
Abstract
Acquired drug resistance poses a challenge for single-target FGFR inhibitors, leading to the development of dual- or multi-target FGFR inhibitors. Sulfatinib is a multi-target kinase inhibitor for treating neuroendocrine tumors, selectively targeting FGFR1/CSF-1R. To elucidate the molecular mechanisms behind its binding and kinase selectivity, we determined the crystal structures of sulfatinib with FGFR1/CSF-1R. The results reveal common structural features and distinct conformational adaptability of sulfatinib in response to FGFR1/CSF-1R binding. Further biochemical and structural analyses disclose sensitivity of sulfatinib to FGFR/CSF-1R gatekeeper mutations. The insensitivity of sulfatinib to FGFR gatekeeper mutations highlights the indispensable interactions with the hydrophobic pocket for FGFR selectivity, whereas the rotatory flexibility may enable sulfatinib to overcome CSF-1RT663I. This study not only sheds light on the structural basis governing sulfatinib’s FGFR/CSF-1R inhibition, but also provides valuable insights into the rational design of dual- or multi-target FGFR inhibitors with selectivity for CSF-1R and sensitivity to gatekeeper mutations.
Similar content being viewed by others


Introduction
The fibroblast growth factor (FGF)-FGF receptor (FGFR) axis plays vital roles in angiogenesis, endocrine functions, wound repair as well as tissue and metabolism homeostasis1,2,3. It regulates multiple downstream signaling pathways, including RAS–RAF–MAPK, PI3K–AKT, signal transducer and activator of transcription (STAT) and phospholipase Cγ (PLCγ), to control cellular proliferation, differentiation, survival, and migration3,4,5. Recently, FGFR gene amplification, activating mutations, and gene rearrangements or fusions, have been documented abundantly in many tumor types, such as cholangiocarcinoma, urothelial carcinoma, and gastric cancer2,6,7. These alterations in FGF/FGFR signaling are closely related to tumorigenesis, tumor progression, and drug resistance, rendering FGFR as a potential target for cancer therapy2,4.
Numerous small-molecule FGFR inhibitors have been clinically approved or have become candidate drugs for cancer therapy. Unfortunately, acquired drug resistance gradually develops after an initial response to these molecules through both “on-target” and “off-target” mechanisms1,8,9. To address the on-target resistance, higher drug dosages are often required to ensure adequate accessibility to tumor tissues, resulting in non-specific targeting and dose-limited toxicity8,9,10. On the other hand, combination therapy is employed to overcome the off-target resistance but increases the risk of drug-drug interaction8,9. Therefore, the development of dual- or multi-target FGFR inhibitors has emerged to improve therapeutic effects and patient compliance, while reducing risks of drug-drug interactions and acquired resistance associated with single-target FGFR inhibitors. However, due to their poor selectivity, these compounds are always related to off-target effects and adverse events, significantly impeding their clinical applications9. Consequently, there is a pressing need to develop dual- or multi-target FGFR inhibitors with high selectivity and potent inhibitory activity.
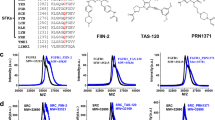
Sulfatinib (Fig. 1A) is a multi-target drug that selectively targets FGFR, collection stimulating factor 1 receptor (CSF-1R), and vascular endothelial growth factor receptor (VEGFR). It has been approved for the treatment of pancreatic and extrapancreatic neuroendocrine tumors (NETs)11,12,13,14. CSF-1R can induce the proliferation, survival, and differentiation of tumor-associated macrophages, which are abundant in various tumor-microenvironment activities, thereby promoting tumor immune evasion and drug resistance15,16,17. Blockade of CSF-1R signaling can reshape the tumor microenvironment and enhance immune responses, making CSF-1R an attractive target combined with FGFR for cancer therapy. Additionally, VEGFR is up-regulated with FGFR genetic changes, promoting tumor angiogenesis that is essential for tumor growth, invasion, and metastasis18,19. Therefore, the encouraging antitumor activity of sulfatinib for NETs is achieved by preventing tumor angiogenesis and tumor immune evasion through simultaneous FGFR, CSF-1R, and VEGFR inhibition11,12,13,14. Sulfatinib exhibits great potency and selectivity for these kinases, with minimal off-target effects8,9,11,12,13,14, making it an ideal template for developing FGFR inhibitors with dual- or multi-targeting capabilities. Thus, understanding how sulfatinib simultaneously targets these kinases is of utmost importance.

A Chemical structures of sulfatinib. Inhibitory effects of sulfatinib on wild-type FGFR1-4 B and CSF-1R C through kinase activity inhibition assays. Error bars represent the standard deviation for at least three independent measurements.
In this study, we aimed to elucidate the structural basis underlying the high potency and selectivity of sulfatinib against FGFR and CSF-1R. To achieve this, we determined the crystal structures of sulfatinib in complex with FGFR1 and CSF-1R using X-ray crystallography. We conducted structural comparisons of sulfatinib bound to different kinases and with other specific inhibitors to reveal common structural features and distinct conformational adaptability of sulfatinib in response to different kinase binding pockets. Furthermore, we evaluated the resistance profiles and mechanisms of sulfatinib inhibition against FGFR and CSF-1R gatekeeper mutations through biochemical and structural analysis. Collectively, our work not only sheds light on the molecular mechanisms of the dual inhibition of FGFR and CSF-1R conferred by sulfatinib, but also provides fundamental information for the rational design of dual- or multi-target FGFR inhibitors with high selectivity for CSF-1R and sensitivity to gatekeeper mutations.
Results
Potent inhibition of FGFRs and CSF-1R by sulfatinib
A kinase inhibition assay was employed to confirm the inhibitory potency of sulfatinib against FGFRs and CSF-1R. Consistent with the previous reports20, sulfatinib demonstrated potent inhibition of FGFR1 and CSF-1R, with IC50 values of 34 and 5 nM, respectively (Fig. 1B, C). The high sequence homology among the FGFRs family indicates that sulfatinib may exhibit similar efficacy against other members. As anticipated, sulfatinib effectively inhibited FGFR2 and FGFR3, comparable to FGFR1, with low nanomolar IC50 values (21 nM and 28 nM, respectively), whereas a reduction in efficacy of approximately 26-fold was observed for FGFR4 (IC50 value of 889 nM). These findings establish sulfatinib as a potent inhibitor of FGFR1-3 and CSF-1R.
Structural basis of the sulfatinib/FGFR1 interaction
To gain structural insights into the mechanism of FGFR inhibition by sulfatinib, we solved the crystal structure of sulfatinib with FGFR1 at 1.988 Å resolution (PDB 8JMZ) (Supplementary Data 1). Data collection and structure refinement statistics are presented in Supplementary Table 1. The overall structure of sulfatinib/FGFR1 complex is described in Fig. 2A, with the activation loop adopting a DFG-in conformation (Fig. 2B). As illustrated in Fig. 2C, the pyrimidine nitrogen and the adjacent phenylamine NH of sulfatinib make a bidentate hydrogen-bonding interaction with Ala564 at the hinge region of FGFR1, whereas the indole group fills the hydrophobic pocket and forms a hydrogen bond with Glu531. The N-(2-dimethylamino-ethyl)-ethanesulfonamide group is exposed to the solvent-exposed region and makes a hydrogen-bond interaction with Asn568. This binding mode is also observed in the FGFR1/infigratinib complex (PDB 3TT0, Supplementary Fig. 1), in which infigratinib is a selective and potent FGFR1-3 inhibitor21. Overall, sulfatinib binds to a DFG-in conformation of FGFR1 by the extensive interaction network in the ATP pocket, corresponding to a type I inhibitor.

A Overall structure of the sulfatinib/FGFR1 complex. B FGFR1 adopts a DFG-in activation loop conformation. C Hydrogen-bond interaction between sulfatinib and FGFR1.
Structural insight into the sulfatinib/CSF-1R interaction
To provide a foundation for understanding the mechanism that enables sulfatinib to govern FGFRs and CSF-1R simultaneously, we then solved the cocrystal structure of CSF-1R with sulfatinib at 1.69 Å resolution (PDB 8JOT) (Supplementary Data 2). Data collection and structure refinement statistics are presented in Supplementary Table 1. The overall CSF-1R/sulfatinib binding pattern is well-presented in Fig. 3A. Specifically, CSF-1R adopts a DFG-out activation loop upon sulfatinib binding (Fig. 3B). Sulfatinib maps in the ATP-binding pocket of CSF-1R with the pyrimidine nitrogen and the adjacent phenylamine NH making a bidentate hydrogen bond with the hinge residue Cys666 and the N-(2-dimethylamino-ethyl)-ethanesulfonamide moiety extending toward the solvent-exposed area (Fig. 3C). Unexpectedly, the indole group of sulfatinib does not occupy the hydrophobic pocket but rotates out in response to the DFG-out flip. These results demonstrate that sulfatinib inhibits CSF-1R by locking the protein in an inactive state, classified as a type II inhibitor.

A Overall structure of the sulfatinib/CSF-1R complex. B CSF-1R adopts a DFG-out activation loop conformation. C Hydrogen-bond interaction between sulfatinib and CSF-1R.
Structural comparison of sulfatinib in complex with FGFR1 and CSF-1R
The kinase domains of FGFR1 and CSF-1R share significant sequence similarity, especially the residues around the ligand-binding pocket22. However, the binding modes of these two kinases bound to sulfatinib are extremely distinct. To figure out the structural basis for the different inhibitory mechanisms, we superposed the two complexes (Fig. 4A). Sulfatinib exhibits dramatic conformational changes upon FGFR1 and CSF-1R except for the hinge-interacting pattern. The indole group of sulfatinib in CSF-1R is flipped out of the hydrophobic pocket via an oxy linker. In contrast, the solubilizing group is rotated by 180° in proximity to the hinge region of CSF-1R, compared with that of FGFR1. These conformational differences in sulfatinib lead to the distinct interaction contacts between FGFR1 and CSF-1R (Supplementary Fig. 2). We supposed that sulfatinib may bind to CSF-1R as the mode with FGFR1, but the indole group of sulfatinib may clash with Lys616 and Phe797 of CSF-1R (Fig. 4B), which means it may have a higher-energy barrier to attain the FGFR1-sulfatinib binding mode (Supplementary Fig. 3A). Thus, the indole group of sulfatinib in CSF-1R rearranges to a less energetically favorable conformation (Supplementary Fig. 3B).

A Comparison of sulfatinib/FGFR1 interaction and sulfatinib/CSF-1R interaction. B FGFR1-bound sulfatinib may clash with Lys616 and Phe797 of CSF-1R when overlaying the sulfatinib/FGFR1 complex and sulfatinib/CSF-1R complex. C The R-spine of FGFR1 is linear and consists of Leu547, Met535, Phe642, His621 and Asp682. D The R-spine of CSF-1R is broken by the substitution of Phe797 with Trp550 from the juxtamembrane domain, which enforces the displacement of Phe797 to the DFG-out conformation. FGFR1 is colored cyan, while CSF-1R is colored salmon.
Apart from the discrepancies in sulfatinib conformation, there is a distinguished conformational difference between FGFR1 and CSF-1R, as the Asp and Phe side chains of the DFG motif swap positions. In the FGFR1 structure, Phe642 is involved in the linear R-spine of FGFR1 that is made up of Leu547, Met535, Phe642, His621, and Asp682 (Fig. 4C), which is a hallmark signature of the active kinase state23,24. As to CSF-1R, the R-spine is broken and Phe797 is displaced by Trp550 from the juxtamembrane domain (JMD) (Fig. 4D), thereby forcing the DFG-out flip and forming an inhibitory R-spine. These results indicate that these conformational alterations are associated with the Asp-Phe swap, showing the flexibility and adaptability of sulfatinib upon different kinase bindings.
Structural comparison of CSF-1R in complex with sulfatinib and vimseltinib
To explore the structural features in ligand binding by the same receptor, we compared the structures of CSF-1R with sulfatinib and vimseltinib (PDB 7MFC), a potent and selective CSF-1R inhibitor (Fig. 5A). Vimseltinib has a similar hydrogen-binding interaction pattern with the hinge residue as sulfatinib25 (Fig. 5B). The indole substituent of sulfatinib and the methylpyridine ring of vimseltinib are both connected by an oxy linker to the hinge-interacting group but present a completely opposite conformation (Fig. 5C). The indole substituent of sulfatinib is flipped out, while the methylpyridine ring coupled with the N-isopropyl pyrimidinone group extends deeply into the hydrophobic pocket, serving as a surrogate for the JMD Trp550 and stabilizing the hydrophobic R-spine. To better accommodate vimseltinib, the Phe797 residue in turn slightly spins out to avoid steric clash. Moreover, the methylpyridine ring of vimseltinib interacts with Asp796 of the DFG motif, which helps vimseltinib firmly anchored in this pocket. These results suggest that the selectivity for CSF-1R could be improved by the occupation of the third position of the R-spine and the hydrogen binding with Asp796.

A Chemical structure of vimseltinib. B Binding mode of vimseltinib in complex with CSF-1R (PDB 7MFC). C Superposition of sulfatinib/CSF-1R (salmon) and vimseltinib/CSF-1R (sky blue).
Efficacy of sulfatinib in response to FGFRs and CSF-1R gatekeeper mutations
The “gatekeeper” residue of protein kinase controls the access of kinase inhibitors to the ATP-binding pocket, by which mutations to a larger hydrophobic residue are responsible for drug resistance. Therefore, it is crucial to counter the problem of the sensitivity of sulfatinib to FGFRs and CSF-1R gatekeeper mutations. Kinase activity inhibition assays were carried out. Unfortunately, sulfatinib cannot overcome any FGFR gatekeeper mutation with IC50 values all over 2 μM for FGFR1V561M, FGFR2V564I/F and FGFR3V555M (Fig. 6A). In order to explain this on-target resistance mechanism, we constructed the binding models of FGFR1V561I/M/F in complex with sulfatinib based on our solved FGFR1/sulfatinib complex (Fig. 6B–D). These structural models predict that the bulkier side chains of isoleucine, methionine and phenylalanine residues may clash with sulfatinib and prevent the extension of sulfatinib into the hydrophobic pocket, thereby leading to the observed loss in potency.

A Inhibitory effects of sulfatinib on FGFR and CSF-1R gatekeeper mutations through kinase activity inhibition assays. B–E Structural models of sulfatinib in complex with FGFR1V561I/M/F and CSF-1RT663I, which are generated by substitution of gatekeeper residues based on our solved FGFR1/sulfatinib (PDB 8JMZ) and CSF-1R/sulfatinib (PDB 8JOT) structures. Error bars represent the standard deviation for at least three independent measurements.
All FGFR kinases possess a valine gatekeeper, in contrast to CSF-1R, c-Kit, PDGFRα and BCR-ABL, which all have a threonine at the gatekeeper position (Supplementary Fig. 4). Although there are no mutations reported upon Thr663 gatekeeper of CSF-1R, c-KitT670I, PDGFRαT674I and BCR-ABLT315I have been found causally linked to drug resistance26, which means that CSF-1R would develop T663I gatekeeper mutation during the clinical usage. The kinase activity inhibition assay results showed that sulfatinib preserved a remarkable inhibitory effect on CSF-1RT663I with IC50 value of 4 nM (Fig. 6A). Here, we proposed the binding model of CSF-1RT663I with sulfaitnib based on our solved CSF-1R/sulfatinib structure. It seems that there is a large space available to accommodate sulfatinib without steric clash with the isoleucine side chain (Fig. 6E), supporting the high sustainability of sulfatinib against CSF-1RT663I mutation as determined by biochemical assay. Collectively, these structural models suggest that sulfatinib cannot overcome FGFR gatekeeper mutations due to the severe hindrance from the bulky isoleucine, methionine and phenylalanine side chains, but exhibit persistent activity against CSF-1RT663I attributing to the distinct conformational adaptation of sulfatinib upon CSF-1R binding.
Discussion
Sulfatinib stands out as a potent and selective multi-target inhibitor, with a lower rate of off-target side effects compared to other reported dual- or multi-target FGFR inhibitors, such as lenvatinib, sunitinib, pazopanib, and nintedanib. This can be attributed to sulfatinib’s high potency and selectivity for FGFR, CSF-1R, and VEGFR over 278 other kinases9,20,27,28,29. As anticipated, we confirmed that sulfatinib has significant potency against FGFR1-3 and CSF-1R with low nanomolar IC50 values (Fig. 1B, C). This finding suggests that sulfatinib has the potential for therapeutic applicability in FGFR-driven malignancies characterized by prominent immune evasion profiles. Tumor immunotherapy remains a highly attractive but not yet fully operational treatment approach30. To date, the FDA has approved thirty-one combinations of tyrosine kinase inhibitors with immunotherapy for various indications, and numerous ongoing clinical trials continue to explore the combinations, reflecting an ever-increasing interest in incorporating immunosurveillance into kinase inhibitors31. Given the significant role of CSF-1R in tumor immune modulation, our focus was primarily on the roles of FGFR and CSF-1R in the context of sulfatinib.
To elucidate the underlying mechanism of inhibitor binding and kinase selectivity for sulfatinib, we solved crystal structures of sulfatinib in complex with FGFR1 and CSF-1R by X-ray crystallization. The N-phenyl-2-pyrimidinamine core scaffold of sulfatinib, forming hydrogen-bound interactions with the hinge region, proves essential for the inhibitory effects on FGFR and CSF-1R, which may serve as an optimal template for the rational design of FGFR inhibitors with CSF-1R selectivity. Notably, the flexibility provided by the oxy linker of sulfatinib emerges as a critical factor for the FGFR and CSF-1R targeting, allowing sulfatinib to adapt and adopt multiple binding conformations in response to different kinase bindings. Furthermore, the occupancy of the indole group of sulfatinib within the hydrophobic pocket appears to enhance the selectivity for FGFR. Introducing bulky isoleucine, methionine, and phenylalanine residues at the gatekeeper position of FGFR leads to severe steric clash with the indole group of sulfatinib. Consequently, the clash disrupts the hydrogen binding with Ala564 of the hinge region and Glu531 surrounding the hydrophobic pocket32,33,34. The flexible oxy linker may assist in shifting the indole group out of this pocket, resulting in a CSF-1R/sulfatinib-like binding model to better accommodate gatekeeper mutation and maintain hydrogen contacts with the hinge residue. However, this conformational change of sulfatinib may lead to a complete loss of inhibitory activity against FGFR, as evidenced by the micromolar range of IC50 values for FGFR1V561M, FGFR2V564I/F, and FGFR3V555M (Fig. 6A). This emphasizes the critical role of the occupancy at the hydrophobic pocket for FGFR selectivity.
Apart from sulfatinib, PRN1371 also had great potency and selectivity against FGFR1-4 and CSF-1R with IC50 values of 0.6, 1.3, 4.1, 19.3, and 8.1 nM, respectively35. We compared the FGFR1/sulfatinib complex with FGFR4/PRN1371 complex (PDB 7F3M)6, and found that these two binding modes are similar, with hydrogen-bound interactions with the hinge region and occupancy of hydrophobic pocket via a hydrogen bond (Supplementary Fig. 5A, B). The bulkier difluoromethoxyphenyl ring of PRN1371 than the indole group of sulfatinib may also clash with bulky isoleucine, methionine, and phenylalanine residues at the gatekeeper position, inducing insensitivity to FGFR gatekeeper mutations. As for CSF-1R binding, we predicted the binding mode of PRN1371 with CSF-1R, and made a comparison with our crystal of sulfatinib/CSF-1R (Supplementary Fig. 5C, D). CSF-1R kept a DFG-out conformation when binding to sulfatinib but tended to adopt a DFG-in conformation in response to PRN1371 binding, in which the latter Asp of DFG forming a hydrogen bond with the difluoromethoxyphenyl ring of PRN1371 firmly anchored this ring into the hydrophobic pocket. In contrast to PRN1371, the structural flexibility of sulfatinib allowed the indole flip out of the pocket even to escape the clash with the bulky gatekeeper residues, which was absolutely not allowed for PRN1371 due to its structural rigidity. Collectively, the binding mode comparison of the two inhibitors bound by FGFR/CSF-1R highlighted the common structural features and the distinct conformational adaptability in the inhibitor-kinase interactions.
Our structure of CSF-1R/FGFR1 with sulfatinib offers significant opportunities for developing dual- or multi-target FGFR inhibitors with improved potency against CSF-1R. For example, the close proximity of the side chain of Asp796 in CSF-1R to the indole group of sulfatinib when occupying the hydrophobic pocket suggests the possibility of hydrogen-bond interactions after proper modifications to anchor this pharmacophore. This observation aligns with the designs of several selective CSF-1R inhibitor36,37,38,39,40,41. Moreover, the residues around the hydrophobic pocket exposed in the DFG-out conformation of CSF-1R show less conserved across kinases, suggesting the possibility of achieving high selectivity for CSF-1R42. Inspired by the structural comparison of CSF-1R with sulfatinib and vimseltinib36,37, incorporating a surrogate for JMD Trp550 or the formation of hydrophobic interactions with Trp550 may stabilize the hydrophobic R-spine and reinforce sulfatinib binding. Currently, pharmacophore-based combination strategies are widely applied in drug design8, which indicates that combining distinct pharmacophores from sulfatinib and vimseltinib or other selective CSF-1R inhibitors is conducive to the rational design of dual- or multi-target FGFR inhibitors with CSF-1R selectivity to improve antitumor efficiency.
To prolong the therapeutic efficacy, further development to overcome gatekeeper mutation-based resistance is warranted. Based on the crystal of FGFR1/CSF-1R with sulfatinib, there are some strategies to preserve sustainable inhibitions against FGFR/CSF-1R gatekeeper mutations. The indole group of sulfatinib could be substituted with other heterocyclic fragments by hydrogen-bond interactions with Asp of DFG motif to move far away from gatekeeper residues, or with other smaller moieties to provide enough space for gatekeeper residues, which may not only avoid steric clash but also meet the requirement of occupying the hydrophobic pocket for FGFR selectivity. Introducing a slim and long linker, such as the alkyne linker in ponatinib43, into the inhibitor core scaffold may also allow avoidance of steric clash and greater spacing to better accommodate a bulky residue at the gatekeeper position. Encouraged by the flexible oxy linker in sulfatinib, exploiting inhibitor flexibility by incorporation of a flexible linker, such as the ethyl linker in AZD454732, may be an effective strategy to curb drug resistance by allowing diverse inhibitor binding modes.
In conclusion, we confirmed that sulfatinib has great potency against FGFR1-3 and CSF-1R by kinase inhibition assay. The structural analysis and comparisons have provided insights into the common structural features and distinct conformational changes in the interaction patterns of sulfatinib with FGFR1 and CSF-1R, shedding light on the binding mechanism and kinase selectivity of sulfatinib. Guided by the structure-based drug design, we provided valuable perspectives for the rational design of dual- or multi-target FGFR inhibitors with enhanced CSF-1R selectivity and gatekeeper mutation sensitivity. These findings pave the way for future advancement in developing more effective therapies targeting FGFR and CSF-1R.
Methods
Expression and purification of the human FGFR kinase domain
FGFRs were prepared as previously described6,34,44,45. Briefly, FGFR1 (residues 458–765), FGFR2 (residues 458–768), FGFR3 (residues 450–758) as well as FGFR4 (residues 445–753), and their mutants, FGFR1C584S, FGFR1V561M, FGFR2V564I, FGFR2V564F and FGFR3V555M, were cloned into a modified pET28a vector in frame with an N-terminal PreScission-cleavable 6×His tag and expressed in E. coli BL21 Rosetta cells. For crystallization, FGFR1C584S was co-expressed with untagged YOPH to induce non-phosphorylated proteins. The harvested cell pellets were lysed in a buffer containing 20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 20 mM imidazole and 0.5 mM TCEP, and FGFRs were purified over Ni-NTA resin followed by enzymatic digestion with PreScission for 6×His-tag cleavage and further purified by anion exchange chromatography (GE Healthcare). For crystallization, FGFR1C584S was further purified by gel filtration chromatography (GE Healthcare). The proteins were concentrated to 5–16 mg/mL and stored at −80 °C.
Expression and purification of the human CSF-1R kinase domain
Wild-type CSF-1R (residues 542–919, △696–741, C677T, C830S, C907T) and CSF-1RT663I carrying an N-terminal 6×His tag was cloned into a pFastBac expression vector, and was expressed in SF9 cells using the Invitrogen Bac-to-Bac Baculovirus Expression System41. For crystallization, CSF-1R was co-expressed with N-terminal GST-tagged YOPH to induce non-phosphorylated proteins. The harvested cell pellets were lysed by sonication in lysis buffer consisting of 40 mM K-phosphate, pH 8.0, 200 mM NaCl, 0.5 mM PMSF, and protease inhibitor mixture (EDTA-free, Roche). After centrifugation, the clarified cell lysate was supplemented with 20 mM imidazole, and incubated with Ni-NTA beads (GE Healthcare). The Ni-NTA bead-bound CSF-1R protein was eluted using 25 mM HEPES, pH 7.0, 150 mM NaCl and 200 mM imidazole, and was subsequently supplemented with 10 mM DTT after elution. CSF-1R was further purified using a cation exchange column (GE Healthcare) followed by a Superdex200 gel filtration column (GE Healthcare) equilibrated in 20 mM HEPES, pH 7.0, 150 mM NaCl and 10 mM DTT.
Crystallization and structure determination
The FGFR1/sulfatinib complex was obtained by micro-seeding as previously described34. FGFR1 was diluted 1:1 with reservoir solution of 18% (w/v) PEG 8000, 0.2 M LiSO4, and 0.1 M MES, pH 6.5, using the hanging drop vapor diffusion method. The apo FGFR1 crystals were allowed to grow for at least one week at 4 °C. For micro-seeding, FGFR1 was incubated with sulfatinib at a ratio of 1:2 at 4 °C overnight, and diluted 1:1 with the same reservoir solution as that of apo FGFR1 with the addition of apo FGFR1 microcrystals. The CSF-1R protein, at a concentration of ~10 mg/mL, was incubated with 1 mM sulfatinib before suspending over a reservoir solution of 0.1 M Tris, pH 7.5, 0.2 M MgCl2 and 20% (w/v) PEG 8000 at 4 °C. Both FGFR1/sulfatinib and CSF-1R/sulfatinib crystals were cryoprotected using their reservoir solution supplemented with 20% glycerol, followed by flash-freezing with liquid nitrogen.
X-ray diffraction data of CSF-1R/sulfatinib crystals were collected at the BL19U1 beamline of Shanghai Synchrotron Radiation Facility (SSRF), while FGFR1/sulfatinib-crystal datasets were collected in our lab with HKL3000 for data processing and scaling46. The structures were solved by molecular replacement using Phaser with the search model of CSF-1R/PLX-647OME (PDB 4HW7) for CSF-1R/sulfatinib and FGFR1/pemigatinib (PDB 7WCL) for FGFR1/sulfatinib32,34,41. Further, the models were refined with Phenix. refine and build with Coot46. The protein–ligand interactions were assessed by LigPlot+ and presented by PyMOL47,48. The structures have been deposited in the Protein Data Bank (PDB 8JOT for CSF-1R/sulfatinib and 8JMZ for FGFR1/sulfatinib).
Kinase inhibition assay
The inhibition by sulfatinib was determined by the measurement of kinase activities following the ADP-Glo assay kit (Promega)6,44,45. Assays were performed in 384-well plates using sulfatinib (triple dilution method), proteins (0.025–0.2 μM), and poly (4:1 Glu, Tyr) peptides (Abcam, UK) (1 mg/mL) in assay buffer (40 mM Tris-HCl, pH 7.5, 20 mM NaCl, 20 mM MgCl2, 1 mM TCEP, 0.1 mg/mL BSA, and 4% DMSO). The reaction was initiated by the addition of ATP into the assay mixture, and then ADP-Glo was added to terminate the reaction and deplete the remaining ATP. Following the addition of detection reagent to convert the generated ADP to ATP, the luminescence was detected by a plate reader (Perkin Elmer). Samples were run in triplicate and the IC50 values were calculated by log[Inhibitor] versus kinase activity ratio in GraphPad Prism 8.0.
Molecular modeling
Computational docking was conducted to evaluate the binding models of sulfatinib with CSF-1R mirroring our solved FGFR1-sulfatinib-like and CSF-1R-sulfatinib-like binding modes. Sulfatinib was redocked into CSF-1R (PDB 8JOT). Gasteiger charges and polar hydrogen were assigned by AutoDock Tools49. Molecular docking was performed with a flexible protein and a flexible ligand. A docking grid was built with the dimensions of 62*40*40 points in the x-, y-, and z-axis directions, which incorporated both FGFR1-bound and CSF-1R-bound sulfatinib conformations. Likewise, PRN1371 was docked into CSF-1R (PDB 3LCD)50. Additionally, the structural models of FGFR and CSF-1R gatekeeper mutations in complex with sulfatinib were prepared by protein mutagenesis based on our solved FGFR1 and CSF-1R structures.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The coordinates and structure factors are deposited in the Protein Data Bank under the accession codes 8JMZ (FGFR1/sulfatinib complex) and 8JOT (CSF-1R/sulfatinib complex). The validation reports are available in Supplementary Data 1 and 2. All other relevant data supporting the key findings of this study are available within the article and its Supplementary Information files. A reporting summary is available as a Supplementary Information file.
References
Yue, S. et al. FGFR-TKI resistance in cancer: current status and perspectives. J. Hematol. Oncol. 14, 23 (2021).
Babina, I. S. & Turner, N. C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 17, 318–332 (2017).
Turner, N. & Grose, R. Fibroblast growth factor signalling: from development to cancer. Nat. Rev. Cancer 10, 116–129 (2010).
Krook, M. A. et al. Fibroblast growth factor receptors in cancer: genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br. J. Cancer 124, 880–892 (2021).
Patani, H. et al. Landscape of activating cancer mutations in FGFR kinases and their differential responses to inhibitors in clinical use. Oncotarget 7, 24252–24268 (2016).
Qu, L. et al. Structural insights into the potency and selectivity of covalent pan-FGFR inhibitors. Commun. Chem. 5, 5 (2022).
Dieci, M. V., Arnedos, M., Andre, F. & Soria, J. C. Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discov. 3, 264–279 (2013).
Sun, D. et al. Dual-target kinase drug design: Current strategies and future directions in cancer therapy. Eur. J. Med. Chem. 188, 112025 (2020).
Liang, Q. et al. Recent advances of dual FGFR inhibitors as a novel therapy for cancer. Eur. J. Med. Chem. 214, 113205 (2021).
Bergers, G. & Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 8, 592–603 (2008).
Xu, J. et al. Surufatinib in advanced extrapancreatic neuroendocrine tumours (SANET-ep): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 21, 1500–1512 (2020).
Xu, J. et al. Surufatinib in advanced pancreatic neuroendocrine tumours (SANET-p): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 21, 1489–1499 (2020).
Xu, J. et al. Surufatinib in advanced well-differentiated neuroendocrine tumors: a multicenter, single-arm, open-label, phase Ib/II Trial. Clin. Cancer Res. 25, 3486–3494 (2019).
Syed, Y. Y. Surufatinib: first approval. Drugs 81, 727–732 (2021).
Chitu, V. & Stanley, E. R. Colony-stimulating factor-1 in immunity and inflammation. Curr. Opin. Immunol. 18, 39–48 (2006).
Morrison, C. Immuno-oncologists eye up macrophage targets. Nat. Rev. Drug Discov. 15, 373–374 (2016).
Mantovani, A., Marchesi, F., Malesci, A., Laghi, L. & Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399–416 (2017).
Giavazzi, R. et al. Distinct role of fibroblast growth factor-2 and vascular endothelial growth factor on tumor growth and angiogenesis. Am. J. Pathol. 162, 1913–1926 (2003).
Liu, X. J. et al. Recent development of multi-target VEGFR-2 inhibitors for the cancer therapy. Bioorg. Chem. 133, 106425 (2023).
Xu, J. M. et al. Sulfatinib, a novel kinase inhibitor, in patients with advanced solid tumors: results from a phase I study. Oncotarget 8, 42076–42086 (2017).
Guagnano, V. et al. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamin o]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J. Med. Chem. 54, 7066–7083 (2011)..
Taylor, S. S. & Kornev, A. P. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem. Sci. 36, 65–77 (2011).
Kornev, A. P., Taylor, S. S. & Ten Eyck, L. F. A helix scaffold for the assembly of active protein kinases. Proc. Natl Acad. Sci. USA 105, 14377–14382 (2008).
Hu, J. et al. Kinase regulation by hydrophobic spine assembly in cancer. Mol. Cell Biol. 35, 264–276 (2015).
Smith, B. D. et al. Vimseltinib: a precision CSF1R therapy for tenosynovial giant cell tumors and diseases promoted by macrophages. Mol. Cancer Ther. 20, 2098–2109 (2021).
Weisberg, E. et al. Discovery of a small-molecule type II inhibitor of wild-type and gatekeeper mutants of BCR-ABL, PDGFRalpha, Kit, and Src kinases: novel type II inhibitor of gatekeeper mutants. Blood 115, 4206–4216 (2010).
Gild, M. L., Bullock, M., Robinson, B. G. & Clifton-Bligh, R. Multikinase inhibitors: a new option for the treatment of thyroid cancer. Nat. Rev. Endocrinol. 7, 617–624 (2011).
Roskoski, R. Jr. Properties of FDA-approved small molecule protein kinase inhibitors: a 2023 update. Pharm. Res 187, 106552 (2023).
Vodopivec, D. M. & Hu, M. I. RET kinase inhibitors for RET-altered thyroid cancers. Ther. Adv. Med Oncol. 14, 17588359221101691 (2022).
Peng, C., Rabold, K., Netea, M. G., Jaeger, M. & Netea-Maier, R. T. Influence of lenvatinib on the functional reprogramming of peripheral myeloid cells in the context of non-medullary thyroid carcinoma. Pharmaceutics 15, https://doi.org/10.3390/pharmaceutics15020412 (2023).
Petrazzuolo, A., Maiuri, M. C., Zitvogel, L., Kroemer, G. & Kepp, O. Trial Watch: combination of tyrosine kinase inhibitors (TKIs) and immunotherapy. Oncoimmunology 11, 2077898 (2022).
Sohl, C. D., Ryan, M. R., Luo, B., Frey, K. M. & Anderson, K. S. Illuminating the molecular mechanisms of tyrosine kinase inhibitor resistance for the FGFR1 gatekeeper mutation: the Achilles’ heel of targeted therapy. ACS Chem. Biol. 10, 1319–1329 (2015).
Gibbons, D. L., Pricl, S., Kantarjian, H., Cortes, J. & Quintas-Cardama, A. The rise and fall of gatekeeper mutations? The BCR-ABL1 T315I paradigm. Cancer 118, 293–299 (2012).
Lin, Q. et al. Characterization of the cholangiocarcinoma drug pemigatinib against FGFR gatekeeper mutants. Commun. Chem. 5, 100 (2022).
Brameld, K. A. et al. Discovery of the Irreversible Covalent FGFR Inhibitor 8-(3-(4-Acryloylpiperazin-1-yl)propyl)-6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)pyrido[2,3-d]pyrimidin-7(8H)-one (PRN1371) for the Treatment of Solid Tumors. J. Med. Chem. 60, 6516–6527 (2017).
Caldwell, T. M. et al. Discovery of vimseltinib (DCC-3014), a highly selective CSF1R switch-control kinase inhibitor, in clinical development for the treatment of Tenosynovial Giant Cell Tumor (TGCT). Bioorg. Med Chem. Lett. 74, 128928 (2022).
Caldwell, T. M. et al. Discovery of acyl ureas as highly selective small molecule CSF1R kinase inhibitors. Bioorg. Med. Chem. Lett. 74, 128929 (2022).
Ikegashira, K. et al. Discovery of a novel azetidine scaffold for colony stimulating factor-1 receptor (CSF-1R) Type II inhibitors by the use of docking models. Bioorg. Med Chem. Lett. 29, 115–118 (2019).
Tap, W. D. et al. Structure-guided blockade of CSF1R kinase in tenosynovial giant-cell tumor. N. Engl. J. Med 373, 428–437 (2015).
Spangenberg, E. et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 10, 3758 (2019).
Zhang, C. et al. Design and pharmacology of a highly specific dual FMS and KIT kinase inhibitor. Proc. Natl Acad. Sci. USA 110, 5689–5694 (2013).
Vijayan, R. S. et al. Conformational analysis of the DFG-out kinase motif and biochemical profiling of structurally validated type II inhibitors. J. Med Chem. 58, 466–479 (2015).
O’Hare, T. et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 16, 401–412 (2009).
Zhou, Z. et al. Characterization of FGF401 as a reversible covalent inhibitor of fibroblast growth factor receptor 4. Chem. Commun. (Camb.) 55, 5890–5893 (2019).
Rezende Miranda, R. et al. Development of a potent and specific FGFR4 inhibitor for the treatment of hepatocellular carcinoma. J. Med Chem. 63, 11484–11497 (2020).
Adams, P. D. et al. The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106 (2011).
Laskowski, R. A. & Swindells, M. B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model 51, 2778–2786 (2011).
Schrödinger, L. L. C. The PyMOL molecular graphics system. Version 1, 8 (2015).
Morris, G. M. et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput Chem. 30, 2785–2791 (2009).
Meyers, M. J. et al. Structure-based drug design enables conversion of a DFG-in binding CSF-1R kinase inhibitor to a DFG-out binding mode. Bioorg. Med Chem. Lett. 20, 1543–1547 (2010).
Acknowledgements
The present study was financially supported by the Natural Science Foundation of China (No. 81974074, 82172654 and 82202920), Hunan Provincial Science and Technology Department (2021 RC4012), Natural Science Foundation of Hunan Province (2021JJ40961) and Graduate Innovation Projects of Central South University (2022ZZTS0839). We thank the staff from the Shanghai Synchrotron Radiation Facility (SSRF) beamline BL17U/BL19U/BL02U/BL10U/BL17B for assistance during diffraction data collection.
Author information
Authors and Affiliations
Contributions
Q.L. performed the experiments; S.D. and L.Q. performed data collection and structure determination. H.L., M.G., and H.W. analyzed the data. Q.L., X.C., and Y.C. prepared the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Xiaoyun Lu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, Q., Dai, S., Qu, L. et al. Structural basis and selectivity of sulfatinib binding to FGFR and CSF-1R. Commun Chem 7, 3 (2024). https://doi.org/10.1038/s42004-023-01084-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-023-01084-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
