
Abstract
The rise of single-cell genomics is an attractive opportunity for data-hungry machine learning algorithms. The scBERT method, inspired by the success of BERT (‘bidirectional encoder representations from transformers’) in natural language processing, was recently introduced by Yang et al. as a data-driven tool to annotate cell types in single-cell genomics data. Analogous to contextual embedding in BERT, scBERT leverages pretraining and self-attention mechanisms to learn the ‘transcriptional grammar’ of cells. Here we investigate the reusability beyond the original datasets, assessing the generalizability of natural language techniques in single-cell genomics. The degree of imbalance in the cell-type distribution substantially influences the performance of scBERT. Anticipating an increased utilization of transformers, we highlight the necessity to consider data distribution carefully and introduce a subsampling technique to mitigate the influence of an imbalanced distribution. Our analysis serves as a stepping stone towards understanding and optimizing the use of transformers in single-cell genomics.
Similar content being viewed by others

Main
Convolutional neural networks (CNNs), generative adversarial networks (GANs), variational autoencoders (VAEs) and graph neural networks (GNNs) have been successful in addressing various data analyses in biomedicine, and genomics in particular. Single-cell genomics data, comprising data from thousands of individual cells, have particularly benefited from the use of neural networks1,2. However, one critical open problem in single-cell data analysis is labelling and discovering individual cell types, which is a non-local problem as the cellular context is important. Yang and colleagues3 have recently proposed the application of ‘transformers’, a state-of-the-art natural language neural-network architecture, for cell-type annotation. By ‘drawing parallels between natural language processing and genomics’ and ‘utilizing self-attention mechanisms’4, transformers can effectively capture long-range dependencies within single-cell genomics data. The scBERT method proposed by Fan Yang et al. can annotate cell types, detect novel cell types and is robust to batch effects.
In this Article we assess the reusability of scBERT, reported by Yang and colleagues3. Although we could largely reproduce their results, our findings indicate that cell-type distribution plays a more important role than initially reported. To validate this observation, we evaluated scBERT on a new dataset. We found that scBERT performs well in cell-type annotation tasks and performs similarly in detecting new cell types, as shown on the datasets reported in the paper. However, our results indicate that cell-type distribution influences the performance of scBERT in annotation and novel cell-type detection tasks on the new dataset. In summary, we anticipate that the use of transformers will be expanded beyond cell-type annotation to include a variety of downstream tasks such as perturbation response prediction, multimodal integration and gene function analysis. On a broader note, our analysis suggests that, when engaging in downstream analysis using transformers, imbalanced data distributions remain an ongoing challenge and should be carefully addressed in each case.
Description of the method and the original experimental validation of scBERT
The scBERT model utilizes the advanced capabilities of the BERT model, which has demonstrated notable performance in natural language-processing tasks. The authors of scBERT have adapted the BERT model for single-cell RNA sequencing (scRNA-seq) data by creating gene embeddings through gene2vec5, which encodes gene embeddings within a predefined vector space to capture semantic similarities between genes. Additionally, the scBERT model incorporates expression embeddings generated through term-frequency analysis to discretize continuous expression variables by binning and converting them into 200-dimensional vectors. These embeddings are utilized as token embeddings within the scBERT model, allowing for consideration of transcription levels of individual genes.
The scBERT model is first pretrained (self-supervised learning) on large amounts of unlabelled scRNA-seq data, providing a general understanding of gene interactions. A fine-tuning process on unseen and user-specific scRNA-seq data for supervised cell-type annotation tasks follows this. In self-supervised learning, unlabelled data were obtained from PanglaoDB6 and utilized in the fine-tuning stage with task-specific data. During the self-supervised pretraining phase, masked expression and gene embeddings are integrated as input and fed into the performer blocks. A reconstructor is employed to generate outputs, with the reconstruction loss being calculated based on the output for masked genes. In the subsequent supervised fine-tuning stage, task-specific scRNA-seq data are input into the previously pretrained encoder.
The performance of scBERT was initially evaluated in comparison to other methods using seven scRNA-seq datasets comprising a comprehensive representation of 17 major organ/tissue systems, 50 cellular subtypes, over 500,000 cells and a variety of mainstream single-cell omics technologies (Drop-seq, 10X, SMART-seq and Sanger-Nuclei). Overall, the benchmark evaluation in ref. 3 comprehensively considered diversity in data size and complexity.
Reproducibility
Using the source code of scBERT from https://github.com/TencentAILabHealthcare/scBERT (git commit 8ac7c1e), we repeated the analyses described in ref. 3 on the two datasets for which scBERT showed the best results, that is, Zheng68k7 and MacParland8. The first is a peripheral blood mononuclear cells (PBMC) dataset, widely used for cell-type annotation performance assessment, and the MacParland dataset profiles 8,444 cells from the human liver (belonging to 20 different hepatic cell populations). The Zheng68k data were available in a preprocessed format (https://github.com/TencentAILabHealthcare/scBERT), whereas the MacParland data were available as a raw count matrix. We used standard preprocessing steps from scanpy9 that the authors have reported in the https://github.com/TencentAILabHealthcare/scBERT repository, that is, (filter, normalize and log1p) to make the MacParland format suitable for model training. We replicated the majority of the reported results. However, to fully assess the utility and efficacy of scBERT, we evaluated its performance on novel datasets, as described in the following (Extended Data Table 1 and Extended Data Fig. 1).
Reusability
To assess the applicability and generalizability of scBERT across new datasets, we investigated its performance on a novel dataset—the NeurIPS dataset—which is a compilation of single-cell multi-omics data collected from mobilized peripheral CD34+ haematopoietic stem and progenitor cells (HSPCs) for cell-type annotation and the detection of novel cell types tasks.
Challenging the scBERT using an additional dataset, NeurIPS
To this end, we explored the application of scBERT on the NeurIPS dataset from the 2022 Kaggle competition, which is accessible via https://www.kaggle.com/competitions/open-problems-multimodal/data, for a cell-type annotation task for predicting seven different cell types (GitHub: scbert-reusability). The data comprise single-cell multi-omics data from mobilized peripheral CD34+ cells (HSPCs) collected from four healthy human donors. This dataset was generated using the 10X Chromium Single Cell Multiome ATAC + Gene Expression technology (Multiome), which allows for the simultaneous measurement of gene expression (RNA) and chromatin accessibility (ATAC) in single cells. Accordingly, we used gene expression (RNA) from this multi-omics data for our experimentation. Specifically, we focused on the gene expression (RNA) data from this multi-omics dataset, which encompassed seven distinct cell types, namely B-cell progenitor (BP, n_cells = 262), erythrocyte progenitor (EryP, n_cells = 3,402), haematopoietic stem cell (HSC, n_cells = 10,757), mast cell progenitor (MasP, n_cells = 2,175), megakaryocyte progenitor (MkP, n_cells = 3,394), monocyte progenitor (MoP, n_cells = 258) and neutrophil progenitor (NeuP, n_cells = 3,663).
Assessing interclass similarity among cell types in the NeurIPS data
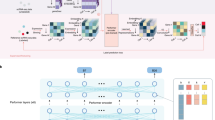
Understanding the similarity among the cell types is key to discerning the nuances of cell-type annotation and the detection of novel cell types. We visualized the different cell types in low two-dimensional uniform manifold approximation and projection (UMAP) plots (Fig. 1a). We also conducted a correlation analysis between cell types to assess scBERT’s robustness in the NeurIPS dataset for cell-type annotation and detection of novel cell types tasks (Fig. 1b), providing both qualitative and quantitative perspectives on the similarity among cell types. These findings indicated a substantial correlation between cell types. Furthermore, the UMAP plots show that individual clusters corresponding to each cell type are not adequately distinct from one another, as depicted in Fig. 1a. However, it is essential to acknowledge that the presence of high interclass similarity in the NeurIPS dataset does not diminish the overall capabilities of scBERT for cell-type annotation and novel cell-type detection, as shown in Fig. 2b,c. scBERT demonstrates a robust performance when applied to datasets with diverse and less homogeneous cell populations.

a, UMAP plot of single-cell expression data, coloured by cell types (ground truth), for the NeurIPS dataset. b, Spearman correlation matrix of single-cell expression data between cell types in NeurIPS.

a, Distribution of cells in the NeurIPS dataset: (1) original dataset (left); (2) subsampling (middle): reduced number of cells (300 cells); (3) oversampling (right): augmented number of cells (4,600 cells) of BP and MoP cell types. b, Heatmaps for the confusion matrices of the prediction results on the NeurIPS dataset (test data, 30%): (1) original dataset (left); (2) subsampling (middle): reduced number of cells (300 cells); (3) oversampling (right): augmented number of cells (4,600 cells) of BP and MoP cell types. c, Performance of scBERT on the discovery of novel n = 7 cell types on the NeurIPS dataset (original, subsampling and oversampling). Process: we removed one cell type in the training process and added it to the predicted dataset. This process was iterated on each cell type. The box plots show the median (centre lines), interquartile range (hinges) and 1.5 times the interquartile range (whiskers). d, Threshold analysis for prediction accuracy in the NeurIPS dataset: (1) original dataset (left), (2) subsampling (middle): reduced number of cells (300 cells), (3) oversampling (right): augmented number of cells (4,600 cells) of BP and MoP cell types.
scBERT shows good performance with NeurIPS data for predicting cell types
The dataset of seven cell types was divided into two subsets, with 70% of the data allocated for training and 30% for testing. An additional split was performed on the training subset to further refine the model’s performance, with 80% of the data utilized for model training and the remaining 20% for validation. We observed that scBERT performed better on this new dataset, with a validation mean accuracy value of 0.8510, compared with Seurat, which achieved a validation mean accuracy of 0.8013 (Fig. 3a). We used Seurat for comparison because Seurat showed the next best performance after scBERT for the cell-type annotation task. However, scBERT showed a slight decrease in mean accuracy value, 0.8397 (Table 1), although this was still better than Seurat, which achieved a mean accuracy value of 0.8160 and F1 score of 0.6395 (Fig. 2b) when applied to the 30% test data. Compared to Seurat, scBERT demonstrated improved performance, as reflected in the mean accuracy values. The P value obtained from a paired t-test was 0.0004, indicating that the performance improvement of scBERT over Seurat is statistically significant. These results demonstrate the potential utility of using pretrained language models such as scBERT for cell-type annotation tasks. Leveraging the pretrained knowledge embedded in these models can improve their performance compared to models trained from scratch. This is consistent with findings reported in the original scBERT paper, where the authors conducted an ablation study. The study demonstrated the value of pretraining in enhancing the model’s downstream performance on cell-type annotation tasks.

a, Left: Performance of scBERT, scBERT (focal loss) and Seurat measured according to accuracy using fivefold cross-validation (n = 5) on the NeurIPS dataset (training data, 70%): (1) original dataset; (2) subsampling: reduced number of cells (300 cells); (3) oversampling: augmented number of cells (4,600 cells) of BP and MoP cell types. The dotted line represents the random accuracy result (1/7 = 0.143). Right: zoomed plot of the accuracy using the original dataset. b, Performance of scBERT, scBERT (focal loss) and Seurat measured by F1 score using fivefold cross-validation (n = 5) on the NeurIPS dataset (training data, 70%): (1) original dataset; (2) subsampling: reduced number of cells (300 cells); (3) oversampling: augmented number of cells (4,600 cells) of BP and MoP cell types. c, Performance of scBERT, scBERT (focal loss) and Seurat in each cell type (n = 7 cell types) measured by F1 score on the NeurIPS dataset (original, subsampling and oversampling) in the training and prediction process. In the box plots, the median and upper and lower quartiles are represented by the centre line and box bounds, respectively. Whiskers display the largest and smallest values within 1.5 times the interquartile range from the quartiles.
scBERT can detect only part of the novel cell types within NeurIPS data
To evaluate the ability of scBERT to detect novel cell types, we performed leave-one-out experiments in which scBERT was trained on all but one cell type and then evaluated on its ability to identify the held-out cell type as a novel type. To this end, we followed the same steps as reported in the original paper of applying a threshold of probability of <0.5 such that cells with a value less than 0.5 be treated as unassigned or novel cell types. We observed that scBERT was only able to detect the neutrophil progenitor (NeuP) cell type as a novel cell type, and it still did not perform well on detecting other cell types as novel cell types (Fig. 2c), with a mean accuracy score of 0.087. It is worth noting that continued work is needed to develop scBERT representations and training procedures using scRNA-seq data to increase the efficacy of detecting novel cell types.
Subsampling improves scBERT’s performance in cell type annotation by balancing the cell-type distribution
As shown in Figs. 2c and 3b,c, scBERT showed poor performance on cell types with fewer cells (that is, with an imbalance in the number of cells). We were thus curious to see how the distribution of cells by cell type affected cell-type annotation. The distribution of cells per cell type is shown in Fig. 2a. In the NeurIPS dataset, we observed that the BP and MoP cells comprised 262 and 258 cells, respectively. We thus subsampled other cell types at 300 cells per type, resulting in a relatively even cell distribution (Fig. 2a). We observed that a balanced distribution of cells by cell type influences the F1 scores. For the original data, the F1 score was 0.6395 (Table 1). In contrast, subsampling enhanced the F1 score to 0.7041 (Fig. 2b and Table 1). Specifically, cells that were previously poorly predicted due to their smaller proportions, such as BP cells, saw an increase in F1 score from 0.0964 to 0.6879 (Fig. 2b), compared to other cell types. We also assessed the robustness of scBERT across various subsampling levels (150, 200, 250 and 350). We found that scBERT still exhibits better performance in terms of F1 score (Fig. 4a), even when we change the subsampling levels and maintain the distribution of the cells by cell type close to each other.

a, Accuracy and F1 score of the prediction performance on the NeurIPS dataset: original, subsampling and oversampling (using SMOTE and random oversampling). b, Performance of scBERT discovery of novel cell types (n = 7) on the NeurIPS dataset: original, subsampling and oversampling (using SMOTE and random oversampling). Process: we removed one cell type in the training process and added it to the predicted dataset. This process was iterated on each cell type (using a probability threshold of 0.5). In the box plots, lower and upper hinges represent the first and third quartiles, the centre lines the median, and whiskers the range of 1.5 times the interquartile.
Balanced cell distribution across various subsampling levels enhances scBERT’s novel cell-type detection performance using NeurIPS data
One possible effect of cell distribution on detecting novel cell types is that it can influence the likelihood of detecting them. We thus asked whether the distribution of cells affects the ability to detect novel cell types using scBERT. We used the same set-up when detecting novel cell types as used in the original NeurIPS dataset, but we used subsampled data, that is, 300 cells per cell type (BP = 262 cells and MoP = 258 cells). We discovered that an even distribution of cells improves the detection of novel cell types. The mean accuracy score improved from 0.087 to 0.3187 (Fig. 2c). This suggests that cell distribution is essential for scBERT when detecting novel cell types. We also evaluated the robustness of scBERT with different subsampling levels (that is, 150, 200 250 and 350). We observed that scBERT still exhibits better performance with different subsampling levels when the distribution of cells is roughly equal across different cell types, as shown in Fig. 4b.
Improved scBERT performance in cell-type annotation and detection of novel cell types across varied oversampling levels using NeurIPS data
Next, we asked whether balancing the cell-type representation by increasing the count of cell types with the fewest cells improves or degrades the performance of scBERT for annotation and detecting novel cell types. To this end, we performed data oversampling utilizing the ‘synthetic minority oversampling technique’ (SMOTE)10. We oversampled the cell types with a low number of cells and increased the mean of the cells with the maximum number of cells (4,600 cells for the BP and MoP cell types). We observed that oversampling slightly improved the F1 score from 0.7041 to 0.7353 (Fig. 4a) compared to when we subsampled to a lower number of samples per cell type (300 cells). Moreover, we examined the robustness of scBERT with different oversampling levels (that is, 1,000, 2,000, 3,000, 4.000 and 5,000 samples) and observed that scBERT consistently performs well for cell-type annotation tasks, even when the sampling levels are changed, as shown in Fig. 4a and Extended Data Fig. 2.
However, scBERT shows a slight improvement in novel cell-type detection compared to when there is a reasonable amount of imbalance in the data (0.087 to 0.181), but not as well as when we subsampled to 300 cells per cell type (0.3187) (Fig. 2c). The rationale for this is that, unlike when we subsampled 300 cells per cell type, which had comparative F1 scores across all cell types, the oversampling raises the F1 score of the enhanced cells and influences the F1 score across other cell types. We further evaluated the efficacy of scBERT across increasing oversampling levels (that is, 1,000, 2,000, 3,000, 4,000 and 5,000 samples). Our observations reveal that scBERT maintains its performance across these different oversampling levels when compared to the performance seen with the original and various subsampled distributions across cell types, as demonstrated in Fig. 4a. These results suggest that scBERT exhibits effective performance across a range of sample sizes.
We also investigated the impact of random oversampling, a type of resampling used for assessing the robustness of scBERT in cell-type annotation and the detection of novel cell types, in addition to SMOTE oversampling. For this purpose, cells from minority classes, BP and MoP, were randomly duplicated with replacements to create a more balanced dataset. This procedure was iteratively executed to augment the BP and MoP cell counts to 1,000, 2,000, 3,000, 4,000 and 5,000, respectively. We examined scBERT’s robustness using random oversampling levels of 1,000, 2,000, 3,000, 4,000 and 5,000. Our observations indicate that scBERT maintains strong performance with an increase in sample size in both cell-type annotation and novel cell-type detection, as depicted in Fig. 4a and Extended Data Fig. 2. These results suggest that scBERT demonstrates substantial robustness across various oversampling levels, indicating its effective handling of increased sample sizes.
No significant improvement in scBERT performance with focal loss for cell-type annotation using NeurIPS data across varied cell distributions
We also assessed the impact of using a focal loss function11 instead of the cross-entropy loss, as utilized in the original publication during the fine-tuning phase. The focal loss function is designed to handle class imbalance issues in classification tasks. It introduces two additional parameters to the traditional cross-entropy loss: alpha, a class balancing weight, and gamma, a factor that adjusts the rate of down-weighting for easy examples. To this end, we trained the models using the focal loss function and observed a slightly inferior performance compared to models trained with cross-entropy loss. The mean validation accuracy was 0.8488 for the focal loss, compared to 0.8510 for the cross-entropy loss in the original dataset, which exhibits an imbalance in the distribution of cells per cell type. Furthermore, in scenarios of subsampling and oversampling, where there is an even distribution of cells per cell type, scBERT with the default loss function (cross-entropy) still outperformed the scBERT trained with the focal loss function (Fig. 3a,b and Extended Data Fig. 2): 0.7646 versus 0.7540. These results suggest that changing the loss function does not substantially improve scBERT’s performance in tackling the uneven distribution of cells per cell-type problem.
Impact of probability threshold on scBERT’s detection of novel cell types on NeurIPS data
To understand the impact of the chosen probability threshold on scBERT’s performance in detecting novel cell types, we conducted a systematic analysis using various sampling strategies (subsampling of 300 cells and oversampling of 4,600 cells) and probability threshold values ranging from 0.0 to 0.9. For novel cells, the accuracy metric was computed as the ratio of cells aptly classified as ‘unassigned’ to the total count of novel cells. In contrast, we used a multi-class accuracy approach for known cells after removing the ‘unassigned’ cells. This calculated accuracy was further adjusted by subtracting the proportion of ‘unassigned’ cells discarded in the previous step, introducing a penalization for misclassification. Figure 2d depicts the mean accuracy for different cell types, both known and novel, across varying probability thresholds. As the threshold increases, we observe a corresponding decrease in the detection accuracy for known cells, while the accuracy for novel cells improves. This trade-off underscores the critical role that threshold settings play in balancing the accurate classification of known cells and the discovery of novel cell types. Notably, when using the default threshold of 0.5, the subsampled dataset outperforms the others, achieving the highest accuracy in novel cell detection (0.3189), closely followed by the oversampled dataset (0.1804), and finally, the original dataset (0.0878). As illustrated in Fig. 2d, we recommend adjusting the threshold upwards to ~0.7 or 0.8 in the original dataset to substantially enhance the detection accuracy of novel cells.
Subsampling with balanced cell distribution improves scBERT’s performance on cell-type annotation on the Zheng68k data
We repeated the same steps for the Zheng68k dataset as used in our analysis using NeurIPS data to validate the observation of the effect of cell distribution per cell type on cell-type annotation. We divided the cell types into subsamples of 2,000 cells each (Fig. 5a). We excluded CD4+ T Helper2 (n = 97) and CD34+ (n = 242) cells. The reason for excluding these two cell types is that the other cell types have over 2,000 cells, and subsampling them to a much smaller number will remove information about the cell types. Using the Zheng68k dataset, we trained the model (fine-tuning the pretrained model) on this equally distributed dataset per cell type and then used this fine-tuned model for the cell-type annotation task. We observed that the distribution impacts the annotation of cell type. We improved the F1 score to 0.6683 and boosted the prediction scores of the cell types for which scBERT demonstrated poor performance when these cell types (CD4+/CD45RA+/CD25− naïve T, dendritic) have a lower proportion of cells than the other cell types.

a, Distribution of cells in the Zheng68k dataset: (1) original dataset (left) and (2) subsampling (right), where we reduce the number of cells (2,000 cells) and remove CD34+ and CD4+ T helper2 cells. b, Performance of scBERT discovery of n = 9 novel cell types on the Zheng68k dataset (original and subsampling). Process: we removed one cell type in the training process and added this to the predicted dataset. This process was iterated on each cell type. Box plots show the median (centre lines), interquartile range (hinges) and 1.5 times the interquartile range (whiskers).
Balanced cell distribution through subsampling improves scBERT’s performance in detecting novel cell types on the Zheng68k dataset
We have demonstrated how scBERT’s efficacy in identifying novel cell types on NeurIPS data is impacted by the distribution of cells across cell types. To further validate this observation, we applied scBERT to the subsampled Zheng68k dataset, for which there is an equal distribution of cells per cell type. scBERT showed improved performance in the mean accuracy value in detecting novel cell types compared to when we have a relatively smaller number of cells in specific cell types (Fig. 5b). This demonstrates that the performance of scBERT relies on the distribution of cells for detecting novel cell types and cell-type annotation.
Discussion
In this study we have demonstrated that reusing the code and data provided by Yang and colleagues3 is sufficient to reproduce the main published results in the cell-type annotation task. However, it was challenging to identify the novel cell types in the MacParland dataset. Ideally, scBERT should have detected all the cell types that were excluded in the training data and included during prediction as novel ones, but we found that it did not perform well in this scenario.
In addition to reproducing the results of ref. 3, we found that the distribution of cells per cell type affects the effectiveness of scBERT in cell-type annotation and detecting novel cell types. This is because, in both the cell-type annotation and the task of recognizing novel cell types, scBERT performs poorly on cell types with fewer cells than other cell types. In our analysis, we conducted two studies, subsampling the number of cells per cell type around the cell type with fewer cells than the other cell types, and, in the second set, augmenting the cell types with fewer cells to see how the distribution affects scBERT’s performance on cell-type annotation and detecting novel cell types. The empirical results show that the distribution affects scBERT’s performance when the cell types are evenly distributed; that is, it improves its performance across cell-type annotation and novel cell-type detection. Furthermore, the performance of scBERT appears sensitive to the degree of skewness of the distributions, an effect that becomes abundantly clear when detecting novel cell types.
For future directions in part based on our analysis, further work is needed to understand the reasons for this distributional sensitivity of transformers in single-cell genomics and to develop methods to mitigate it. Possible directions include examining the effects of class imbalance on the training of transformer models and developing objective functions and training procedures tailored to uneven cell-type distributions. Furthermore, including domain knowledge and appropriate regularizations, which could guide the model towards learning biologically plausible predictions, could potentially aid in detecting novel cell types. Nevertheless, our reusability analysis demonstrates the potential of scBERT in detecting novel cell types and provides insights for future improvements.
More broadly, we envision that transformer models could be applied to various downstream single-cell analysis tasks, such as differential expression analysis, multimodal data integration, gene function analysis and drug-response prediction. With the comprehensive understanding of cellular transcriptional contexts that scBERT learns during pretraining, it can be used for perturbation response prediction. In this scenario, the model will be fine-tuned on perturbation data. The data should consist of paired observations of each cell’s initial state and the state after perturbation, allowing scBERT to learn the relationship between these states. In the context of multimodal data integration, as scBERT is originally pretrained on scRNA-seq data, applying it to multimodal data would require incorporating the additional data modalities into the model’s input. One possible approach might be to concatenate the normalized representations of each data type into a single vector for each cell. The model would then be fine-tuned on the combined data, allowing it to learn correlations between the different data types and thereby enhance the richness of its cell-state representations. Moreover, for gene function analysis, scBERT’s architecture could be used with a change in the output layer to predict the functional category or categories associated with each gene. The model would need to be fine-tuned on gene annotation data, where each gene is associated with one or more functional categories. However, our reusability analysis stresses the need to consider the specific characteristics of the task and data distribution balance when applying a transformer architecture. As with novel cell-type detection, it will be essential to understand and address the effects of potential data imbalances. In general, it appears to be a delicate balance between the power of transformers to detect subtle correlations in datasets versus their sensitivity to skewed data distributions and class imbalances. How this plays out for different tasks in single-cell genomics and biomedicine remains to be investigated.
Methods
Reproducibility experiments
We found a few errors in the syntax of the command line arguments as they were included before the scripts (fine-tuning the pretrained model, predicting using a fine-tuned model, and detecting novel cell types) as stated in the ‘Usage’ section of scBERT’s GitHub repository. However, after rectifying these minor issues, the code was easy to execute to reproduce the results.
Cell-type annotation
Our reproducibility experiments on cell-type annotation followed the steps outlined by the authors3 in the GitHub repository, where they used 100% of the data for training and the same dataset for testing. We ran the experiment in exactly the same way as the original authors, including using the same random seeds specified in the code. We discovered that on the Zheng68k dataset7, our F1 score of 0.677 (Extended Data Table 1) and theirs of 0.691 deviated by 0.014; furthermore, we obtained a mean accuracy value of 0.7802 on the Zheng68k dataset, whereas the original publication reports 0.7590 (0.021 difference). On the MacParland dataset, we obtained an F1 score of 0.9602 as compared to the F1 score reported in the paper of 0.9588, deviating by 0.0014, while our respective mean accuracy value deviated by 0.020 (ours, 0.9558, Fig. 1a; theirs, 0.9760; Extended Data Table 1). Our analysis revealed a slight deviation in the mean accuracy score and F1 score, probably due to excluding a specific immune cell type, CD4+ T Helper2, from the original study. On further investigation, it was observed that this cell type—CD4+ T Helper2—has a lower abundance of cells (n = 97) than the other cell types, resulting in a relatively lower accuracy than the results reported in the original study (Extended Data Fig. 1b).
We also divided the datasets into two subsets, with 70% allocated for training and 30% for testing. An additional split was performed on the training subset to further refine the model’s performance, with 80% of the data utilized for model training and the remaining 20% for validation. To this end, we observed that the prediction accuracy of scBERT decreased in mean accuracy (0.7802 to 0.7551; Extended Data Fig. 1a). However, we found that scBERT continued to perform well in the second phase of this set-up, where we used 70% of the data for training and the remaining 30% for testing, with a mean accuracy score of 0.7533 (Extended Data Fig. 1b) and F1 score of 0.6523.
We performed the same experiments on the MacParland dataset and divided it into 70% for training scBERT and 30% as test data for the prediction. We discovered little difference in mean accuracy values when training and testing scBERT with 70% MacParland data versus 100% MacParland data (0.9532 and 0.9558, respectively; Extended Data Fig. 1a). However, with 30% test data, the mean accuracy score was 0.9372 and the F1 score 0.9400 (Extended Data Fig. 1b).
Detecting novel cell types
In many scenarios, the reference dataset may not include all the different types of cells in the query dataset. This can be a problem for marker-based methods, which rely on pre-selected markers for known cell types and may struggle to identify new, unseen cell types. On the other hand, correlation-based methods often assign novel cell types to the closest available class, which may not always be accurate. In contrast, machine learning-based methods can supposedly automatically detect novel cell types by evaluating the predicted probabilities. This allows for a more precise and flexible classification of cell types.
To this end, as reported in this paper, we tested the reproducibility of scBERT in detecting novel cell types on the MacParland dataset. The cell types excluded for reproducibility were plasma cells, alpha-beta T cells, gamma-delta T cells and mature B cells. We discovered that scBERT predicts only plasma cells as novel cell types. It cannot detect the other cell types (alpha-beta T cells, gamma-delta T cells and mature B cells) as novel cell types (Extended Data Fig. 1c).
Data availability
The NeurIPS data used in our study are available in the figshare repository at https://figshare.com/projects/scbert-reusability/157203. The Zheng68K dataset was downloaded from GitHub, and the MacParland data from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE115469. Source data are provided with this paper.
Code availability
The original scBERT code is available at https://github.com/TencentAILabHealthcare/scBERT. Our GitHub content, with detailed instructions, is available at https://github.com/TranslationalBioinformaticsUnit/scbert-reusability. The code can also be accessed via Zenodo at https://doi.org/10.5281/zenodo.8191571 ref. 12.
References
Ma, Q. & Xu, D. Deep learning shapes single-cell data analysis. Nat. Rev. Mol. Cell Biol. 23, 303–304 (2022).
Wainberg, M., Merico, D., Delong, A. & Frey, B. J. Deep learning in biomedicine. Nat. Biotechnol. 36, 829–838 (2018).
Yang, F. et al. scBERT as a large-scale pretrained deep language model for cell type annotation of single-cell RNA-seq data. Nat. Mach. Intell. 4, 852–866 (2022).
Cui, H., Wang, C., Maan, H., Duan, N. & Wang, B. scFormer: a universal representation learning approach for single-cell data using transformers. Preprint at bioRxiv https://doi.org/10.1101/2022.11.20.517285 (2022).
Du, J. et al. Gene2vec: distributed representation of genes based on co-expression. BMC Genomics 20, 82 (2019).
Franzén, O., Gan, L.-M. & Björkegren, J. L. M. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019, baz046 (2019).
Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
MacParland, S. A. et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 9, 4383 (2018).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Chawla, N. V., Bowyer, K. W., Hall, L. O. & Kegelmeyer, W. P. SMOTE: synthetic minority over-sampling technique. J. Artif. Intell. Res. 16, 321–357 (2002).
Lin, T. Y., Goyal, P., Girshick, R., He, K. & Dollar, P. Focal loss for dense object detection. IEEE Trans. Pattern Anal. Mach. Intell. 42, 318–327 (2020).
Khan, S. A. et al. Translational bioinformatics unit/scBERT-reusability: 2.0.0. Zenodo https://doi.org/10.5281/zenodo.8191571 (2023).
Acknowledgements
King Abdullah University of Science and Technology supported this work, which was also partially funded by the SDAIA-KAUST Center of Excellence in Data Science and Artificial Intelligence.
Author information
Authors and Affiliations
Contributions
S.A.K. and A.M. conducted the reproducibility and reusability experiments, curated the datasets, drafted the first version of the manuscript and integrated all the edits. V.L. and R.L. contributed ideas, and N.A.K., D.G.-C. and J.T. contributed ideas and supervised the overall analysis, manuscript writing and final edits.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Harald Binder, Benjamin Haibe-Kains and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Reproducible performance of scBERT on Zheng68k and MacParland datasets.
a) Performance of scBERT measured by accuracy on Zheng68k and MacParland dataset (size 70% and 100%) using fivefold cross-validation (n = 5). b) Heatmap for the confusion matrices of the prediction results on the Zheng68k and MacParland datasets (test data, 30%). Left side: Zheng68k (accuracy = 0.7533 and F1 score = 0.6523) Right side: MacParland (accuracy = 0.9372 and F1 score = 0.9400) c) Performance of scBERT discovery novel cell types on the MacParland dataset by removing alpha-beta T cell, gamma-delta T cell (gamma-delta_T_Cells_1 and gamma-delta_T_Cells_2), mature B cell, and plasma cell population (denominated as novel) during the training process. Accuracy of detecting n = 15 known and n = 5 novel cell types. In box plots, the lower and upper hinges represent the first and third quartiles, the center lines the median, with whiskers in the range of 1.5-times the interquartile.
Extended Data Fig. 2 Confusion matrices.
Heatmap for the confusion matrices of the prediction results. 1) Subsampling sizes: 150, 200, 250 and 350. 2) SMOTE oversampling sizes: 1000, 2000, 3000, 4000 and 5000. 3) Random-oversampling sizes: 1000, 2000, 3000, 4000 and 5000. 4) Focal-loss: original, subsampling (size = 300) and oversampling using SMOTE (size = 4600). 5) Seurat: original, subsampling (size = 300) and oversampling using SMOTE (size = 4600).
Source data
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khan, S.A., Maillo, A., Lagani, V. et al. Reusability report: Learning the transcriptional grammar in single-cell RNA-sequencing data using transformers. Nat Mach Intell 5, 1437–1446 (2023). https://doi.org/10.1038/s42256-023-00757-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42256-023-00757-8
