (such as UiO-67) owing to its inability to exhibit absorption in the 350–1200 nm region. As an outcome, the plasmonic metal acts as the sole absorber in the LSPR region, and it has evolved as an efficient means of modulating the collective oscillation of free electrons through changes in the local refractive index in response to light irradiation. Such a setup has proven to be robust in terms of chemical and colloidal stability. The most prevalent techniques for synthesizing MOFs are one pot and multistep, with one pot involving temperature initiation by combining all substrates necessary in the same medium.
On the other hand, Chen et al. used a multistep procedure that included numerous seeding strategies49. A maximum yield was obtained with an Au mass loading of 1.9 wt% Au@UiO-66 (Fig. 15a). Both the size of the matrix UiO-66 (146 ± 14 nm) and the size of the Au NPs (2.4 ± 0.6 nm) function efficiently in synergy with the LSPR wavelength at 520 nm in this composition (Fig. 15b). Studies have shown that similar mass loadings of Au embedded in the MOF matrix displayed a higher ammonia evolution rate than with semiconductors such as ZrO2 (1.05 mmol gAu−1 h−1; 2.0 wt% of 3.7 ± 0.7 nm Au NPs) and SiO2 (1.42 mmol gAu−1 h−1; 1.9 wt% of 9.1 ± 1.5 nm Au NPs) as well as unsupported Au NPs (Fig. 15c). Unsupported Au NPs tend to agglomerate during photocatalytic conversion, lowering the production rate. There are numerous ways to improve the efficiency of ammonia synthesis; one typical method is to raise the intensity of visible light, which can factorize the rate by up to six times (Fig. 15d). Despite exhibiting considerable potential, the field of plasmonic MOFs for N2 fixation has remained unexplored.

a Ammonia evolution rate for plasmon-induced photocatalytic NRR (P2NRR) on bare UiO-66 and Au@UiO-66 particles with different mass loadings (0.9- blue, 1.9- green, 3.0- yellow, 4.1- orange, 5.2—red wt% respectively). The highest yield is obtained with 1.9 wt% b AQE (left axis) and Extinction (right axis) of P2NRR on Au@UiO-66 (1.9 wt%) particles at different wavelengths. At 520 nm, the highest ammonia output is obtained, which correlates to the LSPR peak, indicating the relevance of plasmonic metal in catalysis enhancement. c Cycling tests for P2NRR under visible light (>400 nm, 100 mW cm−2) on Au@UiO-66 (1.9 wt%), Au/SiO2 (1.9 wt%), Au/ZrO2 (2.0 wt%), and unsupported Au NPs. Reproducible results over time imply the stability of the matrix. Bare Au NPs, Au@SiO2, and Au@ZrO2 resulted in lower ammonia yield due to the lack of confinement of N2. d Ammonia yield (left axis) and Apparent quantum efficiency (AQE- right axis) of P2NRR on Au@UiO-66 (1.9 wt%) particles under 520 nm irradiation with different intensities. The rate of ammonia evolution exhibits superlinear dependency on intensity49. (Reprinted with permission from ref. 49. Copyright 2021, American Chemical Society).
Recently, Boong et al. developed a novel design to localize and concentrate light intensity, creating a strong field by synthesizing Ag-based plasmon ensembles with dense electromagnetic hotspots146. The group synthesized Ag octahedra and adopted a biphasic self-assembly method to build three unique 2D superlattices (ensemble): square, hexagonal, and disordered (Fig. 16a). The 2D ensemble showed exceptional field enhancement via extensive coupling and eliminated the need for a cocatalyst whatsoever (Fig. 16b). Upon several control experiments and analysis, the Ag-square superlattice observed ~15 fold, ~4 fold, and ~103 fold increase in NH3 production to the hexagonal, disorganized lattice, and traditional catalysts, respectively (Fig. 16c, d). They addressed the poor field enhancement issue and reduced the charge recombination rate. The ensemble remained steady over numerous cycles and demonstrated a rather consistent photocurrent at ~70 μA cm−2 supporting the hot electron flow.

a Graphic illustration of nitrogen fixation over the superlattice. Maximum field confinement was seen at the octahedral tips/edges. Capping with various molecules such as C12SH for square, C4SH for disorganized, and PVP for hexagonal setup was used to modulate lattice build-up through Ag octahedra. Water was used as a hydrogen source, and recombination was mitigated using 1-propanol as a hole scavenger. b SERS (surface-enhanced Raman spectroscopy) enhancement factor is a clear indicator of magnification of the Raman signal of molecules interacting with the surface of plasmonic nanostructures. c Comparison of NH3 generation rate for different ensembles under Ar and N2 atmospheres. d Evidence of N2 fixation via a photocatalytic pathway with significantly less contribution from photothermal effect146 (Reprinted with permission from ref. 146. © 2022 Wiley-VCH GmbH).
One advantage of employing MOFs and plasmon-based ensembles is the capacity to adjust numerous response parameters. However, because of its low yield, these applications for ammonia synthesis are a relatively new sector, making it industrially unusable in the current environment. Nonetheless, we postulate that if further investigated, such plasmon-mediated- macrostructures may open up pathways for nitrogen fixation under environmentally sound settings.
Plasmon-assisted electrochemical reduction
Following plasmon-mediated CO2 reduction and water splitting83,147,148, it remains unknown whether the same might be replicated for nitrogen in classic electrocatalytic systems. In this section, we highlight the synergistic effect of electricity and visible light-induced LSPR of the plasmonic NPs (in particular Au) to accelerate the ammonia production rate. The likelihood of directing the effect of local photothermal heating of an irradiated plasmonic material-coated electrode enables the enhancement of the electrosynthesis of ammonia. This enhancement factor refers to the non-thermal effects of plasmonic excitation, mostly energetic carriers and charged interfaces, and depends on the applied potential. When the electrode is illuminated with light of a wavelength coinciding with the LSPR of plasmonic material, energetic charge carriers are generated through inter and intraband transitions. These photoexcited carriers are responsible for enhanced nitrogen reduction activity. Furthermore, compared to the colloidal solution, the red-shifted LSPR of the surface coated on the electrode accounts for the strong plasmonic coupling among the deposited plasmonic NPs149. Besides the applied potential, oscillating surface potential generated plasmonic NPs further aids in the electrochemical NRR (ENRR).
From our previous discussions regarding the metal-metal composite, we vividly understand the broad applicability of Au. Can we design an antenna reactor type, however, with another plasmon metal functioning as the antenna? Moreover, what if such a design could input solar and electrical energy? Experimenting along the same line, Cheng et al. synthesized Au-Cu hollow beveled nano boxes, where they were successful in highlighting the synergy between solar and electrical energy (Fig. 17)150. The group deposited the hollow nano boxes onto the working electrode and carried out solar-illuminated ENRR. Increase in current density with intermittent light show that the improvement was made possible by an oscillating electric field brought on by plasmonic stimulation. At −0.2 V, current density (3.8 mA cm−2) under irradiation was found to be 2% (3.7 mA cm−2) elevated than in light (Fig. 17a). The rate of ammonia production highlights the synergy between Cu and Au to aid in better NRR (Fig. 17b). Aside from boosting the electric field, the charge transport resistance of the catalysts at the interface of the working electrode in the presence of light was also lowered from 71.08 Ω to 64.36 Ω. Additionally, faradaic efficiency rises by ~0.4% under irradiation precisely points towards the existence of LSPR and its assistance for NRR (Fig. 17c).

a Current density (J) v/s time plot for Au-Cu hollow nano boxes (HNB) catalyst under solar irradiation. Rise in J with light is evidence of electric field enhancement provided by LSPR. b Comparison of ammonia rate production for controls to prove the synergy between Cu and Au to aid in better NRR. c Escalation in current density under the light compared to dark; maximum obtained at −0.2 V v/s RHE150 (a, b, c reprinted with permission from ref. 150. Copyright 2021, American Chemical Society). d Ammonia yield rate or Faradaic efficiency for various types and shapes of NPs at the potential of −0.4 V in 0.5 M LiClO4 aqueous solution152 (d reprinted with permission from152. Copyright 2018, Elsevier B.V. All rights reserved).
The shape and size of Au NPs as an electrode may modify the selectivity and photocatalytic efficiency by tuning the LSPR peaks from 635 to 795 nm. Under ambient circumstances, gold hollow nano cages (AuHNCs) may achieve the maximum Faraday efficiency and ammonia evolution rate. The charge or electron-transfer efficacy during an electrochemical process is referred to as faradaic efficiency. Due to their higher surface area and capacity to contain the reactant molecule, allowing for proximity orientation, AuHNCs show a considerable variation when compared to AuNSs (Nanospheres), AuNCs (Nanocubes), and Au NRs (Nanorods)151 (cage effect) (Fig. 17d)152,153. Amongst all shapes, Nazemi et al. achieved the highest NH3 yield rate (3.7 μg cm−2 h−1) and faradaic efficiency (35.9%) using AuHNCs –750 nm at −0.4 V vs. RHE.
Liang et al. have developed a technique to improve the catalytic efficiency and selectivity of the ENRR154. The group, however, employed NRs as the electrode and achieved an optimum yield. The working electrode was synthesized using the drip coating approach to prevent Au NP aggregation and retain the LSPR capabilities of Au nanorods (Fig. 18a, b).

a Schematic depiction of the fabrication of Au NRs electrode. Drip coating Au NRs on naphthol onto carbon paper prevents Au NPs agglomeration. b Schematic diagram of LSPR enhanced ENRR. c Comparison of extinction spectrum of Au NRs (black line) and ammonia yield rate under different wavelengths (red column). The maximum absorption occurs at the LSPR wavelength (808 nm), which corresponds to the highest ammonia production. d The ammonia yield rate of Au NRs electrode under different laser intensities. Strong linear dependency is observed. e Ammonia yield (red column—under illumination; blank column—in the dark) rate and faradaic efficiency (solid blue—under illumination; dashed blue—in the dark) at various potentials under dark and 808 nm laser illumination154. Reprinted with permission from ref. 154. Copyright 2022, Elsevier B.V. All rights reserved.
The ammonia output surged by 63.3 percent under light compared to dark conditions, verifying the photocatalytic effect. The rate of nitrogen evolution was much higher than the rate of hydrogen yield, further promoting ENRR. Two LSPR peaks, transversal (512 nm) and longitudinal (756 nm), were obtained due to the shape of the Au NRs. Furthermore, the extinction spectrum of Au NRs and Au NR-electrodes were very similar, indicating that Au NRs on carbon paper were dispersed uniformly (Fig. 18a).
While maintaining the LSPR, the uniform distribution allowed for a significant proportion of active catalytic sites. The higher intensity of the extinction spectrum correlates to a stronger electromagnetic field, which generates energetic charge carriers (e-h pairs) under ambient conditions (Fig. 18b). Chronoamperometry measurements were used to determine the NRR performance of the Au NRs electrode and the enhancement impact of illumination. Under illumination, the photocurrent measurement of the Au NRs electrode at various laser intensities may provide information on the mechanistic intricacies, even though the precise process has yet to be investigated. ENRR is influenced by various parameters, including current density, incident light wavelength and intensity, potential and electromagnetic field intensity, shape, size, and composition of electrodes (Fig. 18c–e)155. In one such example, the longitudinal LSPR peak shows a redshift to 808 nm due to some aggregation on the Au NRs electrode, unlike the transversal. The strongest electromagnetic field was localized at both ends at 808 nm. The Irapid/Itotal ratio reaches its maximum value (37.4%) at this wavelength, indicating that more hot electrons are involved, resulting in a greater photocurrent density. \({{{{{{\rm{I}}}}}}}_{{{{{{\rm{rapid}}}}}}}\) and \({{{{{{\rm{I}}}}}}}_{{{{{{\rm{total}}}}}}}\) are defined as the following Eqs. 7 and 8.
Where I1 is the current response after 1 s of laser illumination. \({{{{{{\rm{I}}}}}}}_{{{{{{\rm{off}}}}}}}\) and \({{{{{{\rm{I}}}}}}}_{{{{{{\rm{on}}}}}}}\) are the current responses at the end and the beginning of the laser illumination. Their group recorded a maximum ammonia production of 0.54 μg h−1 cm−2 at 808 nm with a laser intensity of 80 mW cm−2 and a potential of −0.4 V, which is 63.6% higher than that achieved in the dark (0.3 μg h−1 cm−2 at −0.5 V). The ammonia production rate is calculated using the following relation (Eq. 9)154.
Where c = ammonia concentration in μg mL−1; r = rate of production; V = volume of electrolyte in mL; t = time in hour; A = area of working electrode in cm2.
The Au NRs electrode structural stability was also crucial since the ammonia rate after each cycle was identical to that of the fresh electrode. Because of the slow convoluted reaction, the yield of plasmon-enhanced ENRR is inapplicable for industrial uses. Recently, Jain and the group elucidated a gold-based electrocatalyst for ammonia synthesis from nitrate149. Incorporating the light-harvesting capability of Au NPs as a result of LSPR and electrochemical nitrate reduction activity, they increased ammonia synthesis 15-fold over conventional electrocatalysis. This occurs primarily because plasmonic excitation generates nonequilibrium conditions. We predict that further research into this arena, such as employing AuHNCs as electrodes and experimenting with alternative composite structures, might lead to new advancement pathways in plasmon-mediated electrocatalysts.
Using the merits of metal-metal-oxide composites as an electrode can also help advance the electrochemical reduction of N2. For instance, Vu et al. reported a novel photocatalyst by assembling Au NPs with a Fe-doped n-type semiconductor (W18O49 NRs)156. Fe acts to eliminate the bulk defects of W18O49, while the Au-W18O49 composite assists in inducing LSPR as well as reducing the recombination time of the energetic charge carriers. Also, Ag2O, a p-type semiconductor, was also combined with Au in the form of hollow nanocages. The novelty of such a photoelectrode design lies in integrating two light-sensitive species, which increases the density of energetic electrons upon visible light irradiation. The electron from the conduction band of Ag2O can be transferred to the surface of Au NPs, and at the same time, the photogenerated hot electrons in Au can be injected into the Ag2O conduction band. Henceforth, both can act as active catalytic sites and increase the NRR efficiency. It is to be noted that, here, Au also increases the e-h separation by accepting electrons from Ag2O and aids in enhancing the photocatalytic activity50. Furthermore, Niobium (Nb) doped strontium titanate (Nb-SrTiO3) loaded with Au NPs, and Ru cocatalyst was proven highly effective in electrocatalytic nitrogen reduction. Oshikiri et al. designed a two-chamber approach to separate the oxidized and reduced products157. The extended charge separation at the Au/Nb-SrTiO3 interface allows for simultaneous ethanol-water oxidation at the anode and nitrogen reduction on the Ruthenium surface at the cathode. The rate of ammonia synthesis increased linearly with intensity when exposed to visible light with wavelengths longer than 550 nm, confirming the assistance from the LSPR effect of Au NPs. It is to be noted that SrTiO3 does not absorb light with a wavelength longer than 390 nm. On top of that, the nature of the cocatalyst can vary the selectivity and catalytic properties of the reactions; for instance, introducing a thin coating of Zirconium/Zirconium oxide (Zr/ZrOx) instead of Ru improves selectivity by dampening the hydrogen evolution reaction110. As per DFT, the Zr surface preferentially adsorbs N atoms due to the smaller adsorption energy of N (∆N*) value relative to the adsorption energy of H (∆H*), whereas ∆H* value on the Ru and a variety of transitional metal surfaces is smaller than ∆N*, which resulted in a hydrogen production rate that was 15 times greater than that of the ammonia synthesis rate. However, with a Zr/ZrOx cocatalyst, not only did the NH3 generation rate rise by a factor of 6 (1.1–6.2 nmol h−1 cm−2) but the H2 evolution rate was found to be also lower than the NH3 evolution rate. Another two-electrode-based electrochemical approach was reported by Oshikiri et al. consisting of SrTiO3 photoanode coated with Au NPs and Zr coil cathode. The more significant reaction rate of NH3 production can be governed by efficient ion transport, the greater cathode surface area, and the continuous supply of N2 (to avoid air contamination in an open cathode chamber). The novel plasmonic photoanode development could enhance charge separation efficiencies, such as the coupling between plasmons and the other photonic modes158. Electrochemical reduction in coherence with plasmon resonance is a step in the right direction toward lowering the kinetic barrier encountered during nitrogen reduction, but it has drawbacks such as yield and selectivity with regard to hydrogen evolution.
Non-noble metals for plasmon-enhanced nitrogen reduction
Initiating progress in terms of economic viability and abundance, several non-noble metals, such as Al, Mg, Co, Ni, In, Bi, Ga, and Fe have been used, amongst which Bismuth (Bi) has recently attracted significant research attention as a noble metal substitute63. It is a semi-metal (with a work function of 4.5 eV) that inherits the same SPR property near the infrared region as the noble metals. SPR property is more diffused than LSPR and does not necessarily require nanosized particles for its exhibition159. To engineer an efficient composite structure at the nanoscale, Bi can be coupled with several noble metals, semiconductors, or both to form a hybrid that can effectively adsorb and activate the Nitrogen molecule. Focusing first on the Bi-Metal-oxide semiconductors (Bi2O3, TiO2, Cu2O, BiO2(CO3), Bi2WO6160) hybrid nanostructures, higher selectivity for charge separation and migration rates can be achieved.
While various metal-oxide semiconductors such as TiO2 have been highly versatile, the wide bandgap (3.2 eV)160 imposes a challenge of rapid recombination of charge carriers, i.e., the holes and electrons. The separation efficiency can be determined through time-resolved photoluminescence, where a lower PL intensity indicates slow recombination, thereby increasing the lifetime of the charge carriers. The wide bandgap also shifts the energy gap to the far UV region. Therefore, to expand the utility of such semiconductors, they are engineered in composite with other semiconductors (forming a heterojunction), doped with metal having enhanced photocatalytic response, or a narrow bandgap photocatalyst replaces them. Wang et al. proposed the use of InVO4 possessing a narrow bandgap, facilitating the lower separation of the photogenerated charge carriers. The group synthesized homogenized ultrathin sheets of Indium vanadate coupled with 5% Bi161.
Here, the specificity of the reaction, optical response, and surface activation sites are all influenced by the composition of the metal and semiconductor. Numerous techniques such as X-ray diffraction, atomic force microscopy, Transmission electron microscopy (TEM), and Scanning electron microscopy (SEM) can be utilized to confirm the deposition of the metal onto the semiconductor surface, the thickness of the hybrid structure, and accurate nanoscaled sizes. Their hypothesis of employing a low bandgap semiconductor and embedding it with Bi to catalyze the reaction was validated when the yield of NH3 synthesized turned out to be 626 µmol g−1 h−1 (5.2 times higher) on Bi/InVO4 than with pure InVO4.
In contrast to noble metals, Bi has a different mechanism for nitrogen fixation. The conduction band of the semiconductors aligns itself to slightly higher negative energy than Bi’s Fermi level. When the light is irradiated, an enhanced electric field generated due to Bi plasmon resonance influences the movement of the electrons from the valence band to the conduction band of the semiconductor, followed by hole reduction by water molecules eliminate a proton where minor changes in the momentum allow it to be trapped at the metal site162. Nitrogen adsorbed at metal surfaces can get activated through these hot electrons. N2 adsorbed in associative pathways where arises two possibilities- distal and alternating pathways163. Although this cascade of charge transfer has boosted ammonia synthesis, several minute details must be taken care of while designing the nanostructure. Even though the plasmonic effect is increased many folds at the nanoscale leading to a higher extinction cross-section, hydrogen evolution remains the main challenge. The proton released through water oxidation via holes can form radical species combining further to evolve hydrogen gas.
Semiconductor depositions are commonly used to prolong photogenerated charge carrier lifetime. Dong et al. used Bi as a photocatalyst using BSC microspheres and discovered improved catalytic activity when exposed to visible light. This further demonstrated the SPR impact of Bi164. Another semiconductor-based heterojunction witnessed is via graphitic-C3N4, having a unique 2D framework and viable synthetic methodology165. Owing to the high work function, Bi decoration allows for the facile transfer of electrons from g-C3N4 to Bi (Fig. 19a). Bi decoration on g-C3N4, shows a bathochromic shift compared to g-C3N4 (Fig. 19b). It aids in increment in adsorption sites, validated from the DFT calculation. The adsorption energy of Bi/g-C3N4 (−2.3 eV) was found to be 7.6 times higher greater than that of pure g-C3N4 (−0.3 eV), clearly indicating thermodynamic accessibility of N2 on active sites (Fig. 19c). While Bi decoration enhances the visible light absorption via SPR, its excess loading can lower the NH3 production rate. This arises from thermodynamic competition between electron transfer from Bi/g-C3N4 to inert N2 and Bi. The maximum yield (1025 μmol L−1 g−1 h−1) was obtained at 30% mass loading of Bi on g-C3N4 (Fig. 19d).

a Schematic depiction of nitrogen fixation aided by the surface plasmon effect (SPR) of the Bi. b UV-DRS spectra, Bi/g-C3N4, show a bathochromic shift compared to g-C3N4. Higher absorption plainly indicates more exciton generation. c N2 adsorption-desorption isotherms imply an abundance of active sites. d Ammonia yield achieved the maximum in 30% mass loading of Bi165. Reprinted with permission from ref. 165. Copyright 2021, American Chemical Society.
Aside from semiconductor deposition, in situ Bi reduction methods have been used to build composite structures such as Bi/Bi2WO6160. Chen and the group used preferential arrangements to prolong charge carrier separation upon realizing the lattice match between Bi and Bi2WO6. Pure Bi2WO6 showed zero response to nitrogen fixation, whereas oriented-Bi/Bi2WO6 produced a significant amount of NH3 (86 μmol g−1 h−1).
Bi provides a hurdle since it quickly oxidizes in air to Bi2O3, despite its adaptable qualities and technological advancements. Bi-based nonstoichiometric catalysts may exhibit remarkable selectivity due to their unique photochemical characteristics. Still, they can also facilitate the alteration of band structures and the formation of catalytic defects such as oxygen vacancies.
In accordance with the prior-mentioned examples, we may lay the groundwork for points to consider when developing any photocatalyst that captures the essence of Bi’s SPR. Interestingly, the plasmonic attribute of another non-noble metal, namely Al (abundant, highly stable, and non-toxic), has been exploited to perform NRR at room temperature when clubbed with a semiconductor (vide supra)80,166,167.
Another intriguing insight would be the employment of Cu due to the low cost and tendency to induce surface plasmon resonance, which has prompted researchers to synthesize composites incorporating Cu metal. Fe can also be used with Cu to photo-fixate nitrogen molecules. Porous Cu-Fe hybrids with different molar ratios were synthesized by selectively etching Fe from Cu21Fe79 with varied quantities of H2SO487. The formation of an active nitrogen-containing complex on the Fe surface is supported by an increase in the valence state of Fe and coordination number during the catalytic process. This active nitrogen-containing complex on the Fe atoms produces ammonia with the help of energetic charge carriers generated from Cu. The porous network further enhances the catalytic process by providing much better stability than the individual NPs. These factors lead to a high ammonia production rate of 342 μmol gcat –1 h–1 for Cu96Fe4 without utilizing any sacrificial agent. These heterogeneous catalysts have a pronounced absorption peak at 550 nm, which reflects plasmon-induced absorption. However, one of the most significant obstacles in this arena is the use of Cu, which may get oxidized in both air and water.
2D-Nanosheets: defects induced plasmonic catalysis
The limited capacity of the noble plasmonic metals (Au, Ag) to effectively adsorb nitrogen and the heterogeneous interface’s inability to allow for charge transport (attributable to the Schottky barrier or any unwanted charge trapping site) prompted the development of a single plasmonic nanostructure with a rich number of vacancies as active sites. Wu et al. developed semiconducting plasmonic MoO3-x (x essentially denotes the excess of Mo or O site left uncoordinated) that outperformed four earlier reported catalysts (TiO2, W18O49, BiOBr11, and g-C3N4)156,168 at wavelength >580 nm, emphasizing the benefit of employing MoO3-x for performing solar energy-driven chemical transformations. Low-valence Mo moieties and intrinsic OVs boost the NRR on the nanosheets in pure water. The OVs increase the longevity of highly energetic charge carriers while impeding electron–hole recombination. A significant quantity of NH3 generation rate, 328 μM gcat−1 h−1 was found under the complete sun spectrum. Low-valence Mo facilitates N2 chemisorption by electron-back donation. The introduction of excess electrons by the OVs in the conduction band improves the LSPR throughout a broad spectrum (vis to NIR), which provides hot electrons. Low-valence Mo5+ species were speculated to be produced by reducing Mo6+ by free-electron trapping. It should be noted that, despite the enhanced separation, a large number of hot charge carriers are lost upon passing the Schottky barrier allowing random movement of the carriers. With this knowledge, Bai et al. proposed a Schottky barrier-free semiconducting plasmonic MoO3-x spheres with reduced recombination rates and hot charge carrier loss. MoO3-x spheres were synthesized using the aerosol-spray method under an oxygen-deficient environment for incomplete precursor oxidation169. The three inequivalent oxygen sites in the MoO3 lattice, namely O1, O2, and O3 (Fig. 20a), allow for different combinations of OVs, causing defect states to form near the conduction band. Their analysis suggests that the Fermi level is above the conduction level, implying the metallic nature of MoO3-x. In comparison to typical metal-semiconductor devices, the barrier-less nature allows for the unfettered flow of hot electrons. This model achieved the highest apparent quantum efficiency of 1.2% generating 435.6 µmol g−1 h−1 of NH3.

a Crystal structure of oxygen vacancy-rich MoO3-x single nanostructures showing three inequivalent oxygen vacancies169 (a reprinted with permission from ref. 169. © 2021 Wiley‐VCH GmbH). b The proposed nitrogen fixation mechanism by LSPR excitation in SrMoO4 nanostructure. Abundant OVs source both the formation of add-on states near the Fermi level and collective oscillations of unpaired electrons responsible for generating LSPR. Plasmonic excitations from these add-on states to the conduction band or nearby higher energy level and the usual band excitations suffice for NRR170 (b reprinted with permission from ref. 170. Copyright 2021, Elsevier B.V. All rights reserved).
Recently, plasmonic SrMoO4 with OVs has been a potential candidate for NRR under the UV, vis, and near-infrared range (λ > 420 nm)170. The pure SrMoO4 was prepared by the facile solvothermal method, followed by introducing OVs by annealing the pure SrMoO4 in H2/Ar (5%/95%), reducing the atmosphere to 500 °C for 10 min. When pure SrMoO4 was irradiated with visible light, no NRR was detected, proving the role of OVs. The innate band excitation of SrMoO4 releases electrons from the valence band, leaving holes behind. Evanescent bands form near the Fermi level due to the abundance of OVs. Excitation causes plasmonic hot electrons to move from these evanescent bands to the conduction band or higher energy levels, allowing for the effective harvesting of visible and near-infrared light (Fig. 20b). Electrons from the valence band excitation and the plasmonic excitations (LSPR) contribute to the thermodynamic photocatalytic reduction of N2 to NH3 in ultrapure water with a yield of 3.9 μmol gcat–1 h–1.
MXenes mentioned in “Noble metal-mediated plasmonic catalysts for NRR” section also belong in this category, which can either be used as a semiconductor or plasmonic material. Clubbing all properties (such as effective adsorption of the substrate, enhanced photon absorption, fulfilling thermodynamic energy requirement, etc.) to build effective single nanostructures is still in its infancy, making it difficult to forecast the specific mechanistic pathway.
TiO2 integrated plasmonic composites for N2 reduction
Titanium dioxide (TiO2) is a superior choice within the industry and fundamental research, necessitating a whole section devoted to it. An optical bandgap near the UV region (~380 nm) shows efficient photoactivity (although limited to UV), high stability, and low cost. It is an n-type semiconductor and is extensively used as a photocatalyst because of its high electron-accepting ability (amplified by introducing defects—TiO2-x) and surface tunability. However, the lack of sufficient photogenerated electrons accessible at the TiO2 surface impedes both the reaction rate and the product yield. Noble and non-noble metals both are often anchored to the TiO2 surface to bypass the aforesaid drawback and prolong the lifetime of the excitons, thus aiding in the optimization of the catalytic activity171. Activated charge carriers could be generated close to the surface of the semiconductor as a consequence of interactions between the metal and the augmented localized electromagnetic field produced by the illumination of the metal center. These charge carriers can now reach the surface catalytic sites, enabling spatially separated redox reactions. This section highlights recent advances in TiO2-based plasmonic composites for the ambient N2 reduction reaction. The following examples demonstrate how to promote the reaction yield by combining the merits of doping, defect state-controlled catalysis, and electrochemical catalysis.
Yang et al. devised a nitrogen reduction catalyst employing Au and OV-containing TiO2 nanosheets. It was proven effective in reducing nitrogen to ammonia, with a 66.2% increase in the production rate compared to bare Au NPs. Au/TiO2 systems have been the most researched for nitrogen fixations. Fast electron injection (<50 fs) from the gold NP to TiO2 was seen in all Au-TiO2 systems172,173. The OVs and Au work in tandem to fixate nitrogen. Hot electrons generated by plasmonic Au nanocrystals cross the Schottky barrier and inject into the TiO2-OV conduction band. The injected hot electrons diffuse and become entrapped at the transient OV defect states (Fig. 21a, b).

a Schematic illustration of photo fixation of N2 with Au/TiO2-OV catalyst upon the irradiation of visible light. The hot electrons generated due to the LSPR effect of Au nanocrystals are further transferred to the transient electronic states induced by the OVs, which are subsequently injected into the LUMO of the N2 molecule. b Schematic depiction of plasmon-induced N2 photo fixation on Au/TiO2-OV surface, c Adsorption (left) and charge density difference (right) of N2 molecules at the oxygen vacancies (OVs) on the TiO2 surface. The light blue depicts the charge accumulation, and the yellow portion shows the charge depletion in space. d Temperature-programmed desorption (TPD) of four different Au-based catalysts. For TiO2 and Au/TiO2 catalysts, only one peak was observed at low temperatures owing to the physisorption of N2 molecules. In addition, a peak at higher temperatures was observed for both Au/TiO2-OV and TiO2/OV samples; this is attributed to the chemisorption of N2 molecules at the OVs of TiO2. (e) Adding an electron-capturing agent (Cr2O72−) and electron transport inhibitor (SiO2 shell) significantly lowers the ammonia production rate, suggesting the role of electron generation and transport in photocatalytic nitrogen reduction51. Reprinted with permission from ref. 51. Copyright 2018, American Chemical Society.
The trapped hot electrons convert OV-activated N2 to NH3. Furthermore, the adsorption efficiency of the catalytic surface was traced by a DFT-monitored clear charge density difference for the defect-incorporated TiO2 surface, unlike the OV-free TiO2 surface (Fig. 21c). In temperature-programmed desorption (TPD) analysis, pristine, defect-free TiO2, and Au/TiO2 surfaces only displayed one peak, which was assigned to physisorption. In contrast, the peak for TiO2-OV and Au/TiO2-OV surfaces at elevated temperatures was attributed to N2 chemisorption at the OVs on the TiO2 surface (Fig. 21d). Impediment of NH3 production rate by the addition of an electron-capturing agent (i.e., Cr2O72−) or an electron transport inhibitor (CTAB or SiO2 shell) to the catalyst reinforced the necessity of energetic electron generation as well as transport (Fig. 21e)51. Finely tuned OV-induced catalytic reduction of N2 molecule could also be achieved by using an amorphous TiO2 (a-TiO2) layer on plasmon-enhanced surface oxygen vacancy modified rutile TiO2/Au photoelectrode112. The formation energy of such OVs in the amorphous structure of TiO2 is 3 to 4 eV lower than that of crystalline TiO2129. It has grabbed much attention owing to its ability to possess much more OVs (higher carrier concentrations) than its crystalline counterparts. The interfacial electron transfer from Au particles and bulk rutile TiO2 to the π* LUMO of N2 to produce NH3 is facilitated by introducing transient electronic states in the a-TiO2 bandgap. SPR of Au NPs augments light absorption and synergistically enhances the photocatalytic N2 reduction on TiO2 photoelectrodes. The successive protonation leading to an associative mechanism of the adsorbed N2 on an a-TiO2 surface has been supported by DFT studies. The UV-Vis spectra of TiO2/Au and TiO2/Au/a-TiO2 samples exhibit a substantial bathochromic shift at 544 nm compared to TiO2 and TiO2/a-TiO2 samples that display a strong band at 410 nm, indicating that the surface plasmons are offering photo-induced electrons that accelerate N2 reduction174. Au NPs and an amorphous TiO2 layer, when encumbered onto the TiO2 NRs, exhibit a 2.6-fold increase in the production of NH3 (rate = 13.4 nmol cm−2 h−1) compared to bare TiO2 (rate = 5.1 nmol cm−2 h−1). Such rates for different photoelectrodes were measured by plunging in N2 saturated water with continuous N2 bubbling, then illuminated with visible light, eventually generating NH3. No such detection of NH3 was observed when the reaction was carried out in the dark and with argon instead of N2, corroborating that N2 bubbling was the sole source of NH3.
Parallel to the Au/TiO2 hybrid, a K/Ru/TiO2-x Hx (3 wt% Ru) catalyst has been developed175, where the electron trapping property of OVs and the proton trapping property of reversibly incorporated H atoms (Hinc) work in synergy to accelerate solar energy-driven N2 reduction at room temperature (Fig. 22a, b, c).

a Schematic depiction of K/Ru/TiO2-xHx based solar-driven ammonia synthesis b Comparison of ammonia synthesis rates of 4 Ru catalyst at 360 °C. The highest ammonia production rate (black bar) was obtained with K/Ru/TiO2-xHx, which is about 140.7 and 6.7 times those of K/Ru/Al2O3 and K/Ru/MgO. The TOF (Turnover frequency, red bar) value was also enhanced with a higher flow rate. This result is comparable to the conventional Haber-Bosch process. c Energy alignment of the Fermi levels (Ef) for TiO2, TiO2-xHx, and Ru. The Fermi level of TiO2-xHx is above the Fermi level of Ru, so the localized electron could be transferred to the Ru center, unlike the Fermi level of pure TiO2, where it lies below the Fermi level of Ru175. Reprinted with permission from ref. 175. Copyright 2017, Elsevier B.V. All rights reserved.
With 300 W Xe lamp illumination, they attained an NH3 yield of 112.6 μmol g−1 h−1 at 360 °C temperature175. In a similar line, leveraging the plasmonic properties of Ag metal, a heterostructure composed of Ag/PW12/Zr-mTiO2 was reported by Feng et al. (Fig. 23)176. They used interfacial engineering and doping approaches to target faster recombination rates and improve charge transfer. Zr doping in TiO2 modulates the overall porosity, which eventually augments the active sites for N2 chemisorption (Fig. 23a). Phosphotungstic acid (PW12 optimal 10 wt%) and Ag NPs (optimal 1 wt%) interface allow for smooth hot electron transfer averting the exciton combination probability (confirmed by photocurrent response) (Fig. 23d). An intriguing prospect relating to this scheme is flexibility in both light (324.2 μmol gcat−1 h−1) and electrochemical conditions (55.0 μg mgcat−1 h−1 at −0.6 V v/s RHE) individually (Fig. 23b, c).

a Mechanistic details of Nitrogen Reduction b NH3/NH4+ yield under simulated light irradiation for different controls. c UV-Vis Diffuse Reflectance spectra under monochromatic light irradiation. d Transient photocurrent responses showing separation efficiency for photogenerated electron-hole pairs for different controls; maximum for Ag/PW12/Zr-mTiO2176. Reprinted with permission from ref. 176. © 2021 Wiley-VCH GmbH.
The fundamentals behind each composite design are the appropriate wt% loading of each element. The same group simultaneously came up with another hybrid design-Ag/PW10V2/am-TiO2-X, however, explicitly for solar177. Engineering vanadium substituted PW drew its inspiration from band matching with TiO2-x, which allows TiO2-x to act as the electron reservoir. Photogenerated electrons from LUMO of PW10V2 to VB of TiO2-x combine with the holes, thus reducing recombination considerably. Hot electrons formed as a result of Ag NPs LSPR, pool themselves into the conduction band of TiO2-x. Another critical aspect is the amorphous TiO2-x, which generates localized electronic trap states close to it, allowing for copious electron input.
Despite all the encouraging optical and catalytic properties, Ag and Au are unstable under high temperatures. With this viewpoint, Thangamuthu et al. engineered an Al-based composite providing a cheap, efficient, and scalable method80. For the first time, the group used aluminum nanotriangles (AlNTs) embedded in TiO2 photoelectrodes on Indium tin oxide (ITO). Plasmonic near-field coupling to TiO2 prolongs the charge carrier generation efficiency over bare TiO2.
In “Noble metal-mediated plasmonic catalysts for NRR” section, we have witnessed the use of Ti3C2Tx MXenes as a semiconductor; however, due to substantial charge carrier densities on their surface, they were also seen to exhibit surface plasmon excitations at near-infrared regions (NIR) frequencies178. Its optimal utilization of plasmonic properties coupled with robust charge dynamics in TiO2 yielded a 422 μmol gcat−1 h−1 179. Low-energy infrared light was sufficient to produce hot holes on the surface of Ti3C2Tx and N2 adsorption was taken care of by defect sites on TiO2.
We have addressed the plenitude of examples majorly (Table 1); however, there is one additional peculiar approach for N2 fixation put to trial by Mao and the group53. They proposed the dual temperature zone-based catalysis producing high-temperature N2 activation on Fe and low-temperature NH3 generation on TiO2-x. The approach considers the plasmon heating effect of Fe and amplifies it with TiO2. TiO2’s broad application in many forms and its inherent synergy with most hybrids contribute to its appeal.
Conclusion and prospects
In this review, we delve into recent advancements in plasmonic catalysis-directed nitrogen fixation, providing a comprehensive guide for systematically designing potent photocatalysts. Beginning with the initiation of chemical reactions on single noble metal surfaces, this field has evolved to incorporate a diverse array of hybrid structures, showcasing incremental yet significant progress over time.
This exploration is particularly crucial as plasmonic catalysis in nitrogen fixation holds the potential to significantly impact global sustainability, addressing key issues related to food security and environmental protection. The technology offers an energy-efficient and environmentally friendly approach to ammonia production, potentially enhancing agricultural productivity while reducing the carbon footprint associated with fertilizers. By exploring alternative materials and optimizing catalysts, plasmonic catalysis shows promise in mitigating resource constraints. Moreover, its capacity for distributed production challenges the conventional centralized model, potentially improving fertilizer accessibility in regions with limited infrastructure. In summary, the broader implications of plasmonic catalysis position it as a transformative solution with substantial benefits for addressing critical global challenges.
The effectiveness of surface plasmons in expediting diverse processes underscores the critical need for selecting compatible plasmonic metals prior to fabrication. Specifically, plasmonic metals with LSPR in the UV-Vis range are essential for efficiently utilizing solar energy as a light source. Metals with a low imaginary dielectric constant enhance photon absorption, resulting in increased radiant flux (\({\sigma }_{{ext}}\), aiming to generate long-lifetime, highly energetic charge carriers (e-h) crucial for driving challenging reactions. Overcoming obstacles such as the inert nature of N2 and various factors affecting reaction dynamics requires strategic approaches, including hybrid constructions integrating plasmonic metals with semiconductors or metals. This integration amplifies near-field effects or extends exciton lifetimes.
However, the reported semiconductors suffered from several limitations, namely, the inability to absorb visible light or maintain long-term catalytic performance. The two primary objectives of synthesizing any photocatalyst, besides enhancing the rate of chemical processes, are (i) effective adsorption of reactants on the catalyst surface and (ii) absorption of incident light to generate excitons. The concurrent effect of plasmon metals and semiconductors can remarkably improve photocatalytic activity. This core structure of the photocatalyst was designed for plasmon metals to direct energy flow, concentrate the electric field intensity, and transfer energetic charge carriers to the adsorbed nitrogen molecules.
Semiconductor selection mainly depends on bandgap alignment, which triggers electron flow from the metal to the semiconductor or vice versa depending on the Fermi level position; band bending at the plasmon metal-semiconductor interface enables the creation of a Schottky barrier. Although this enhances the overall energetic charge dynamics, a large proportion of hot electrons is lost while crossing the Schottky barrier. In addition, interfacial energy loss also climbs as the number of components in the hybrid system increases. Furthermore, the high work function of noble metals (Au- 5.1 eV, Ag- 4.3 eV) stimulates a competitive side electron transfer, functioning as an electron sink. Hence, there is only a threshold to which plasmonic materials can be incorporated into a hybrid, after which the catalytic response gets lowered. Optimal plasmon loading provides viable degrees of freedom to tune the LSPR properties within the visible range.
To target the exceptionally high e-h recombination rate (~fs), surface imperfections, including oxygen vacancies (OVs), nitrogen vacancies (NVs), and sulfur vacancies (SVs), have been introduced in semiconductors. They provide transient electronic states that serve as electron-trapping sites. These surfaces are also paramount for enhancing nitrogen adsorption per unit area. Note that NVs are more susceptible to N2 bond elongation than OVs and SVs. Nevertheless, surface defects have the downside of being very reactive. For example, OVs that interact with water and oxygen can readily poison them. Hence, the inadequacies of heterostructures spurred a further exploration into integrating plasmonic properties, such as enhanced LSPR and efficient photon absorption, with the traits of a semiconductor, i.e., effective adsorption sites into single nanostructures.
While offering appealing characteristics, plasmon-aided N2 reduction also presents significant challenges. The concentration of solar energy on less reactive noble metals and the limited window for LSPR tuning are major setbacks. Researchers seek novel approaches to extend the plasmonic resonance wavelength. For instance, dendritic Ni-based black Au plasmonic colloidosomes enable plasmonic excitation across the entire visible spectrum, independent of a specific wavelength for catalytic activity180. Nanoparticle clustering offers a solution for LSPR broadening20,181. Despite advancements, the scarce abundance and high cost of noble metals limit their industrial applications. Non-noble plasmonic metals (NNPMs) provide an alternative due to their availability, low cost, and broader spectral SPR tuning. However, NNPMs require further modifications to maintain LSPR functionality. Reaction parameters subtly influence ammonia production rate, including solvent, temperature, N2 pressure, pH, light intensity, and irradiation time. Higher reaction rates are observed under N2-saturated atmospheres. Catalysts utilizing solar energy and electricity can promote NRR. Mechanistic routes and analyses contribute to developing effective plasmonics-mediated catalysts. Spectroscopic methods such as IR and SERS improve under plasmonic excitation, revealing mechanistic details. Combining these measurements with electrochemistry offers insights into catalytic activity’s influence.
A deep understanding of catalyst topology and underlying mechanisms greatly influences next-generation catalyst fabrication. Theoretical assessments, including correlational analyses and in situ ab initio calculations, contribute significantly to improving plasmonic catalysts. Various computational approaches alleviate hydrogen evolution bottlenecks, while DFT experiments effectively evaluate stability of adsorption sites in diverse materials. These methodologies enhance catalyst morphology understanding, leading to precise breakdowns of reaction mechanisms. This precision contributes to more specific and selective product formation compared to current approaches. For instance, in a recent 2023 report, Herring et al. used real-time time-dependent DFT (RT-TD-DFT) coupled with Ehrenfest dynamics to demonstrate that both charge transfer and near-field enhancement drive N2 dissociation on Au or Ag NPs182.
Post-reaction identification and quantification of ammonia are crucial to assess photocatalyst efficiency and specificity. Spectrometric methods such as indophenol blue and Nessler’s reagent (K2HgI4) are commonly used for NH3 quantification. However, due to Hg toxicity in Nessler’s reagent, faster photodegradation of indophenol dye, and ion chromatographic methods have become more reliable for real-time monitoring. UC Berkeley chemists recently designed a new variety of MOF comprising Cu (linked via cyclohexane dicarboxylate) to selectively bind and release ammonia without compromising structural integrity, offering novel pathways for precise ammonia separation.
Overall, we can affirm that plasmonic catalysis holds promise for nitrogen fixation. However, its transition from laboratory to industrial-scale applications demands careful consideration of key factors. Scalability relies on effectively addressing challenges related to cost, catalyst stability, and reactor design. The inherent cost of noble metals in plasmonic catalysts presents an economic obstacle, necessitating strategies such as exploring alternative materials or optimizing catalyst composition.
Ensuring catalyst stability is pivotal for sustained performance, especially in the demanding conditions of industrial processes. Exploring strategies such as protective coatings, novel catalyst formulations, or recycling methods becomes crucial to enhance stability and extend the lifespan of plasmonic catalysts.
Optimizing reactor design plays a central role in achieving efficiency and scalability. Essential adjustments in configuration include uniform light distribution, efficient mass transfer, and scalability. Moreover, integrating considerations for potential by-products and waste management into reactor design is imperative for environmental sustainability.
To replicate the Haber-Bosch process using light, a photocatalyst must possess several key characteristics: broadband absorption, the ability to generate highly energetic charge carriers, extended excited state lifetime, numerous active sites (as unsupported nanoparticles agglomerate during the reaction, leading to catalyst degradation), increased selectivity, efficiency, and stability over multiple catalytic cycles. In Fig. 24, we present a scheme that considers these desired attributes and the latest developments in the field of plasmon-assisted NRR, while also highlighting the limitations hindering further progress toward the creation of potent photocatalysts.

The heart of this matter appears to be centered on the judicious selection of materials and a deeper understanding of the underlying fundamental aspects of plasmonic chemistry. Identifying structure-function relationships in plasmonic catalysis is crucial for activity optimization and developing a design-driven approach for targeted activity with high selectivity. Currently, the ammonia production rate, ranging from micrograms to milligrams per hour per unit area, presents the primary challenge for commercialization. However, with advancements in mechanistic understanding, structure-function relationships, and synthetic chemistry, we anticipate future plasmonic catalyst designs to be more promising than ever.
References
Hu, C. et al. Surface plasmon enabling nitrogen fixation in pure water through a dissociative mechanism under mild conditions. J. Am. Chem. Soc. 141, 7807–7814 (2019).
Wang, S., Ichihara, F., Pang, H., Chen, H. & Ye, J. Nitrogen fixation reaction derived from nanostructured catalytic materials. Adv. Funct. Mater. 28, 1803309 (2018).
Chen, J. G. et al. Beyond fossil fuel–driven nitrogen transformations. Science 360, eaar6611 (2018).
Erisman, J. W., Sutton, M. A., Galloway, J., Klimont, Z. & Winiwarter, W. How a century of ammonia synthesis changed the world. Nat. Geosci. 1, 636–639 (2008).
Qing, G. et al. Recent advances and challenges of electrocatalytic N2 reduction to ammonia. Chem. Rev. 120, 5437–5516 (2020).
Guo, X., Du, H., Qu, F. & Li, J. Recent progress in electrocatalytic nitrogen reduction. J. Mater. Chem. A 7, 3531–3543 (2019).
Tanabe, Y. & Nishibayashi, Y. Developing more sustainable processes for ammonia synthesis. Coord. Chem. Rev. 257, 2551–2564 (2013).
Hoffman, B. M., Lukoyanov, D., Yang, Z.-Y., Dean, D. R. & Seefeldt, L. C. Mechanism of nitrogen fixation by nitrogenase: the next stage. Chem. Rev. 114, 4041–4062 (2014).
Dixon, R. & Kahn, D. Genetic regulation of biological nitrogen fixation. Nat. Rev. Microbiol. 2, 621–631 (2004).
Brown, K. A. et al. Light-driven dinitrogen reduction catalyzed by a CdS:nitrogenase MoFe protein biohybrid. Science 352, 448–450 (2016).
Wang, L. et al. Greening ammonia toward the solar ammonia refinery. Joule 2, 1055–1074 (2018).
Seefeldt, L. C., Hoffman, B. M. & Dean, D. R. Mechanism of Mo-dependent nitrogenase. Annu. Rev. Biochem. 78, 701–722 (2009).
Watanabe, K., Menzel, D., Nilius, N. & Freund, H.-J. Photochemistry on metal nanoparticles. Chem. Rev. 106, 4301–4320 (2006).
Hartland, G. V., Besteiro, L. V., Johns, P. & Govorov, A. O. What’s so hot about electrons in metal nanoparticles? ACS Energy Lett. 2, 1641–1653 (2017).
Yuan, Y. et al. Earth-abundant photocatalyst for H2 generation from NH3 with light-emitting diode illumination. Science 378, 889–893 (2022).
Khurgin, J. B. Hot carriers generated by plasmons: where are they generated and where do they go from there? Faraday Discuss. 214, 35–58 (2019).
Brown, A. M., Sundararaman, R., Narang, P., Goddard, W. A. & Atwater, H. A. Nonradiative plasmon decay and hot carrier dynamics: effects of phonons, surfaces, and geometry. ACS Nano 10, 957–966 (2016).
Zhang, X., Chen, Y. L., Liu, R.-S. & Tsai, D. P. Plasmonic photocatalysis. Rep. Prog. Phys. 76, 046401 (2013).
Narang, P., Sundararaman, R. & Atwater, H. A. Plasmonic hot carrier dynamics in solid-state and chemical systems for energy conversion. Nanophotonics 5, 96–111 (2016).
Rao, V. G., Aslam, U. & Linic, S. Chemical requirement for extracting energetic charge carriers from plasmonic metal nanoparticles to perform electron-transfer reactions. J. Am. Chem. Soc. 141, 643–647 (2019).
Boerigter, C., Aslam, U. & Linic, S. Mechanism of charge transfer from plasmonic nanostructures to chemically attached materials. ACS Nano 10, 6108–6115 (2016).
Ahlawat, M., Mittal, D. & Govind Rao, V. Plasmon-induced hot-hole generation and extraction at nano-heterointerfaces for photocatalysis. Commun. Mater. 2, 1–15 (2021).
Boerigter, C., Campana, R., Morabito, M. & Linic, S. Evidence and implications of direct charge excitation as the dominant mechanism in plasmon-mediated photocatalysis. Nat. Commun. 7, 10545 (2016).
Wang, M., Ye, M., Iocozzia, J., Lin, C. & Lin, Z. Plasmon‐mediated solar energy conversion via photocatalysis in noble metal/semiconductor composites. Adv. Sci. 3, 1600024 (2016).
Zhao, W. et al. Single Mo1(Cr1) atom on nitrogen-doped graphene enables highly selective electroreduction of nitrogen into ammonia. ACS Catal. 9, 3419–3425 (2019).
Martirez, J. M. P. & Carter, E. A. Prediction of a low-temperature N2 dissociation catalyst exploiting near-IR–to–visible light nanoplasmonics. Sci. Adv. 3, eaao4710 (2017).
Monyoncho, E. A. & Dasog, M. Photocatalytic plasmon‐enhanced nitrogen reduction to ammonia. Adv. Energy Sustain. Res. 2, 2000055 (2021).
Yang, J., Guo, Y., Lu, W., Jiang, R. & Wang, J. Emerging applications of plasmons in driving CO2 reduction and N2 fixation. Adv. Mater. 30, 1802227 (2018).
Puértolas, B., Comesaña‐Hermo, M., Besteiro, L. V., Vázquez‐González, M. & Correa‐Duarte, M. A. Challenges and opportunities for renewable ammonia production via plasmon‐assisted photocatalysis. Adv. Energy Mater. 12, 2103909 (2022).
Oshikiri, T. & Misawa, H. Plasmonic catalysis for N2 fixation. Plasmon. Catal. Fundam. Appl. 6, 165–189 (2021).
Hao, E. & Schatz, G. C. Electromagnetic fields around silver nanoparticles and dimers. J. Chem. Phys. 120, 357–366 (2004).
Halas, N. J., Lal, S., Chang, W.-S., Link, S. & Nordlander, P. Plasmons in strongly coupled metallic nanostructures. Chem. Rev. 111, 3913–3961 (2011).
Chavez, S., Rao, V. G. & Linic, S. Unearthing the factors governing site specific rates of electronic excitations in multicomponent plasmonic systems and catalysts. Faraday Discuss. 214, 441–453 (2019).
Linic, S., Christopher, P. & Ingram, D. B. Plasmonic-metal nanostructures for efficient conversion of solar to chemical energy. Nat. Mater. 10, 911–921 (2011).
Kim, Y., Smith, J. G. & Jain, P. K. Harvesting multiple electron–hole pairs generated through plasmonic excitation of Au nanoparticles. Nat. Chem. 10, 763–769 (2018).
Hartland, G. V. Optical studies of dynamics in noble metal nanostructures. Chem. Rev. 111, 3858–3887 (2011).
Khurgin, J. B. Fundamental limits of hot carrier injection from metal in nanoplasmonics. Nanophotonics 9, 453–471 (2020).
Linic, S., Chavez, S. & Elias, R. Flow and extraction of energy and charge carriers in hybrid plasmonic nanostructures. Nat. Mater. 20, 916–924 (2021).
Link, S. & El-Sayed, M. A. Spectral properties and relaxation dynamics of surface plasmon electronic oscillations in gold and silver nanodots and nanorods. J. Phys. Chem. B 103, 8410–8426 (1999).
Kreibig, U. & Vollmer, M. Optical Properties of Metal Clusters (Springer-Verlag, 1995).
Brongersma, M. L., Halas, N. J. & Nordlander, P. Plasmon-induced hot carrier science and technology. Nat. Nanotechnol. 10, 25–34 (2015).
Blanco-Rey, M., Alducin, M., Juaristi, J. I. & De Andres, P. L. Diffusion of hydrogen in Pd assisted by inelastic ballistic hot electrons. Phys. Rev. Lett. 108, 115902 (2012).
Bernardi, M., Mustafa, J., Neaton, J. B. & Louie, S. G. Theory and computation of hot carriers generated by surface plasmon polaritons in noble metals. Nat. Commun. 6, 7044 (2015).
Liu, G., Zhen, C., Kang, Y., Wang, L. & Cheng, H.-M. Unique physicochemical properties of two-dimensional light absorbers facilitating photocatalysis. Chem. Soc. Rev. 47, 6410–6444 (2018).
Jacoboni, C. Theory of Electron Transport in Semiconductors: A Pathway from Elementary Physics to Nonequilibrium Green Functions (Springer-Verlag, 2010).
Xia, Y., Xiong, Y., Lim, B. & Skrabalak, S. E. Shape‐controlled synthesis of metal nanocrystals: simple chemistry meets complex physics? Angew. Chem. Int. Ed. 48, 60–103 (2009).
Mourdikoudis, S., Pallares, R. M. & Thanh, N. T. Characterization techniques for nanoparticles: comparison and complementarity upon studying nanoparticle properties. Nanoscale 10, 12871–12934 (2018).
Baig, N., Kammakakam, I. & Falath, W. Nanomaterials: a review of synthesis methods, properties, recent progress, and challenges. Mater. Adv. 2, 1821–1871 (2021).
Chen, L.-W. et al. Metal–organic framework membranes encapsulating gold nanoparticles for direct plasmonic photocatalytic nitrogen fixation. J. Am. Chem. Soc. 143, 5727–5736 (2021).
Nazemi, M. & El-Sayed, M. A. Plasmon-enhanced photo(electro)chemical nitrogen fixation under ambient conditions using visible light responsive hybrid hollow Au-Ag2O nanocages. Nano Energy 63, 103886 (2019).
Yang, J. et al. High-efficiency “working-in-tandem” nitrogen photofixation achieved by assembling plasmonic gold nanocrystals on ultrathin titania nanosheets. J. Am. Chem. Soc. 140, 8497–8508 (2018).
Li, Y., Chen, J., Cai, P. & Wen, Z. An electrochemically neutralized energy-assisted low-cost acid-alkaline electrolyzer for energy-saving electrolysis hydrogen generation. J. Mater. Chem. A 6, 4948–4954 (2018).
Mao, C. et al. Beyond the thermal equilibrium limit of ammonia synthesis with dual temperature zone catalyst powered by solar light. Chem 5, 2702–2717 (2019).
Chavez, S., Aslam, U. & Linic, S. Design principles for directing energy and energetic charge flow in multicomponent plasmonic nanostructures. ACS Energy Lett. 3, 1590–1596 (2018).
Chowdhury, I. H., Gupta, S. & Rao, V. G. Covalent organic framework: an emerging catalyst for renewable ammonia production. ChemCatChem 15, e202300243 (2023).
Ma, X.-L., Liu, J.-C., Xiao, H. & Li, J. Surface single-cluster catalyst for N2-to-NH3 thermal conversion. J. Am. Chem. Soc. 140, 46–49 (2018).
Jia, H.-P. & Quadrelli, E. A. Mechanistic aspects of dinitrogen cleavage and hydrogenation to produce ammonia in catalysis and organometallic chemistry: relevance of metal hydride bonds and dihydrogen. Chem. Soc. Rev. 43, 547–564 (2014).
Biehl, H. & Stuhl, F. Vacuum-ultraviolet photolysis of N2H2: Generation of NH fragments. J. Chem. Phys. 100, 141–145 (1994).
Comer, B. M. & Medford, A. J. Analysis of photocatalytic nitrogen fixation on rutile TiO2 (110). ACS Sustain. Chem. Eng. 6, 4648–4660 (2018).
Shilov, A. E. Catalytic reduction of molecular nitrogen in solutions. Russ. Chem. Bull. 52, 2555–2562 (2003).
Shipman, M. A. & Symes, M. D. Recent progress towards the electrosynthesis of ammonia from sustainable resources. Catal. Today 286, 57–68 (2017).
Mubeen, S. et al. An autonomous photosynthetic device in which all charge carriers derive from surface plasmons. Nat. Nanotechnol. 8, 247–251 (2013).
Sayed, M., Yu, J., Liu, G. & Jaroniec, M. Non-noble plasmonic metal-based photocatalysts. Chem. Rev. 122, 10484–10537 (2022).
Kok, S. H. W., Lee, J., Tan, L.-L., Ong, W.-J. & Chai, S.-P. MXene─a new paradigm toward artificial nitrogen fixation for sustainable ammonia generation: synthesis, properties, and future outlook. ACS Mater. Lett. 4, 212–245 (2022).
El-Demellawi, J. K., Lopatin, S., Yin, J., Mohammed, O. F. & Alshareef, H. N. Tunable multipolar surface plasmons in 2D Ti3C2Tx MXene flakes. ACS Nano 12, 8485–8493 (2018).
Nerl, H. C. et al. Probing the local nature of excitons and plasmons in few-layer MoS2. Npj 2D Mater. Appl. 1, 2 (2017).
Butun, S., Tongay, S. & Aydin, K. Enhanced light emission from large-area monolayer MoS2 using plasmonic nanodisc arrays. Nano Lett. 15, 2700–2704 (2015).
Zhu, Z. et al. Near‐infrared plasmonic 2D semimetals for applications in communication and biology. Adv. Funct. Mater. 26, 1793–1802 (2016).
Low, T. & Avouris, P. Graphene plasmonics for terahertz to mid-infrared applications. ACS Nano 8, 1086–1101 (2014).
Wachsmuth, P., Hambach, R., Benner, G. & Kaiser, U. Plasmon bands in multilayer graphene. Phys. Rev. B 90, 235434 (2014).
Grigorenko, A. N., Polini, M. & Novoselov, K. S. Graphene plasmonics. Nat. Photonics 6, 749–758 (2012).
Huber, M. A. et al. Femtosecond photo-switching of interface polaritons in black phosphorus heterostructures. Nat. Nanotechnol. 12, 207–211 (2017).
Pan, C. T. et al. Nanoscale electron diffraction and plasmon spectroscopy of single- and few-layer boron nitride. Phys. Rev. B 85, 045440 (2012).
Jauffred, L., Samadi, A., Klingberg, H., Bendix, P. M. & Oddershede, L. B. Plasmonic heating of nanostructures. Chem. Rev. 119, 8087–8130 (2019).
Linic, S., Christopher, P., Xin, H. & Marimuthu, A. Catalytic and photocatalytic transformations on metal nanoparticles with targeted geometric and plasmonic properties. Acc. Chem. Res. 46, 1890–1899 (2013).
Aslam, U., Rao, V. G., Chavez, S. & Linic, S. Catalytic conversion of solar to chemical energy on plasmonic metal nanostructures. Nat. Catal. 1, 656–665 (2018).
Guo, W. et al. Cooperation of hot holes and surface adsorbates in plasmon-driven anisotropic growth of gold nanostars. J. Am. Chem. Soc. 142, 10921–10925 (2020).
Swaminathan, S., Rao, V. G., Bera, J. K. & Chandra, M. The pivotal role of hot carriers in plasmonic catalysis of C−N bond forming reaction of amines. Angew. Chem. Int. Ed. 60, 12532–12538 (2021).
Christopher, P., Xin, H. & Linic, S. Visible-light-enhanced catalytic oxidation reactions on plasmonic silver nanostructures. Nat. Chem. 3, 467–472 (2011).
Thangamuthu, M., Santschi, C. & Martin, O. J. Photocatalytic ammonia production enhanced by a plasmonic near-field and hot electrons originating from aluminium nanostructures. Faraday Discuss. 214, 399–415 (2019).
Long, R., Li, Y., Song, L. & Xiong, Y. Coupling solar energy into reactions: materials design for surface plasmon‐mediated catalysis. Small 11, 3873–3889 (2015).
Shirhatti, P. R. et al. Observation of the adsorption and desorption of vibrationally excited molecules on a metal surface. Nat. Chem. 10, 592–598 (2018).
Mittal, D., Ahlawat, M. & Govind Rao, V. Recent progress and challenges in plasmon-mediated reduction of CO2 to chemicals and fuels. Adv. Mater. Interfaces 9, 2102383 (2022).
Mascaretti, L. & Naldoni, A. Hot electron and thermal effects in plasmonic photocatalysis. J. Appl. Phys. 128, 041101 (2020).
Wu, H. et al. Plasmon-driven N2 photofixation in pure water over MoO3-x nanosheets under visible to NIR excitation. J. Mater. Chem. A 8, 2827–2835 (2020).
Li, S. et al. Recent advances in plasmonic nanostructures for enhanced photocatalysis and electrocatalysis. Adv. Mater. 33, 2000086 (2021).
Hou, T. et al. Porous CuFe for plasmon-assisted N2 photofixation. ACS Energy Lett. 5, 2444–2451 (2020).
Khurgin, J. B. How to deal with the loss in plasmonics and metamaterials. Nat. Nanotechnol. 10, 2–6 (2015).
Cortés, E. et al. Challenges in plasmonic catalysis. ACS Nano 14, 16202–16219 (2020).
Clavero, C. Plasmon-induced hot-electron generation at nanoparticle/metal-oxide interfaces for photovoltaic and photocatalytic devices. Nat. Photonics 8, 95–103 (2014).
Kim, Y., Dumett Torres, D. & Jain, P. K. Activation energies of plasmonic catalysts. Nano Lett. 16, 3399–3407 (2016).
Furube, A., Du, L., Hara, K., Katoh, R. & Tachiya, M. Ultrafast plasmon-induced electron transfer from gold nanodots into TiO2 nanoparticles. J. Am. Chem. Soc. 129, 14852–14853 (2007).
Du, L., Furube, A., Hara, K., Katoh, R. & Tachiya, M. Ultrafast plasmon induced electron injection mechanism in gold–TiO2 nanoparticle system. J. Photochem. Photobiol. C. Photochem. Rev. 15, 21–30 (2013).
Sheldon, M. T., Van De Groep, J., Brown, A. M., Polman, A. & Atwater, H. A. Plasmoelectric potentials in metal nanostructures. Science 346, 828–831 (2014).
Zhang, Y. et al. Surface-plasmon-driven hot electron photochemistry. Chem. Rev. 118, 2927–2954 (2018).
Khan, M. R., Chuan, T. W., Yousuf, A., Chowdhury, M. N. K. & Cheng, C. K. Schottky barrier and surface plasmonic resonance phenomena towards the photocatalytic reaction: study of their mechanisms to enhance photocatalytic activity. Catal. Sci. Technol. 5, 2522–2531 (2015).
Stefancu, A. et al. Fermi level equilibration at the metal–molecule interface in plasmonic systems. Nano Lett. 21, 6592–6599 (2021).
Greiner, M. T. et al. Universal energy-level alignment of molecules on metal oxides. Nat. Mater. 11, 76–81 (2012).
Zhang, C., Jia, F., Li, Z., Huang, X. & Lu, G. Plasmon-generated hot holes for chemical reactions. Nano Res. 13, 3183–3197 (2020).
Dong, B., Fang, Y., Chen, X., Xu, H. & Sun, M. Substrate-, wavelength-, and time-dependent plasmon-assisted surface catalysis reaction of 4-nitrobenzenethiol dimerizing to p,p ′-dimercaptoazobenzene on Au, Ag, and Cu films. Langmuir 27, 10677–10682 (2011).
Singh, M. R., Xiang, C. & Lewis, N. S. Evaluation of flow schemes for near-neutral pH electrolytes in solar-fuel generators. Sustain. Energy Fuels 1, 458–466 (2017).
Holec, D., Dumitraschkewitz, P., Vollath, D. & Fischer, F. D. Surface energy of Au nanoparticles depending on their size and shape. Nanomaterials 10, 484 (2020).
Zhao, Y. et al. Layered‐double‐hydroxide nanosheets as efficient visible‐light‐driven photocatalysts for dinitrogen fixation. Adv. Mater. 29, 1703828 (2017).
Yu, C. et al. Novel hollow Pt-ZnO nanocomposite microspheres with hierarchical structure and enhanced photocatalytic activity and stability. Nanoscale 5, 2142–2151 (2013).
Knight, M. W. et al. Embedding plasmonic nanostructure diodes enhances hot electron emission. Nano Lett. 13, 1687–1692 (2013).
Jo, Y. K., Lee, J. M., Son, S. & Hwang, S.-J. 2D inorganic nanosheet-based hybrid photocatalysts: design, applications, and perspectives. J. Photochem. Photobiol. C. Photochem. Rev. 40, 150–190 (2019).
Quidant, G. B. R. Nanoplasmonics for chemistry. Chem. Soc. Rev. 43, 3898–3907 (2014).
Cushing, S. K. et al. Photocatalytic activity enhanced by plasmonic resonant energy transfer from metal to semiconductor. J. Am. Chem. Soc. 134, 15033–15041 (2012).
Zhou, L. et al. Quantifying hot carrier and thermal contributions in plasmonic photocatalysis. Science 362, 69–72 (2018).
Oshikiri, T., Ueno, K. & Misawa, H. Selective dinitrogen conversion to ammonia using water and visible light through plasmon‐induced charge separation. Angew. Chem. Int. Ed. 55, 3942–3946 (2016).
Jia, H. et al. Site-selective growth of crystalline ceria with oxygen vacancies on gold nanocrystals for near-infrared nitrogen photofixation. J. Am. Chem. Soc. 141, 5083–5086 (2019).
Schneider, J. et al. Understanding TiO2 photocatalysis: mechanisms and materials. Chem. Rev. 114, 9919–9986 (2014).
Li, B. et al. (Gold core)@(ceria shell) nanostructures for plasmon-enhanced catalytic reactions under visible light. ACS Nano 8, 8152–8162 (2014).
Schrauzer, G. N. Photoreduction of nitrogen on TiO2 and TiO2-containing minerals. In Energy Efficiency and Renewable Energy Through Nanotechnology (Springer-Verlag, 2011).
Medford, A. J. & Hatzell, M. C. Photon-driven nitrogen fixation: current progress, thermodynamic considerations, and future outlook. ACS Catal. 7, 2624–2643 (2017).
Li, H., Shang, J., Ai, Z. & Zhang, L. Efficient visible light nitrogen fixation with BiOBr nanosheets of oxygen vacancies on the exposed {001} facets. J. Am. Chem. Soc. 137, 6393–6399 (2015).
Xiao, C., Hu, H., Zhang, X. & MacFarlane, D. R. Nanostructured gold/bismutite hybrid heterocatalysts for plasmon-enhanced photosynthesis of ammonia. ACS Sustain. Chem. Eng. 5, 10858–10863 (2017).
Zhang, X. et al. Controlled morphogenesis and self-assembly of bismutite nanocrystals into three-dimensional nanostructures and their applications. J. Mater. Chem. A 2, 2275–2282 (2014).
Peng, S. et al. Monodispersed Ag nanoparticles loaded on the PVP-assisted synthetic Bi2O2CO3 microspheres with enhanced photocatalytic and supercapacitive performances. J. Mater. Chem. A 1, 7630–7638 (2013).
Cen, W., Xiong, T., Tang, C., Yuan, S. & Dong, F. Effects of morphology and crystallinity on the photocatalytic activity of (BiO)2CO3 nano/microstructures. Ind. Eng. Chem. Res. 53, 15002–15011 (2014).
Christopher, P., Xin, H., Marimuthu, A. & Linic, S. Singular characteristics and unique chemical bond activation mechanisms of photocatalytic reactions on plasmonic nanostructures. Nat. Mater. 11, 1044–1050 (2012).
Xing, P. et al. New application and excellent performance of Ag/KNbO3 nanocomposite in photocatalytic NH3 synthesis. ACS Sustain. Chem. Eng. 7, 12408–12418 (2019).
Jiang, W. et al. Free‐standing nanoarrays with energetic electrons and active sites for efficient plasmon‐driven ammonia synthesis. Small 18, 2201269 (2022).
Jia, H. et al. Steric hindrance-induced selective growth of rhodium on gold nanobipyramids for plasmon-enhanced nitrogen fixation. Chem. Sci. 14, 5656–5664 (2023).
Aslam, U., Chavez, S. & Linic, S. Controlling energy flow in multimetallic nanostructures for plasmonic catalysis. Nat. Nanotechnol. 12, 1000–1005 (2017).
Huang, J. et al. High-efficiency and stable photocatalytic hydrogen evolution of rhenium sulfide co-catalyst on Zn0.3Cd0.7S. Mater. Adv. 1, 363–370 (2020).
Ali, M. et al. Nanostructured photoelectrochemical solar cell for nitrogen reduction using plasmon-enhanced black silicon. Nat. Commun. 7, 11335 (2016).
Chang, B. et al. Plasmon-enabled N2 photofixation on partially reduced Ti3C2 MXene. Chem. Sci. 12, 11213–11224 (2021).
Pham, H. H. & Wang, L.-W. Oxygen vacancy and hole conduction in amorphous TiO2. Phys. Chem. Chem. Phys. 17, 541–550 (2015).
Li, H. et al. New reaction pathway induced by plasmon for selective benzyl alcohol oxidation on BiOCl possessing oxygen vacancies. J. Am. Chem. Soc. 139, 3513–3521 (2017).
Li, H., Li, J., Ai, Z., Jia, F. & Zhang, L. Oxygen vacancy‐mediated photocatalysis of BiOCl: reactivity, selectivity, and perspectives. Angew. Chem. Int. Ed. 57, 122–138 (2018).
Li, H., Shang, J., Shi, J., Zhao, K. & Zhang, L. Facet-dependent solar ammonia synthesis of BiOCl nanosheets via a proton-assisted electron transfer pathway. Nanoscale 8, 1986–1993 (2016).
Gao, X., Shang, Y., Gao, K. & Fu, F. Plasmon sensitized heterojunction 2D ultrathin Ag/AgI-δ-Bi2O3 for enhanced photocatalytic nitrogen fixation. Nanomaterials 9, 781 (2019).
Yuan, J., Yi, X., Tang, Y., Liu, M. & Liu, C. Efficient photocatalytic nitrogen fixation: enhanced polarization, activation, and cleavage by asymmetrical electron donation to N≡N bond. Adv. Funct. Mater. 30, 1906983 (2020).
Wu, S. et al. Chemisorption‐induced and plasmon‐promoted photofixation of nitrogen on gold‐loaded carbon nitride nanosheets. ChemSusChem 13, 3455–3461 (2020).
Guo, Y. et al. Au nanoparticle-embedded, nitrogen-deficient hollow mesoporous carbon nitride spheres for nitrogen photofixation. J. Mater. Chem. A 8, 16218–16231 (2020).
Zheng, G., Pastoriza‐Santos, I., Pérez‐Juste, J. & Liz‐Marzán, L. M. Plasmonic metal‐organic frameworks. SmartMat 2, 446–465 (2021).
Wang, M., Tang, Y. & Jin, Y. Modulating catalytic performance of metal–organic framework composites by localized surface plasmon resonance. ACS Catal. 9, 11502–11514 (2019).
Choi, K. M. et al. Plasmon-enhanced photocatalytic CO2 conversion within metal–organic frameworks under visible light. J. Am. Chem. Soc. 139, 356–362 (2017).
Han, X., Yang, S. & Schröder, M. Metal–organic framework materials for production and distribution of ammonia. J. Am. Chem. Soc. 145, 1998–2012 (2023).
Koh, C. S. L. et al. Plasmonic nanoparticle-metal–organic framework (NP–MOF) nanohybrid platforms for emerging plasmonic applications. ACS Mater. Lett. 3, 557–573 (2021).
Zheng, G. et al. Encapsulation of single plasmonic nanoparticles within ZIF‐8 and SERS analysis of the MOF flexibility. Small 12, 3935–3943 (2016).
Yang, Y. et al. Nanoporous gold embedded ZIF composite for enhanced electrochemical nitrogen fixation. Angew. Chem. Int. Ed. 58, 15362–15366 (2019).
Wu, H. et al. Unusual and highly tunable missing-linker defects in zirconium metal–organic framework UiO-66 and their important effects on gas adsorption. J. Am. Chem. Soc. 135, 10525–10532 (2013).
Winarta, J. et al. A decade of UiO-66 research: a historic review of dynamic structure, synthesis mechanisms, and characterization techniques of an archetypal metal–organic framework. Cryst. Growth Des. 20, 1347–1362 (2020).
Boong, S. K. et al. Superlattice‐based plasmonic catalysis: concentrating light at the nanoscale to drive efficient nitrogen‐to‐ammonia fixation at ambient conditions. Angew. Chem. Int. Ed. 62, e202216562 (2023).
Zhang, W. et al. Progress and perspective of electrocatalytic CO2 reduction for renewable carbonaceous fuels and chemicals. Adv. Sci. 5, 1700275 (2018).
Ingram, D. B. & Linic, S. Water splitting on composite plasmonic-metal/semiconductor photoelectrodes: evidence for selective plasmon-induced formation of charge carriers near the semiconductor surface. J. Am. Chem. Soc. 133, 5202–5205 (2011).
Contreras, E. et al. Plasmon-assisted ammonia electrosynthesis. J. Am. Chem. Soc. 144, 10743–10751 (2022).
Talukdar, B. et al. Enhancement of NH3 production in electrochemical N2 reduction by the Cu-rich inner surfaces of beveled CuAu nanoboxes. ACS Appl. Mater. Interfaces 13, 51839–51848 (2021).
Chen, H., Shao, L., Li, Q. & Wang, J. Gold nanorods and their plasmonic properties. Chem. Soc. Rev. 42, 2679–2724 (2013).
Nazemi, M., Panikkanvalappil, S. R. & El-Sayed, M. A. Enhancing the rate of electrochemical nitrogen reduction reaction for ammonia synthesis under ambient conditions using hollow gold nanocages. Nano Energy 49, 316–323 (2018).
Nazemi, M. & El-Sayed, M. A. Electrochemical synthesis of ammonia from N2 and H2O under ambient conditions using pore-size-controlled hollow gold nanocatalysts with tunable plasmonic properties. J. Phys. Chem. Lett. 9, 5160–5166 (2018).
Liang, W. et al. Localized surface plasmon resonance enhanced electrochemical nitrogen reduction reaction. Appl. Catal. B Environ. 301, 120808 (2022).
You, H., Yang, S., Ding, B. & Yang, H. Synthesis of colloidal metal and metal alloy nanoparticles for electrochemical energy applications. Chem. Soc. Rev. 42, 2880–2904 (2013).
Vu, M.-H., Nguyen, C.-C. & Do, T.-O. Synergistic effect of Fe doping and plasmonic Au nanoparticles on W18O49 nanorods for enhancing photoelectrochemical nitrogen reduction. ACS Sustain. Chem. Eng. 8, 12321–12330 (2020).
Oshikiri, T., Ueno, K. & Misawa, H. Plasmon‐induced ammonia synthesis through nitrogen photofixation with visible light irradiation. Angew. Chem. Int. Ed. 53, 9802–9805 (2014).
Oshikiri, T., Ueno, K. & Misawa, H. Ammonia photosynthesis via an association pathway using a plasmonic photoanode and a zirconium cathode. Green Chem. 21, 4443–4448 (2019).
Bousiakou, L. G., Gebavi, H., Mikac, L., Karapetis, S. & Ivanda, M. Surface enhanced Raman spectroscopy for molecular identification-A review on surface plasmon resonance (SPR) and localised surface plasmon resonance (LSPR) in optical nanobiosensing. Croat. Chem. Acta 92, 479–494 (2019).
Zhou, S. et al. Formation of an oriented Bi2WO6 photocatalyst induced by in situ Bi reduction and its use for efficient nitrogen fixation. Catal. Sci. Technol. 9, 5562–5566 (2019).
Wang, J., Hua, C., Dong, X., Wang, Y. & Zheng, N. Synthesis of plasmonic bismuth metal deposited InVO4 nanosheets for enhancing solar light-driven photocatalytic nitrogen fixation. Sustain. Energy Fuels 4, 1855–1862 (2020).
Chen, L. et al. Recent advances in bismuth-containing photocatalysts with heterojunctions. Chin. J. Catal. 37, 780–791 (2016).
Huang, Y., Zhang, N., Wu, Z. & Xie, X. Artificial nitrogen fixation over bismuth-based photocatalysts: fundamentals and future perspectives. J. Mater. Chem. A 8, 4978–4995 (2020).
Dong, F., Li, Q., Sun, Y. & Ho, W.-K. Noble metal-like behavior of plasmonic Bi particles as a cocatalyst deposited on (BiO)2CO3 microspheres for efficient visible light photocatalysis. ACS Catal. 4, 4341–4350 (2014).
Yuan, C., Liang, S., Lu, Z., Hao, W. & Teng, F. Bismuth semimetal-boosted electron transfer, nitrogen adsorption, and nitrogen photoreduction activity of Bi/g-C3N4 nanosheets. Energy Fuels 35, 16232–16240 (2021).
Zhou, L. et al. 3D self-assembly of aluminium nanoparticles for plasmon-enhanced solar desalination. Nat. Photonics 10, 393–398 (2016).
Gérard, D. & Gray, S. K. Aluminium plasmonics. J. Phys. Appl. Phys. 48, 184001 (2014).
Hafeez, H. Y., Lakhera, S. K., Shankar, M. V. & Neppolian, B. Synergetic improvement in charge carrier transport and light harvesting over ternary InVO4-g-C3N4/rGO hybrid nanocomposite for hydrogen evolution reaction. Int. J. Hydrog. Energy 45, 7530–7540 (2020).
Bai, H. et al. A Schottky‐barrier‐free plasmonic semiconductor photocatalyst for nitrogen fixation in a “one‐stone‐two‐birds” manner. Adv. Mater. 34, 2104226 (2022).
Li, Q. et al. Tunable and stable localized surface plasmon resonance in SrMoO4 for enhanced visible light driven nitrogen reduction. Chin. J. Catal. 42, 1763–1771 (2021).
Ranjit, K. T., Varadarajan, T. K. & Viswanathan, B. Photocatalytic reduction of nitrite and nitrate ions to ammonia on Ru/TiO2 catalysts. J. Photochem. Photobiol. Chem. 89, 67–68 (1995).
Du, L. et al. Plasmon-induced charge separation and recombination dynamics in gold−TiO2 nanoparticle systems: dependence on TiO2 particle size. J. Phys. Chem. C. 113, 6454–6462 (2009).
Kowalska, E., Mahaney, O. O. P., Abe, R. & Ohtani, B. Visible-light-induced photocatalysis through surface plasmon excitation of gold on titania surfaces. Phys. Chem. Chem. Phys. 12, 2344–2355 (2010).
Li, C. et al. Promoted fixation of molecular nitrogen with surface oxygen vacancies on plasmon‐enhanced TiO2 photoelectrodes. Angew. Chem. Int. Ed. 57, 5278–5282 (2018).
Mao, C., Yu, L., Li, J., Zhao, J. & Zhang, L. Energy-confined solar thermal ammonia synthesis with K/Ru/TiO2-xHx. Appl. Catal. B Environ. 224, 612–620 (2018).
Feng, C. et al. Fabricating Ag/PW12/Zr‐mTiO2 composite via doping and interface engineering: an efficient catalyst with bifunctionality in photo‐ and electro‐driven nitrogen reduction reactions. Adv. Sustain. Syst. 6, 2100307 (2022).
Feng, C. et al. Amorphization and defect engineering in constructing ternary composite Ag/PW10V2/am-TiO2- x for enhanced photocatalytic nitrogen fixation. N. J. Chem. 46, 1731–1740 (2022).
Mauchamp, V. et al. Enhanced and tunable surface plasmons in two-dimensional Ti3C2 stacks: Electronic structure versus boundary effects. Phys. Rev. B 89, 235428 (2014).
Hou, T. et al. Near-infrared light-driven photofixation of nitrogen over Ti3C2Tx/TiO2 hybrid structures with superior activity and stability. Appl. Catal. B Environ. 273, 119072 (2020).
Verma, R. et al. Nickel-laden dendritic plasmonic colloidosomes of black gold: forced plasmon mediated photocatalytic CO2 hydrogenation. ACS Nano 17, 4526–4538 (2023).
Jain, P. K., Lee, K. S., El-Sayed, I. H. & El-Sayed, M. A. Calculated absorption and scattering properties of gold nanoparticles of different size, shape, and composition: applications in biological imaging and biomedicine. J. Phys. Chem. B 110, 7238–7248 (2006).
Herring, C. J. & Montemore, M. M. Mechanistic insights into plasmonic catalysis by dynamic calculations: O2 and N2 on Au and Ag nanoparticles. Chem. Mater. 35, 1586–1593 (2023).
Acknowledgements
V.G.R. acknowledges financial support from the Ministry of Human Resource Development (MHRD), Government of India, under the STARS scheme (Grant No. STARS/APR2019/CS/257/FS), and the Indian Institute of Technology Kanpur for infrastructure and initial lab setup funding. P.A. thanks the Indian Institute of Technology Kanpur for the research fellowship.
Author information
Authors and Affiliations
Contributions
V.G.R. conceived the idea and supervised the project. V.G.R. and A.C. jointly conceptualized the proposal. A.C. and A.H. made equal contributions to writing the first draft of the manuscript. V.G.R. and P.A. revised the manuscript. All authors provided valuable feedback during the revision stage.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests. V.G.R. is an Editorial Board Member for Communications Materials and was not involved in the editorial review, or the decision to publish, this review.
Peer review
Peer review information
Communications Materials thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Jet-Sing Lee. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Choudhary, A., Halder, A., Aggarwal, P. et al. Plasmonic chemistry for sustainable ammonia production. Commun Mater 5, 69 (2024). https://doi.org/10.1038/s43246-024-00510-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-024-00510-7
