
Abstract
The 5′ untranslated region (UTR), a regulatory region at the beginning of a messenger RNA (mRNA) molecule, plays a crucial role in regulating the translation process and affects the protein expression level. Language models have showcased their effectiveness in decoding the functions of protein and genome sequences. Here, we introduce a language model for 5′ UTR, which we refer to as the UTR-LM. The UTR-LM is pretrained on endogenous 5′ UTRs from multiple species and is further augmented with supervised information including secondary structure and minimum free energy. We fine-tuned the UTR-LM in a variety of downstream tasks. The model outperformed the best known benchmark by up to 5% for predicting the mean ribosome loading, and by up to 8% for predicting the translation efficiency and the mRNA expression level. The model was also applied to identifying unannotated internal ribosome entry sites within the untranslated region and improved the area under the precision–recall curve from 0.37 to 0.52 compared to the best baseline. Further, we designed a library of 211 new 5′ UTRs with high predicted values of translation efficiency and evaluated them via a wet-laboratory assay. Experiment results confirmed that our top designs achieved a 32.5% increase in protein production level relative to well-established 5′ UTRs optimized for therapeutics.
Similar content being viewed by others

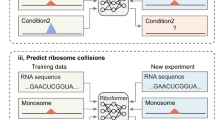

Main
The 5′ untranslated region (5′ UTR) is a region at the beginning of an mRNA that precedes the coding sequence of the protein. It plays a critical role in regulating the translation from mRNA to proteins, as it can influence the stability, localization and translation of the mRNA molecule1. There has been a substantial amount of research2,3,4,5,6,7,8,9 exploring the biology of the 5′ UTR, including its secondary structure (SS)2, RNA-binding proteins that may interact with it3 and the effect of mutations within the 5′ UTR on the gene expression4. The complex functions of mRNA and their potential implications for human health underscore the necessity for more universally applicable computational approaches.
Investigation into the role of 5′ UTRs encompasses various aspects of translational control. With the growing interest in studying and designing 5′ UTRs, various computational tools5,6,7,8,9,10,11 have been developed to study its functions. For example, the ribosome load measures the number of ribosomes engaged in translating a given mRNA at a given time. Supervised machine learning models were shown to predict the mean ribosome loading (MRL)5,6,9,10 on the basis of the UTR sequence or its biological features. Additionally, RNABERT12 and RNA-FM10 are language models specific to RNA sequences and have been shown to be useful for predicting the MRL. Scientists also tried to predict the mRNA translation efficiency (TE)7,9 that quantifies the rate of translation into proteins and the mRNA expression level (EL)7,9 that reflects the relative abundance of the mRNA transcript in the cell. MTtrans9 uses a multi-task model that harnesses diverse data types for translation rate prediction. While there exist specialized models for individual prediction tasks, a unified methodology to decode functions of 5′ UTR is lacking.
In this study, we adopted the principled approach of a language model to extract meaningful semantic representations from UTRs of raw mRNA sequences and further map them to predict functions of interest. Specifically, we developed a semisupervised language model, which we refer to as the UTR-LM, trained using sequences of 5′ UTR from multiple data sources (Fig. 1). The transformer-based model is pretrained to extract representations from the raw sequences via nucleotide masking and reconstruction. It also incorporated supervised information such as the SS and the minimum free energy (MFE). We applied the UTR-LM and fine-tuned it for a variety of downstream tasks, such as predicting the MRL, the mRNA TE and the mRNA EL. Experimental results showed that the UTR-LM accurately predicts these regulatory functions. When compared with existing baselines in each downstream task5,6,7,9,10,12,13, the UTR-LM demonstrated state-of-the-art performances across modalities and test sets. In particular, the UTR-LM outperforms the MTtrans9 by 5% for MRL prediction, and outperforms the Cao et al.7 (Cao-RF) by up to 8% for TE and EL predictions in terms of the Spearman R score. The model generalized well to unseen data, especially human 5′ UTRs with varying lengths. Additionally, we adapted the UTR-LM to identify unannotated internal ribosome entry sites (IRESs)13,14,15,16, which are sequences within mRNAs that enable ribosomes to initiate translation internally, bypassing the traditional cap-dependent mechanism. The UTR-LM outperforms IRESpy13 by 0.15 in terms of the area under the precision–recall (AUPR) score.

a, The input of the proposed pretrained model is the 5′ UTR sequence, which is fed into the transformer layer through a randomly generated 128-dimensional embedding for each nucleotide and a special [CLS] token. The pretraining phase uses a combination of masked nucleotide (MN) prediction, 5′ UTR SS prediction and 5′ UTR MFE prediction. b, Following pretraining, the [CLS] token is used for downstream task-specific training. c, The UTR-LM is fine-tuned for downstream tasks such as predicting MRL, TE, mRNA EL and IRES. d, Designing an in-house library of 5′ UTRs with highly predicted TE and the wet-laboratory experimental validation using mRNA transfection and luciferase assays. Panels a,c and d created with BioRender.com.
Given the vital role of UTRs of mRNA on the translation process, artificially designing the 5′ UTR holds potential to improve the TE and optimize the protein production6. Leveraging this biological principle, we designed an in-house library of 211 new 5′ UTRs with high predicted values of TE. We conducted experiments of mRNA transfection and luciferase assay to evaluate our designs. Wet-laboratory experiments revealed that top 5′ UTRs in our design library achieved up to 32.5% increase in protein production efficiency, when compared to the benchmark NCA-7d-5′UTR17, which was optimized for encoding SARS-CoV-2 antigens and elicited strong immunity when delivered using liquid nanoparticle for in vivo vaccination. The observed increase in protein production efficiency suggests that the newly designed 5′ UTRs could have practical applications in various biotechnological processes. Further, we used the in-house data as an independent set to test UTR-LM for zero-shot fitness prediction, and showed that the model outperformed other methods by up to 51% in terms of the Spearman R score.
Additionally, we analysed the pretrained embedding and the attention score of the UTR-LM. The embedding of UTR-LM is able to differentiate between species and captures features such as the minimal fold energy. We also sought to identify motif patterns using an attention-based detection pipeline. In particular, the presence of the Kozak consensus sequence (KCS) with higher GC content was found to be important, a finding that aligns with previous research18,19. Further, attention score analysis also identified that upstream ATGs (uATGs) were depleted in high-expression UTRs and enriched in low-expression ones, consistent with the biological insight that uATG driving an upstream open reading frame could compete and thus lower downstream TE and protein expression20. In summary, our study presents the UTR-LM, a new self-supervised language model for studying UTRs of mRNA and decoding its functions. This research holds promising implications for advancing our understanding of gene regulation and innovating therapeutic interventions.
Results
UTR-LM predicts the ribosome loading
Ribosome loading refers to the number of ribosomes that are actively translating a specific mRNA molecule at any given time. It is a measure of how efficiently a particular mRNA is being translated into protein, and can be measured by various experimental techniques such as Ribo-seq or polysome profiling21. The ribosome loading can influence the rate of protein production and is influenced by factors such as the 5′ UTR sequence, SSs within the mRNA and the availability of ribosomes. Designing the 5′ UTR can affect ribosome loading, which in turn can be used to optimize protein ELs for various applications, including biotechnology and therapeutic protein production. Scientists had attempted to predict the effect of 5′ UTR sequence on the MRL. Several machine learning models were developed for this specific task, including Optimus6, FramePool5 and MTtrans9. RNA language models namely RNABERT12 and RNA-FM10 were also tested using this task.
In our study, we used the pretrained foundation model for 5′ UTR and further fine-tuned it for the task of MRL prediction. The baseline model is pretrained on unlabelled 5′ UTR sequences from five species within the Ensembl database, via the masked nucleotides task, and subsequently fine-tuned for MRL prediction. As shown in Fig. 2a, we tested variants of the model with additional training, including raw sequences from the downstream library, supervised information such as SS, MFE and other biology features. Figure 2b reveals that the baseline model alone attains satisfactory performance, and minor differences are observed among UTR-LM variants. For the final model, we chose the version that incorporates the downstream library, SS and MFE, and we will refer to it as UTR-LM MRL in the rest of the subsection. To investigate the effect of pretraining, we also compared the pretraining and fine-tuning strategy with other strategies that do not use pretrained weights or do not fine tune the full model. As shown in Fig. 2d, our UTR-LM model showed the highest performance, notably outperforming other baselines. This aligns with our expectation and demonstrates the effectiveness of our pretraining and fine-tuning strategy (more details in Supplementary DiscussionB.5).

a, Illustration of the UTR-LM framework, which includes variants integrating downstream library (DSL), SS and MFE tasks during pretraining, along with biological features (BF) during downstream prediction. b, Ablation study of UTR-LM hyperparameters under the U1 library (a sublibrary of synthetic 50-nucleotide 5′ UTRs, see Methods for details) with rank splitting. For subsequent experiments, we used the baseline UTR-LM enhanced by DSL, SS and MFE, termed UTR-LM MRL. The final model choice is marked in bold. c, Comparison of various pretrained methods followed by either a simple MLP with one hidden layer or a complex 32-layer residual network (ResNet) under U1 library with rank splitting. d, Ablation study on the training strategy. We compared the pretraining + fine-tuning strategy with other training strategies that use random weights and/or frozen weights (U1 library with rank splitting). e,f, Evaluation of various methods across eight libraries with random 50-nucleotide 5′ UTRs: rank splitting (e) and random splitting (f). g,h, Evaluation of various methods using independent tests. In independent tests, we fine-tuned UTR-LM MRL and retrained baselines on 76,319 random 5′ UTRs (25–100 bp), and tested models on 7,600 random (g) and 7,600 human 5′ UTRs (h).
We tested the UTR-LM MRL model with two downstream predictors: a simple multilayer perceptron (MLP) with one hidden layer and a much more complex 32-layer residual network (ResNet). In Fig. 2c, UTR-LM MRL followed by both predictors achieves similar performances, outperforming two other RNA foundation language models, that is, RNA-FM and RNABERT, on this same task. Moreover, RNA-FM and RNABERT worked well with a deep ResNet predictor, but performed poorly with the simpler MLP predictor. This observation hints that RNA-FM and RNABERT rely on deep neural networks to learn the sequence-to-MRL relation. By contrast, our pretrained UTR-LM MRL model does not rely on such complex predictors and achieves robustly high performances, indicating that it has extracted better semantic embeddings from 5′ UTR sequences.
We compared the UTR-LM MRL model with benchmark methods from the MRL prediction literature5,6,9,10 across eight synthetic libraries. This analysis focused on the 50 bp segment within the 5′ UTR closest to the start codon. Variants within this region are believed to be subject to stronger negative selection, probably because they can have more immediate effects on the gene’s ability to produce proteins22. We tested two different splitting strategies (Supplementary DiscussionB.3): rank splitting6 selects the 5′ UTRs with the highest read counts as the test data and uses the rest for training; random splitting splits data randomly into training and test sets. As illustrated in Fig. 2e,f, UTR-LM MRL consistently shows higher performance compared to other methods across all these tests. In particular, the UTR-LM MRL outperforms Optimus by up to 0.08, outperforms FramePool by up to 0.07 and outperforms RNA-FM by up to 0.43, in terms of the Spearman R score (additional metrics and results are reported in Supplementary DiscussionD.1–D.3).
We aimed to assess whether our model, specifically trained on 50 bp synthetic sequences, could predict the regulatory functions of human 5′ UTR sequences with varying lengths. While human 5′ UTR sequences can span from tens to thousands of nucleotides, a mere 13% of them are less than 50 bp long. We used two datasets that were originally proposed by Sample et al.6 and later as independent tests in several studies5,6,9,10. They allow evaluating the model’s adaptability from the training data (including only synthetic 5′ UTRs of 50 bp length) to human 5′ UTRs of varying lengths. These datasets encompass both synthetic 5′ UTRs and human 5′ UTRs, with their lengths ranging from 25 to 100 bp, and they do not overlap with our training data. By following the length-based held-out testing approach suggested by Optimus6, we fine-tuned the UTR-LM MRL model and retrained available baselines5,6,9,10 on the 76,319 random 5′ UTRs with 25 to 100 bp. Then, we tested and compared UTR-LM MRL and baselines on 7,600 random and 7,600 human 5′ UTRs. Further details can be found in Methods and Supplementary DiscussionB.4. Our results are illustrated in Fig. 2g,h. They show that the UTR-LM MRL, when paired with a ResNet downstream predictor, outperforms all other methods in both tests, while UTR-LM MRL with a simpler MLP exhibits similarly competitive performance. In addition, we analysed the prediction results across various sequence lengths. The results demonstrate that UTR-LM MRL can be effectively extended to both longer and shorter 5′ UTRs. Notably, our model shows the state-of-the-art performance on human 5′ UTRs (Fig. 2h). In particular, the UTR-LM MRL outperforms Optimus, FramePool and RNA-FM by about 1–6%, in terms of Spearman R score. It highlights the generalizability of UTR-LM MRL for decoding functions of endogenous 5′ UTRs.
UTR-LM predicts mRNA TE and expression
Protein production involves two primary processes: transcription and translation. The level of protein expression is highly dependent on the mRNA EL and also the TE of the transcripts23,24. EL is measured on the basis of the relative abundance of the mRNA transcript in the cell and is quantified using RNA sequencing (RNA-seq) RPKM7, where RPKM denotes reads per kilobase of transcript per million mapped reads. On the other hand, the TE of a gene, reflecting the rate of mRNA translation into protein, is calculated by dividing the Ribo-seq RPKM (indicative of ribosomal footprints on the mRNA) by the RNA-seq RPKM7.
In this section, we applied the pretrained UTR-LM and fine-tuned it for TE and EL prediction via tenfold cross-validation. We used three endogenous datasets7 gathered from human muscle tissue (muscle), human prostate cancer cell line PC3 (PC3) and human embryonic kidney (HEK) 293T cell line for training and testing. For benchmarking, we compared our method with a random forest model based on 3,000+ handcrafted biological features by Cao et al.7 (Cao-RF), and several other sequence-based models including Optimus6, FramePool5, MTtrans9, RNABERT12 and RNA-FM10. Here note that the Optimus, FramePool, RNABERT and RNA-FM were not developed for the mRNA TE and EL tasks, thus for comparison we retrained these models using TE and EL datasets.
For the prediction of TE, we conducted ablation studies using various UTR-LM variants. Unlike the MRL task (as shown in Fig. 2b), the TE task (illustrated in Fig. 3a–c) reveals notable performance discrepancy among different UTR-LM variants. When augmented with downstream library and MFE loss, the fine-tuned model demonstrates substantially higher performance across various datasets. We observed that the use of the downstream library alone or SS could lead to performance degradation, possibly because we had to truncate long sequences in the model and it might have introduced noticeable bias. We also observed that the additional use of biological features only affects the performance very minorly, so we chose to not include it in the final model. In the rest of the section, we chose the UTR-LM enhanced by the downstream library and MFE for the tasks TE and EL prediction. We will refer to this variant as UTR-LM TE for brevity. We also adapted the UTR-LM TE architecture to predict EL, which we refer to as UTR-LM EL.

Sources for these datasets include human muscle tissue (muscle), PC3 and HEK293T cells. a–c, Ablation study of UTR-LM hyperparameters on TE tasks, including the downstream library’s 5′ UTRs (DSL), a downstream task-independent SS and a downstream task-related MFE during the pretraining phase, and the inclusion of biological features (BF) during the downstream prediction phase: muscle (a), PC3 (b) and HEK (c). For the subsequent experiments, we used the baseline UTR-LM enhanced by DSL and MFE as the final model. d, For the TE prediction, the UTR-LM model outperforms Cao-RF by up to 5% and outperforms Optimus by up to 27% in terms of Spearman R. e, For the EL prediction, the UTR-LM model outperforms Cao-RF by up to 8% and outperforms Optimus by up to 47% in terms of Spearman R. e, In d,e, data are presented as mean values ± s.d. obtained via tenfold cross-validation. Paired t-tests showed that UTR-LM outperforms other benchmarks significantly (P < 0.05).
For the prediction of TE and EL, we used UTR-LM TE and UTR-LM EL and compared their performances with benchmark methods5,6,7,9,10,12. Figure 3d,e illustrates that UTR-LM TE and UTR-LM EL performs competitively with Cao-RF and outperforms other methods. Specifically, for Spearman R, UTR-LM model outperforms Cao-RF by up to 5 and 8% for TE and EL tasks, and outperforms Optimus by up to 27 and 47% for TE and EL tasks. While Cao-RF proves effective in predicting TE and EL, it relies on more than 3,000 handcrafted features including k-mer frequency, RNA folding energy, 5′ UTR length and number of open reading frameworks, and its random forest model that may encounter scalability issues with larger datasets. By contrast, the training of a language model offers a more principled solution to modelling 5′ UTR sequences and only uses information (for example, MFE) that can be easily computed. Thus, we believe that the UTR-LM provides a more robust and generalizable model for understanding 5′ UTR sequences.
URR-LM identifies IRESs
IRESs are unique RNA sequences, most located within the 5′ UTR of mRNAs. Unlike the typical cap-dependent translation initiation that starts at the 5′ end of an mRNA, IRESs enables ribosomes to initiate translation directly at the internal sites. Approximately 10% of cellular and viral mRNA are believed to use IRESs for translation initiation25. However, there existed only a limited number of verified IRESs, and most of them remain unannotated.
In this study, we applied the pretrained UTR-LM model to identify unannotated IRESs. We assembled a library of 46,774 sequences including both viral and cellular mRNAs, sourced from multiple databases14,26,27,28,29 (see details in Methods), with 37,602 sequences labelled as non-IRESs and 9,172 as IRESs. Building on the pretrained UTR-LM, we developed a contrastive learning approach to train a downstream IRES classifier (Methods).
Several baseline methods exist for predicting IRES, such as IRESfinder15, IRESpred14, IRESpy13 and DeepCIP16. See Supplementary DiscussionC for more details about these methods. For our comparison, we selected IRESpy as the benchmark because it represents the most recent method for both viral and cellular IRES detection and has shown advantages over IRESpred. We also compared with convolutional neural network baselines that were reported as top performers in previous literature5,6. As illustrated in Fig. 4, the UTR-LM IRES classifier substantially outperforms the best known benchmark, improving the AUPR from 0.37 to 0.52.

Our method, the UTR-LM IRES classifier, achieves higher test accuracy than best known baselines for this task. All models were trained and tested on a dataset of 46,774 sequences via tenfold cross-validation. Here we chose the metric AUPR because it is more suitable for imbalanced data. AUPR, area under the precision-recall curve; AUC, area under the receiver operating characteristic curve; CNN, convolutional neural network.
New 5′ UTR designs validated in wet-laboratory experiments
Finally, we conducted a set of wet-laboratory experimental assay to validate our prediction model and generate new 5′ UTR designs with high TE. We designed a library of 211 distinct 5′ UTR sequences with high predicted values of TE. In the experiment, we used expression of a luciferase reporter gene to measure mRNA translation in human cells (Fig. 5a). We first cloned all designed 5′ UTRs upstream of the same, standard luciferase reporter gene and then transfected the synthesized mRNAs into C2C12 cells, followed by quantitative luciferase assay (Fig. 5b). Specifically, we measured the relative light units (RLU) that quantifies the luciferase activity. This allowed us to assess the protein production level from each mRNA, which provided a direct measurement of how the designed 5′ UTRs might affect the protein synthesis process. We also compared the predicted values of MRL and TE of the designed library with the background distribution (that is, distribution of training data in each task). As shown in Fig. 5c,d, the UTR-LM model predicts that these in-house designed 5′ UTRs exhibit notably higher MRL and TE values than the background.

A library of 211 in-house 5′ UTRs with high TE was designed and validated through wet-laboratory experiments. These sequences were subsequently used as an independent test set to verify the efficiency of our model. a, The mRNA structure with in-house designed 5′ UTR. b, Experimental workflow for mRNA transfection and luciferase assay. c,d, The in-house 5′ UTRs had higher predicted values of MRL (c) and TE (d) compared to the background. e, Wet-laboratory results: relative TE of the top 20 designed 5′ UTRs compared to the benchmarks 5′ UTRs NCA-7d-5′UTR and HBB-5′UTR. f, Using the wet-laboratory results as an independent test set, the UTR-LM gave substantially more accurate zero-shot predictions of the RLU compared to the benchmark. Panels a and b created with BioRender.com.
We next compared the efficiencies of the top in-house designed 5′ UTRs with well-established UTRs used for mRNA therapeutics. For benchmarks, we measured our designs against two well-known 5′ UTRs, namely the HBB-5′UTR (ref. 30) and the NCA-7d-5′UTR (ref. 17). The HBB-5′UTR, a 5′ UTR from the human haemoglobin subunit beta, is commonly used in studies of mRNA translation and stability30. The NCA-7d-5′UTR, an optimized 5′ UTR for protein-coding mRNAs, has demonstrated effective delivery via lipid-derived TT3 nanoparticles, resulting in pronounced expression of potential SARS-CoV-2 antigens17. Our experiment validated that top candidates in our designed library achieved a marked improvement in protein production level (Fig. 5e). In particular, the top 5′ UTR sequence found in our assay had a 32.5% increase in protein production level compared to NCA-7d-5′UTR. Full details of the experimental design and methods are given in Supplementary DiscussionA.6.
Next, we evaluated our UTR-LM for zero-shot fitness prediction using our in-house design library and wet-laboratory results as an independent test set. In this test, we do not have measurements of TE or MRL, but instead we need to predict a new target RLU, measured by the fold change of log2-transformed RLU relative to that of NCA-7d-5′UTR. For zero-shot fitness prediction, we transferred the learned models UTR-LM MRL and UTR-LM TE to the new test set for predicting RLU, without any additional training or fine-tuning. For comparison, we also tested the benchmark model, Optimus6, which was trained on MRL. As illustrated in Fig. 5f, UTR-LM MRL and UTR-LM TE notably outperformed the benchmark Optimus on this task of zero-shot prediction of RLU. In particular, UTR-LM TE’s accuracy exceeds that of Optimus by more than onefold, demonstrating the transferability of our model across tasks and modalities.
Further, we applied the pretrained UTR-LM embedding, trained a new RLU predictor using our in-house data and tested it via tenfold cross-validation. As illustrated in Fig. 5f, our UTR-LM RLU model substantially outperformed the benchmark Optimus and the zero-shot models. The outcome underscores the potential of the language model approach in modelling genome sequences, showcasing its robustness and ability to generalize in predicting wet-laboratory outcomes.
Discussion
Attention scores reveal proven genome patterns
We examine the 5′ UTR embeddings across the five species used for pretraining. In Fig. 6a, we illustrated the frequency of each nucleotide at each base, per species. In Fig. 6b, we reported the silhouette scores, a metric that measures the separation between clusters, for pairs of species using both the UTR-LM embedding and the 4-mer representation. They showed that the UTR-LM embeddings achieved notably higher silhouette scores than the 4-mer representation. It suggests that the pretrained transformer provides more meaningful representations for differentiating between species. Figure 6c showed that the UTR-LM embedding of 5′ UTR captured most variation in the MFE value, while the traditional 4-mer representation struggled. See also Supplementary DiscussionE.1, E.2 for additional visualizations.

a, Sequence logo of the five species. b, Silhouette scores for pairs of species suggest the language model gives better differentiation between species on the basis of 5′ UTR. c, Uniform manifold approximation and projection (UMAP) visualization of 4-mer representation and UTR-LM embeddings, coloured by the MFE. d, Attention score of an instance human 5′ UTR, where a higher score means the corresponding site could have higher influence on the target function measured by ribosome loading. e, Average attention scores per position and nucleotide across 15,555 human 5′ UTRs revealed the KCS and its conserved pattern CCACC, which is a known motif that largely affects the ribosome loading.
The attention mechanism in the transformer architecture helps to draw connections between any parts of the sequence. In Fig. 6d, we visualize the attention scores of an instance of human 5′ UTR associated with the MRL prediction. A high attention score for a specific site suggests it could be influential in determining the target function. See Supplementary DiscussionE.3 for more examples and visualizations. Next, we analysed the average attention score per each position and per nucleotide across all human 5′ UTRs, with the hope to identify potential motif patterns. As shown in Fig. 6e, we saw that high attention scores in positions 1–6 align with the proven conserved nucleotide patterns31 of the KCS18,19. KCS is a nucleotide motif that functions as the protein translation initiation site in most mRNA transcripts. More specifically, our analysis found a most common conserved pattern CCACC32 (Fig. 6e) among the KCS variations. Finally, we screened all 3–12-mer fragments, filtered and ranked them by their attention scores to identify new potential motifs; see Supplementary DiscussionE.4 for the top found motifs. Notably, attention scores identified uATG being critical for UTR design, and a potential positional effect that inspires future investigation into the influence of uATG (and thus upstream open reading frame) within the 5′ UTR. While these motif patterns call for biological validation and further research, they hold the potential to unlock new understanding into mRNA translation and de novo design.
Summary
We introduced a language model for 5′ UTRs that integrates sequence, SS and MFE. The UTR-LM model has learned meaningful semantic representations from 5′ UTR sequences that pertain to the mRNA translation process. It applies to predicting the MRL, TE, mRNA EL and IRES and outperforms the best known baseline in each task. Also, we generated new 5′ UTR designs with highly predicted TE and measured their performances via wet-laboratory experiments. Our experiment revealed highly efficient new 5′ UTR designs with potential therapeutic value. While our model’s current performance is not limited by sequence length, it will be an exciting future direction to consider sparse transformers for modelling longer RNA sequences and more complex biological functions.
Methods
Overview of UTR-LM
We developed a unified foundation language model to provide meaningful and rich representations for 5′ UTRs. The model adopts a transformer architecture and it is trained using multi-modal data, including raw sequences, SS and MFE. The model is pretrained in a self-supervised learning manner via mask reconstruction, SS prediction and MFE prediction. It is later fine-tuned for a variety of downstream function prediction tasks, where it is shown to improve the state-of-the-art performances in each task.
We trained UTR-LM on two computing clusters. The first cluster, hosted on the Amazon Web Services cloud platform, was equipped with four Tesla V100-SXM2 graphical processing units, each boasting 16 GB of high-bandwidth memory. The second cluster, the Stanford University’s Sherlock high-performance computing system, used four TESLA_P100_PCIE graphical processing units, each with 32 GB of memory. We configured the training to a maximum of 200 epochs and 2 days.
Datasets
For pretraining the language model, we collected unlabelled 5′ UTR sequences from three sources: the Ensembl database33, synthetic libraries from Sample et al.6 and endogenous human 5′ UTR data analysed by Cao et al.7. We preprocessed the raw data to keep only high-quality and well-defined 5′ UTRs, details are shown in Supplementary DiscussionA.1. After the preprocessing steps, the final large-scale dataset from several sources is obtained:
We obtained 214,349 unlabelled 5′ UTR sequences from the Ensembl database33, spanning five species: human, rat, mouse, chicken and zebrafish.
We obtained eight synthetic libraries of random 5′ UTRs truncated to 50 nucleotides long from Sample et al.6 These libraries are sorted into two groups on the basis of their coding sequence: six libraries are linked to the enhanced green fluorescent protein (eGFP) and two are linked to mCherry. The eGFP-linked libraries consist of two unmodified uridine (U) libraries (U1 and U2), two pseudouridine (Ψ) libraries (Ψ1 and Ψ2) and two 1-methyl pseudouridine (m1Ψ) libraries (m1Ψ1 and m1Ψ2). Each eGFP library contains approximately 280,000 distinct 5′ UTRs. The mCherry-linked libraries, named mC-U1 and mC-U2, each contain around 200,000 unique 5′ UTRs.
We obtained three endogenous human 5′ UTR datasets analysed by Cao et al.7, each originating from a distinct cell line or tissue type: HEK293T, PC3 and human muscle tissue (muscle). The HEK, PC3 and muscle datasets comprised 14,410, 12,579 and 1,257 sequences, respectively.
In addition, we also included unlabelled raw sequences from datasets of downstream tasks in the pretraining. For more in-depth description of each dataset, please see Supplementary DiscussionA.
Architecture and pretraining of UTR-LM
We developed a specialized language model for studying 5′ UTRs, called UTR-LM (as illustrated in Fig. 1). The main architecture comprises an encoder block and a predictor block. The encoder block consists of a six-layer transformer34 with 16 multi-heads for self-attention. The layer normalization and residual connections are applied before and after each encoder block. The predictor block is a two-layer feed-forward neural network.
In our UTR-LM model, a 5′ UTR sequence of length L is input as a series of nucleotide tokens (such as ‘A’, ‘G’, ‘C’, ‘T’), along with a special [CLS] token. These tokens are first converted into 128-dimensional vectors via an embedding layer, forming an (L + 1) × 128 matrix. This matrix then goes through the encoder block, generating a representation for each nucleotide token in the sequence.
In the pretraining stage, we used a mix of self-supervised learning and supervised learning. For the self-supervised training part, we followed the masked language modelling (MLM)35, where we randomly masked 15% of the nucleotide tokens in the 5′ UTR sequence. The model is then trained to predict these masked tokens by minimizing the cross-entropy loss to encourage accurate reconstruction. We refer to this part as the masked nucleotide (MN) training task. This training objective function is
In equation (1), we randomly select a set of indices, denoted as M, from each input sequence x (making up 15% of the entire sequence). The token at each index i is replaced with the mask token [MASK]. The objective is to minimize the negative log-likelihood of the correct reconstruction of each xi, when the unmasked part \({x}_{/M}\) is given as context.
For the supervised training part, we used two labels to provide auxiliary supervision. First, we included the SS of 5′ UTR for training, which is calculated using the software ViennaRNA36. We represented the SS using the ‘dot-bracket’ notation36, where paired nucleotides are given by ‘(‘ and ’)’ characters and unpaired nucleotides are given by a ‘.’. For instance, the SS of the sequence AUGCAUGCGAUCAGC is given by ‘(((‥)))‥((.))’. To use the SS, we introduced an MLM-inspired task (which we call the SS task) to predict the SS symbols associated with masked nucleotides. The training objective function is
Equation (2) differs from equation (1) in that it uses the SS s as input. It attempts to reconstruct the masked SS, when the unmasked part \({x}_{/M}\) is given as context. The set of mask indices M is the same as for the masked nucleotides task. Second, we used the MFE of the 5′ UTR as an additional target of prediction, because a proven correlation exists between the MFE and the TE6. We refer to this step as the MFE training task, where the predictor block uses the [CLS] token’s representation to estimate the MFE value. We used the ViennaRNA software36 to calculate the actual MFE. The training objective is to minimize the mean squared error:
where MFEx and \(\hat{{\mathrm{MFE}}_{\rm{x}}}\) are the actual and predicted MFE of the input sequence x.
Downstream training
We used the learned representations from UTR-LM for several downstream tasks, as illustrated in Fig. 1b,c. We extended the predictor block of the pretrained UTR-LM from predicting MFE to predicting the downstream label. Instead of using UTR-LM merely as a static encoder to generate embeddings, we fine-tuned the model using the pretrained base model as the initial point. We provided a full chart of model sizes and runtime needed for fine-tuning the proposed and benchmark models on the synthesis library in Supplementary DiscussionB.6.
Prediction of MRL
Ribosome loading, a measure of mRNA TE influenced by factors such as the 5′ UTR, is experimentally assessed through techniques such as polysome profiling to derive the MRL. Understanding and designing the 5′ UTR can optimize MRL, thereby enhancing protein production for applications such as biotechnology and therapeutics. Here, we attempted to predict MRL on the basis of the 5′ UTR sequence.
First, we used eight libraries of randomized 50-nucleotide oligomer 5′ UTRs6, which are U1, U2, Ψ1, Ψ2, m1Ψ1, m1Ψ2, mC-U1 and mC-U2. These libraries include the measured MRL for each 5′ UTR sequence. Details and statistics can be found in Supplementary DiscussionA.3. Following Sample et al.6, we fine-tuned the UTR-LM for each downstream library, which we collectively refer to as UTR-LM MRL.
We performed independent tests on a dataset containing 83,919 random 5′ UTRs and another one containing 15,555 human 5′ UTRs, both with varying lengths from 25 to 100 bp. We used two testing strategies for independent tests. The first strategy, proposed by Optimus6, used a length-based held-out testing approach. Each test set contains 7,600 random 5′ UTRs or 7,600 human 5′ UTRs with the most reads. The remaining 76,319 random 5′ UTRs were used as the training set for both independent tests. The second strategy is to use a tenfold cross-validation. Because the U1 is commonly used in previous UTR research5,6, we started with the UTR-LM MRL model that was trained on U1 as the initial model and then fine-tuned it using the training set.
Prediction of mRNA EL and TE
Protein production is influenced by mRNA EL, measured in RNA-seq RPKM7, and TE, calculated as the ratio of Ribo-seq to RNA-seq RPKM7. Both factors are essential for understanding how the 5′ UTR influences the rate at which mRNA is translated into protein.
We used three different human datasets7 for the TE and the EL tasks. These datasets are named muscle, PC3 and HEK, and together they contain 41,446 unique 5′ UTRs. Each sequence of these datasets provides measurements of TE and EL. In alignment with the limitation of commercially available single-stranded DNA template biosynthesis7, a fixed 5′ UTR length of 100 bp was chosen for training. More detailed statistics can be found in Supplementary DiscussionA.4.
For each dataset, we trained a TE prediction model and EL prediction model, respectively. The model is fine-tuned on the basis of the parameter of the UTR-LM model pretrained on both Ensembl database and 41,446 human 5′ UTRs as an initial point.
While our 5′ UTR model was able to achieve reasonable performance to predict mRNA EL, 3′ UTR is thought to be more critical in determining mRNA stability and other properties. Nonetheless, recent work showcased that 5′ UTR may also contain structured RNA elements that influence mRNA stability37. Thus, we note that the EL is ultimately determined by a combination of factors that included 5′ and 3′ UTRs, and requires further investigation.
Identification of IRES with contrastive learning
IRESs are unique sequences that are often found within the 5′ UTRs of mRNAs. These specialized sequences enable cap-independent translation initiation and are thought to regulate translation for a subset of cellular and viral mRNAs25.
We assembled an unbalanced dataset sourced from several studies, including Weingarten-Gabbay et al.26, IRESbase27, IRESite28, Rfam29 and IRESpred14. This assembled dataset contains 46,774 sequences, with 37,602 sequences labelled as non-IRESs and 9,172 identified as IRESs. More detailed statistics can be found in Supplementary DiscussionA.5.
We adopted a contrastive learning approach to distinguish between IRES and non-IRES. First, we constructed contrastive samples where each sample consists of a pair of IRES and non-IRES sequences. For each sequence in the training set, we made one contrastive sample by randomly selecting another sequence with the opposite label to pair with it.
Although many IRESs are often found in the 5′ UTR of mRNA, they can also appear elsewhere. To address this, we transferred the UTR-LM model to fine tune using the IRES labelled data. We started by using the U1-trained version of UTR-LM as the initial base model. Then, we fine-tuned this model by minimizing a combination of three loss functions. The first one is the cross-entropy loss of the masked nucleotides task (equation (1)). The second one is binary cross-entropy loss for IRES versus non-IRES classification, as follows:
where yx and \({\hat{y}}_{\rm{x}}\) stand for the ground-truth label and the predicted probability of the label being IRES for the sequence x, respectively. The third one is a specially designed contrastive loss that measures the difference between the predicted probabilities for sequences labelled as non-IRES and sequences labelled as IRES. This loss function is computed over contrastive samples, that is, pairs of an IRES and a non-IRES. It is given by
where the pair (xi, xj) consists of two sequences selected from the set of contrastive samples, denoted by C. Since the pair comes from contrastive samples, their ground-truth labels are not equal \({y}_{{x}_{\rm{i}}}\ne {y}_{\rm{{x}_{j}}}\). The variables \({\hat{y}}_{\rm{{x}_{i}}}\) and \({\hat{y}}_{\rm{{x}_{j}}}\) represent the predicted probabilities of the label being IRES for sequences xi and xj, respectively.
Prediction of RLU
The RLU quantifies the level of luciferase activity, providing a direct measurement of how the designed 5′ UTRs affects the protein synthesis process. In our experiment, we designed a library of 211 unique 50-bp 5′ UTR sequences, and obtained the RLU by measuring the fold change of log2-transformed RLU relative to the well-established benchmark NCA-7d-5′ UTR17.
We fine-tuned the UTR-LM for the in-house designed library, termed UTR-LM RLU. Furthermore, we explored the model generalization ability in zero-shot fitness prediction. For this, we applied the learned UTR-LM MRL and UTR-LM TE models to predict RLU, without any additional training or fine-tuning.
Implementation of benchmarks
For comparisons, We retrained benchmark models including Optimus6, FramePool5, MTtrans9, RNABERT12, RNA-FM10 and Cao-RF7 using the same settings as used for training UTR-LM. Each model’s parameters and hyperparameters adhered to their respective GitHub implementations, such as learning rate and batch size and so on. For RNA-FM and RNABERT, the generated embeddings are of dimensions (sequence_length, 640) and (sequence_length, 120), respectively. These 2D embeddings were averaged into 1D (size 640 and 120) and then fed into an MLP downstream predictor.
Attention-based motif detection
Attention scores provide insights into the contribution of specific positions in the input sequence to the model’s prediction. Analysis of the attention scores of UTR-LM allows us to pinpoint important regions and potential motifs in 5′ UTRs. At the level of individual sequences, we examined the attention scores to find regions that highly contribute to the predictive result. At the position level, we averaged attention scores from the same nucleotide positions across human sequences, revealing potential motif patterns. The computation workflow is given in Extended Data Fig. 1.
To do this, we adopted a methodology inspired by ESM38. We generate an initial attention matrix for each sequence, which has dimensions (no. layers × no. heads, L, L), where no. layers and no. heads are hyperparameters of the self-attention layer and L is the sequence length. We first make the attention matrix symmetrical by taking the sum of it and its transpose. We then normalize the symmetrized matrix across the (L, L) dimension using the formula:
Equation (6) describes how to perform element-wise subtraction to compute a normalized matrix xnormalized from an input matrix x. Here, \(\odot\) denotes element-wise multiplication, and 1 is a matrix of ones with dimensions matching x. This allows the average value to be broadcasted across all elements of x during subtraction. Specifically, the average value is calculated by taking the product of the sums of the rows i and columns j of x and dividing it by the sum of all elements in x. Next, we sum the normalized matrix across the no. layers × no. heads dimension, thereby reducing the dimensions to (L, L). Finally, we sum along the rows of the resulting (L, L) matrix to generate the final attention vector.
To analyse attention at the positional level, we generated attention vectors for 15,555 human sequences. Across these different sequences, we computed the average attention score for each nucleotide type at each specific position. This provides insight into how specific nucleotides at particular positions contribute to the overall sequence behaviour.
Web server availability
The web server is freely available at https://huggingface.co/spaces/yanyichu/UTR_LM (ref. 39).
Data availability
The datasets are available and can be downloaded at https://codeocean.com/capsule/6711822 (ref. 39). This link includes training data for the pretrained model as well as datasets for various downstream tasks. Detailed statistics for these datasets are provided in Supplementary DiscussionA. Source data are provided with this paper.
Code availability
The code is freely available at https://github.com/a96123155/UTR-LM (ref. 40) under the GNU General Public Licence Version 3 and the implemented demo can be found at https://codeocean.com/capsule/6711822 (ref. 39).
References
Araujo, P. R. et al. Before it gets started: regulating translation at the 5′ UTR. Comp. Funct. Genomics 2012, 475731 (2012).
Miao, Z., Tidu, A., Eriani, G. & Martin, F. Secondary structure of the SARS-CoV-2 5′-UTR. RNA Biol. 18, 447–456 (2021).
Li, X., Kazan, H., Lipshitz, H. D. & Morris, Q. D. Finding the target sites of RNA-binding proteins. Wiley Interdiscip. Rev. RNA 5, 111–130 (2014).
Zeraati, M. et al. Cancer-associated noncoding mutations affect RNA G-quadruplex-mediated regulation of gene expression. Sci. Rep. 7, 708 (2017).
Karollus, A., Avsec, Ž. & Gagneur, J. Predicting mean ribosome load for 5′ UTR of any length using deep learning. PLoS Comput. Biol. 17, e1008982 (2021).
Sample, P. J. et al. Human 5′ UTR design and variant effect prediction from a massively parallel translation assay. Nat. Biotechnol. 37, 803–809 (2019).
Cao, J. et al. High-throughput 5′ UTR engineering for enhanced protein production in non-viral gene therapies. Nat. Commun. 12, 4138 (2021).
Barazandeh, S., Ozden, F., Hincer, A., Seker, U. O. S. & Cicek, A. E. UTRGAN: learning to generate 5′ UTR sequences for optimized translation efficiency and gene expression. Preprint at bioRxiv https://doi.org/10.1101/2023.01.30.526198 (2023).
Zheng, W. et al. Discovery of regulatory motifs in 5′ untranslated regions using interpretable multi-task learning models. Cell Syst. 14, 1103–1112.e6 (2023).
Chen, J. et al. Interpretable RNA foundation model from unannotated data for highly accurate RNA structure and function predictions. Preprint at https://doi.org/10.48550/arxiv.2204.00300 (2022).
Ozden, F., Barazandeh, S., Akboga, D., Seker, U. O. S. & Cicek, A. E. RNAGEN: a generative adversarial network-based model to generate synthetic RNA sequences to target proteins. Preprint at bioRxiv https://doi.org/10.1101/2023.07.11.548246 (2023).
Akiyama, M. & Sakakibara, Y. Informative RNA base embedding for RNA structural alignment and clustering by deep representation learning. NAR Genom. Bioinform. 4, lqac012 (2022).
Wang, J. & Gribskov, M. IRESpy: an XGBoost model for prediction of internal ribosome entry sites. BMC Bioinf. 20, 409 (2019).
Kolekar, P., Pataskar, A., Kulkarni-Kale, U., Pal, J. & Kulkarni, A. IRESPred: web server for prediction of cellular and viral internal ribosome entry site (IRES). Sci. Rep. 6, 27436 (2016).
Zhao, J. et al. IRESfinder: identifying RNA internal ribosome entry site in eukaryotic cell using framed k-mer features. J. Genet. Genomics 45, 403–406 (2018).
Zhou, Y. et al. DeepCIP: a multimodal deep learning method for the prediction of internal ribosome entry sites of circRNAs. Comput. Biol. Med. 164, 107288 (2023).
Zeng, C. et al. Leveraging mRNA sequences and nanoparticles to deliver SARS-CoV-2 antigens in vivo. Adv. Mater. 32, e2004452 (2020).
Babendure, J. R., Babendure, J. L., Ding, J.-H. & Tsien, R. Y. Control of mammalian translation by mRNA structure near caps. RNA 12, 851–861 (2006).
Hinnebusch, A. G., Ivanov, I. P. & Sonenberg, N. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 352, 1413–1416 (2016).
Calvo, S. E., Pagliarini, D. J. & Mootha, V. K. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl Acad. Sci. USA 106, 7507–7512 (2009).
Zuccotti, P. & Modelska, A. Studying the translatome with polysome profiling. Post-Transcriptional Gene Regulation (ed Dassi, E.) 59–69 (2016).
Whiffin, N. et al. Characterising the loss-of-function impact of 5′ untranslated region variants in 15,708 individuals. Nat. Commun. 11, 2523 (2020).
Kozak, M. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 15, 8125–8148 (1987).
Kozak, M. Downstream secondary structure facilitates recognition of initiator codons by eukaryotic ribosomes. Proc. Natl Acad. Sci. USA 87, 8301–8305 (1990).
Stoneley, M. & Willis, A. E. Cellular internal ribosome entry segments: structures, trans-acting factors and regulation of gene expression. Oncogene 23, 3200–3207 (2004).
Weingarten-Gabbay, S. et al. Comparative genetics. Systematic discovery of cap-independent translation sequences in human and viral genomes. Science 351, aad4939 (2016).
Zhao, J. et al. IRESbase: a comprehensive database of experimentally validated internal ribosome entry sites. Genom. Proteom. Bioinform. 18, 129–139 (2020).
Mokrejs, M. et al. IRESite–a tool for the examination of viral and cellular internal ribosome entry sites. Nucleic Acids Res. 38, D131–D136 (2010).
Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 49, D192–D200 (2021).
Leppek, K. et al. Combinatorial optimization of mRNA structure, stability, and translation for RNA-based therapeutics. Nat. Commun. 13, 1536 (2022).
Gleason, A. C., Ghadge, G., Chen, J., Sonobe, Y. & Roos, R. P. Machine learning predicts translation initiation sites in neurologic diseases with nucleotide repeat expansions. PLoS ONE 17, e0256411 (2022).
Hernández, G., Osnaya, V. G. & Pérez-Martínez, X. Conservation and variability of the AUG initiation codon context in eukaryotes. Trends Biochem. Sci. 44, 1009–1021 (2019).
Cunningham, F. et al. Ensembl 2022. Nucleic Acids Res. 50, D988–D995 (2022).
Vaswani, A. et al. Attention is all you need. In 31st Conference on Neural Information Processing Systems (NIPS, 2017).
Sinha, K. et al. Masked language modeling and the distributional hypothesis: order word matters pre-training for little. In Proceedings of the 2021 Conference on Empirical Methods in Natural Language Processing 2888–2913 (2021).
Lorenz, R. et al. ViennaRNA package 2.0. Algorithms Mol. Biol. 6, 26 (2011).
Leppek, K., Das, R. & Barna, M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 19, 158–174 (2018).
Rao, R. M., Meier, J., Sercu, T., Ovchinnikov, S. & Rives, A. Transformer protein language models are unsupervised structure learners. International Conference on Learning Representations (ICLR, 2020).
Chu, Y. et al. A 5′ UTR language model for decoding untranslated regions of mRNA and function predictions. Zenodo https://doi.org/10.5281/zenodo.10621605 (2024).
Chu, Y. et al. UTR-LM GitHub https://github.com/a96123155/UTR-LM (2024).
Acknowledgements
This paper is partially supported by National Science Foundation grant no. 1953686 and partially supported by RVAC Medicines.
Author information
Authors and Affiliations
Contributions
Y.C. developed the UTR-LM model. D.Y. performed experimental validation. Y.L. produced in-house data. K.H. reviewed both the code and manuscript. Y.S. developed the web server. L.C. contributed to manuscript preparation. J.Z. initiated the experimental part of the project. M.W. led the entire project. All authors contributed to manuscript preparation.
Corresponding author
Ethics declarations
Competing interests
RVAC Medicines has submitted patent applications related to the designed UTR sequences. D.Y., Y.L. and Y.S. are affiliated with RVAC Medicines. J.Z. is affiliated with Zipcode Bio. Other authors have declared no conflicts of interest.
Peer review
Peer review information
Nature Machine Intelligence thanks Joshua W. K. Ho, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1
The computation flow of identifying patterns in 5′ UTR sequences based on attention scores.
Supplementary information
Supplementary Information
Supplementary Discussion, Figs. 1–10 and Tables 1–6.
Source data
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chu, Y., Yu, D., Li, Y. et al. A 5′ UTR language model for decoding untranslated regions of mRNA and function predictions. Nat Mach Intell 6, 449–460 (2024). https://doi.org/10.1038/s42256-024-00823-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42256-024-00823-9
