
Abstract
The depletion of fossil resources as well as environmental concerns contribute to an increasing focus on finding more sustainable approaches for the synthesis of polymeric materials. In this work, a synthesis route towards non-isocyanate polyurethanes (NIPUs) using renewable starting materials is presented. Based on the terpenes limonene and carvone as renewable resources, five-membered cyclic carbonates are synthesized and ring-opened with allylamine, using thiourea compounds as benign and efficient organocatalysts. Thus, five renewable AA monomers are obtained, bearing one or two urethane units. Taking advantage of the terminal double bonds of these AA monomers, step-growth thiol-ene polymerization is performed using different dithiols, to yield NIPUs with molecular weights of above 10 kDa under mild conditions. Variation of the dithiol and amine leads to polymers with different properties, with Mn of up to 31 kDa and Tg’s ranging from 1 to 29 °C.
Similar content being viewed by others
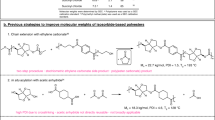


Introduction
With a current global market volume of 25 million tons1, polyurethanes are an indispensable class of polymers with versatile applications in our daily life, such as coatings, foams and adhesives2. With regard to several hazards related to their production, including the use of toxic phosgene3 for the synthesis of isocyanates, which are hazardous themselves4, scientific effort within the last years has been put increasingly into the development of isocyanate-free routes to polyurethanes5,6,7,8,9. One possibility to access these non-isocyanate polyurethanes (NIPUs) is the ring-opening of cyclic carbonates with amines, as was reviewed extensively10,11.
Apart from the development towards less hazardous synthesis routes, the use of renewable feedstock for the production of polymeric materials, including polyurethanes, is highly desirable with regard to overall sustainable procedures12,13,14. Suitable monomers can be obtained from different renewable resources, such as oleochemicals15,16, tannin17 or terpenes18,19,20. As an example, cyclic carbonates derived from epoxidized soybean oil have been used for the synthesis of a variety of polymeric materials containing urethane units21,22,23.
Due to their structural diversity and their occurrence as waste products in the chemical industry24,25,26, terpenes have gained increasing research interest for the production of fine chemicals27, pharmaceutical products28 and polymeric materials29,30,31,32,33,34,35. In works by the group of Mülhaupt36,37 as well as by Della Monica and Kleij et al.38, the use of terpene-derived dicarbonates as AA monomers for the step-growth synthesis of NIPUs was investigated. However, in all cases, oligomers with limited molar masses were obtained due to viscosity reasons, thus requiring further reaction steps for the use of these pre-polymers.
The ring-opening of cyclic carbonates with amines can be catalyzed by the addition of Lewis acids39 and also by organocatalysts such as 1,5,7-triazabicyclo[4.4.0]dec-5-ene40,41,42, which was also already applied in polymer synthesis43. Another possible compound group that can be used for the aminolysis of cyclic carbonates are thioureas40,42,44,45, which coordinate to carbonyl groups46 and thus can activate cyclic carbonates via hydrogen bonding47,48. For efficient hydrogen bonding, electron-withdrawing substituents are beneficial, further, aromatic groups can increase the activation by preorganization49,50. As such, the 3,5-bis(trifluoromethyl) phenyl group is often used in thiourea catalysts50, but can also be substituted by other electron-withdrawing groups51,52,53,54,55. With the aim to develop more sustainable processes, it is possible to synthesize such thiourea compounds via a multicomponent reaction from an isocyanide, an amine and base-activated sulfur55,56,57. With this, thiourea organocatalysts are possibly attractive with respect to the generation of urethane moieties within polyurethanes.
A possibility to obtain polyurethanes is by already implementing the urethane motif into a monomer58. In previous work of our group59, polyurethanes were synthesized not by polyaddition of amines and cyclic carbonates, but by synthesizing monomers that contained a urethane functional group and two terminal double bonds. The urethane moieties were obtained via a Lossen rearrangement60 starting from fatty acid-based hydroxamic acids. The terminal double bonds could then react with renewable dithiols in a step-growth polymerization via thiol-ene reaction. This enabled the accessibility of renewable polyurethanes with high molar masses under comparably mild conditions.
Extending this approach, we synthesized different renewable urethane monomers from terpenes, bearing terminal double bonds for polymerization using a thiol-ene approach. To this end, we made use of previously described cyclic carbonates from terpenes in order to open them with amines containing a double bond. For the aminolysis, the effect and possible use of thiourea catalysts was investigated thoroughly.
Results
Synthesis of carbonate monomers
The epoxides 3-5 and thereof derived carbonates 6-8, respectively derived from limonene 1 and carvone 2, were synthesized on the basis of previous studies61,62,63,64. With limonene containing two double bonds, it is possible to oxidize either both double bonds62 using common epoxidizing agents or to oxidize the higher substituted double bond selectively by reaction with N-bromo succinimide (NBS) and sodium hydroxide61. Also in the case of carvone 2, due to the additional keto group when compared to limonene, the two double bonds show different reactivity, and it is possible to address them selectively62.
Based on this, the respective cyclic carbonates 6-8 were synthesized from the respective epoxides by insertion of CO2 using tetrabutylammonium chloride (TBACl) as catalyst63. Despite recent findings showing high yields for the formation of cyclic carbonates when using aluminum catalysts63, it was decided to constrain the use of catalytic substances to the halogenide for a simpler approach. Indeed, no additional catalyst was necessary for a sufficient conversion of the epoxides. The synthesis of the cyclic carbonates is shown in Fig. 1. In the case of the two double bonds of carvone 2, only compound 8 bearing an exocyclic carbonate could be isolated selectively.

After selective oxidation of the terpene double bonds, epoxides 3–5 are converted into the respective cyclic carbonates by CO2 insertion.
It must be noted that the reactivity of the endocyclic and exocyclic terpene epoxides varies depending on the substitution pattern. In limonene dioxide 4, this could be observed in the formation of a monocarbonate intermediate 7a, in which the epoxide attached to the ring was not yet converted into the carbonate (see Fig. 2).

Formation of dicarbonate 7 and monocarbonate 7a were observed by gas chromatography.
By isolating the intermediate 7a, the ratio of 7 to 7a could be determined and compared with respect to different reaction conditions (see Supplementary Table 1). As catalysts for the carbonate formation, tetrabutylammonium chloride (Supplementary Table 1, entries 1 and 4), bromide (entry 2) and iodide (entry 3) were used and the ratio of 7 to the starting material 4 and the intermediate product 7a was compared using GC-FID measurements. It was shown that, in order to obtain full conversion of the limonene dioxide 4, stirring at 130 °C for three days with TBACl as catalyst was necessary. This low reactivity of the internal epoxide was not observed in the case of 3, a difference that could be explained by a lower reactivity of 4 as well as by an increase in viscosity after the carbonation of one epoxide group in 7a, thus making longer stirring and higher temperature necessary for further carbonation towards the final product 7.
When using NBS and sodium hydroxide for the oxidation of limonene, one diastereomer of 3 is formed selectively, fitting to previous findings that the trans isomer was mainly observed61. The diastereomeric ratio was determined to be 93:7 by GC-FID and NMR spectroscopy (see Supplementary Figs. 19 and 20). During the carbonate formation, the stereocenter is retained, thus also in compound 6 one main diastereomer is observed (see Supplementary Figs. 23 and 24). On the other hand, oxidation of carvone to the epoxide 5 led to the formation of two diastereomers in nearly equimolar ratio (see Supplementary Figs. 37 and 38), which was also preserved in the carbonate formation (see Supplementary Figs. 41 and 42). For the formation of 7, a commercially available diastereomeric mixture of limonene dioxide 4 was used, and in compound 7, two isomers were observed by NMR spectroscopy in a ratio of 51:49 (see Supplementary Fig. 34).
Ring-opening of carbonates with amine
Opening of endocyclic carbonate group
To test if thiourea catalysts can effectively promote the ring opening of cyclic carbonates 6–8 with an amine, four different thiourea catalysts 9–12 were compared with respect to their capability to activate the cyclic carbonate (see Fig. 3). All of these catalysts are accessible via a multicomponent approach, which is significantly more sustainable than classic synthesis approaches, from the respective aromatic isocyanide, elemental sulfur and cyclohexyl amine. Depending on the residue on the aromatic ring, they vary in their hydrogen bonding ability, with catalysts 9 and 10 showing the strongest affinity towards carbonyl groups due to the presence of strong electron-withdrawing groups55.

The choice of substituents determines the hydrogen bonding ability of these thiourea catalysts.
Opening of the cyclic carbonates described above with amines containing a double bond leads to monomers containing a urethane moiety and two terminal double bonds, thus making them suitable building blocks for potential NIPU synthesis.
For the investigation of a possible catalytic effect upon opening the synthesized carbonates with amines, limonene monocarbonate 6 was chosen as model compound to analyze the opening to the endocyclic carbonate adjacent to the terpene ring structure separately. As nucleophile, allylamine was used. Investigation started using thiourea 9, as it is easily accessible from the commercially available isothiocyanate and was shown to be a promising compound for the activation of carbonyl compounds50.
As in the synthesis of the carbonate 6 a high conversion was achieved, the crude reaction mixture could directly be used for the urethane synthesis after washing with brine, without the need of a purification via column chromatography in between. Further, no solvent was necessary for both the carbonate and the urethane formation, thus contributing to the overall sustainability of the synthesis. The ratio of the urethane monomer 13 to the carbonate 6 was determined via GC-FID over time, as no other signals were observed, and is shown in Fig. 4 (for exact values, see Supplementary Table 2).

The GC-FID fraction of 13 is obtained by dividing the GC integral of the signal associated with 13 by the sum of the integrals of the signals assigned to 6 and 13. Only one of two formed regioisomers of 13 is shown for clarity.
It can be stated that the reaction is significantly more effective in the presence of thiourea 9, indicating a catalytic effect of the thiourea that positively influences the reaction efficiency. During purification of the urethane monomer 13, the thiourea 9 could be recovered, corresponding to 100% of the starting amount, thus further contributing to the sustainability of the presented approach. The presence of a high percentage of intact thiourea in the final reaction mixture indicates that the thiourea compound is indeed acting as a catalyst. The monomer 13 could be isolated in a yield of 85%.
The unsymmetric carbonate 6 can be opened in two ways, thus two regioisomers are possibly formed during the reaction. 2D NMR spectra confirm the presence of two regioisomers (see Supplementary Figs. 27 and 28). From GC-FID measurements, their ratio was determined to be 53:47, indicating low regioselectivity. Thus, only one of the two regioisomers of 13 is shown in Fig. 4 for clarity.
Variation of reaction conditions
To optimize the reaction conditions, equivalents and temperature were varied (see Table 1, entries 1–5). Further, alternative thiourea compounds 10–12 were tested next to compound 9, displaying a range of stronger and weaker electron-withdrawing groups (entries 6–8, Table 1).
As can be seen from entry 2 (Table 1), it is possible to reduce the amount of catalyst used, but higher yields were obtained for 5 mol%, thus further experiments were carried out using this catalyst loading. Reduction to only one equivalent of allylamine led to a lower conversion (entry 3, Table 1), probably due to the volatility of the amine. Further, the initial choice of 70 °C (entry 1, Table 1) proved to be the most suited for the investigated reaction if compared to temperatures of 60 and 80 °C (see entries 4 and 5, Table 1).
The use of alternative catalysts 10–12 also led to the formation of product, with electron withdrawing sulfone (entry 6, Table 1) and ester groups (entry 7, Table 1) resulting in higher conversions than in the case of a simple phenyl residue (entry 8, Table 1). However, catalyst 9 bearing two CF3 groups still showed the highest activity. This confirmed the choice of catalyst 9 for further experiments, yet catalysts 10 and 11 remain valid options that can be obtained more sustainably.
The investigations proved the initial reaction conditions from entry 1 (Table 1) to be the most suited for the opening of the endocyclic carbonate group. As this functionality is assumed to be the less reactive one, the same conditions were chosen for a closer look into the opening in the exocyclic carbonate groups in the terpene derivatives 7 and 8.
Opening of exocyclic carbonate groups
To analyze the influence of the thiourea catalyst on the exocyclic carbonate group found in compounds 7 and 8, the carbonate 8 based on carvone was chosen as model substrate as it does not contain an additional endocyclic carbonate group.
For the opening of the carbonate, the optimized conditions of the opening of 6 (see Table 1, entry 1) were used. The conversion was detected via GC-FID as in the case before and the comparison of the reactions with and without the presence of thiourea 9 is shown in Table 2.
In general, higher conversions to the respective urethane 14 are observed, as expected, than in the case of the endocyclic carbonate (see Table 1), which can be attributed to a lower steric hindrance. Already without the addition of thiourea, high conversions are obtained (entry 2, Table 2). Addition of thiourea does not significantly influence the conversion (see entry 1, Table 2), thus no activation is necessary to obtain the urethane monomer 14 from carbonate 8.
Similar to compound 6, the carbonate 8 can in principle be opened on two different sides, leading to regioisomers. Based on NMR data (see Supplementary Figs. 43 and 44), mainly one regioisomer of 14 is formed. This can be related to the two sides of the carbonate group in 8 differing stronger in steric demand than in the case of 6. Still, two different diastereomers are distinguishable via GC-FID and NMR measurements (see Supplementary Figs. 45 and 46).
Compound 7 bears two carbonate groups, which show different reactivities as demonstrated before. Therefore, thiourea catalyst 9 was used for the activation of especially the endocyclic carbonate. The general reaction conditions for the opening of 7 were chosen as in Table 1 (entry 1), using the double amount of allylamine and thiourea 9 (see Fig. 5).

The SEC chromatogram was measured after 2 h reaction time, still showing presence of the intermediate 15a. The signal at 20.7 min is a system peak and does not correspond to any compound found in the mixture. In the chemical equation, only one regioisomer of 15 is shown for clarity, respectively.
As intermediate, the monourethane 15a (see Fig. 5) could be isolated, confirming the observation that the endocyclic carbonate group is less reactive. The chemical similarity between the monourethane 15a and the diurethane 15 results in a difficult separation via column chromatography, requiring a gradient column that hampers the recyclability of the solvent mixture. In order to facilitate the work-up, the reaction progress was monitored over time (see Supplementary Table 3), also investigating whether it is possible to push the reaction to a quantitative conversion of 7 and 15a by adding further allylamine after a certain amount of time. The low volatility of 15 required the use of SEC instead of GC-FID for monitoring, nevertheless enabling a qualitative observation of the presence of unreacted 15a within the mixture. In entries 1b-7b (Supplementary Table 3), an additional equivalent of allylamine was added after 6 h. However, no difference in yield was observed compared to the batch where no allylamine was added at a later stage (see entries 1a–7a, Supplementary Table 3), and in both cases, full conversion of 15a was not observed. The occurring of this structurally similar intermediate, which is not straightforward to isolate from the product, represents a drawback of the bifunctional monomer 15 when compared to monomer 13. When stopping the reaction after one day, most of the intermediate was converted and after purification via column chromatography the product was isolated in 85% yield.
Regarding the regioselectivity of the reaction, an analogous trend was observed in the endocyclic and exocyclic carbonate groups as in compounds 13 and 14. The exocyclic carbonate is opened selectively on the sterically less demanding side, whereas in the case of the endocyclic carbonate, both regioisomers are formed in a ratio of 45:55 as determined by NMR measurements (see Supplementary Fig. 34). Since compound 15 was not volatile enough to analyze the product mixture via GC-FID, the ratio can be determined with less accuracy due to overlapping signals in the NMR spectrum, however, the observations match the previous findings for 13 and 14.
Fatty acid-based amine
Considering the toxicity of allylamine as well as solubility issues in the case of the substrate 15, it is desirable to look for alternative amines containing a double bond. As such, fatty acid-based derivatives are desirable, being still simple in their structure. In previous work, it was shown that fatty acid-derived undecenoic acid can be used as starting material for the synthesis of decenyl amine (see Fig. 6) via esterification, substitution with hydroxyl amine, Lossen rearrangement and subsequent saponification65. The amine was thus synthesized according to the procedure described in the literature. The introduction of this moiety into the urethane monomers was first tried using limonene monocarbonate 6 (see Fig. 6). Analogous reaction conditions as in the case of allylamine were chosen (see Table 1, entry 1). Also in this case, two equivalents of the amine were used to enable higher conversion, and thiourea 9 was added to activate the endocyclic carbonate. The monomer 18 was obtained in a yield of 55%, the carbonate opening was thus less effective in comparison to the reaction with allylamine, which can be attributed to the lower reactivity of the amine due to its higher molecular weight66.

Only one of two formed regioisomers of 18 and 19 is shown for clarity, respectively.
Further, limonene dicarbonate 7 was opened with 17 using analogous reaction conditions, yielding the diurethane monomer 19 in a yield of 15%. The low yield can be attributed to both the carbonate and the amine being less reactive than their respective counterparts, as discussed above.
Synthesis of linear NIPUs
To demonstrate a possible application of the synthesized urethane monomers for polymer synthesis, the substrates 13–15, 18, and 19 were reacted with dithiols in a step-growth thiol-ene polymerization to obtain linear NIPUs. To promote a radical formation, 2,2-dimethoxy-2-phenylacetophenone (DMPA) was added as initiator and the reaction mixture was irradiated with UV light of 365 nm.
As promising substrate, limonene mono-urethane 13 was chosen as it showed good solubility in various solvents. Among commercially available dithiols, 1,10-decanedithiol 20 was chosen for first test reactions. It contains a linear spacer that is expected to keep the polar urethane moieties at a sufficient distance to enable a certain degree of solubility. Test reactions were performed in chloroform, a solvent shown suitable in previous studies59, and 2-methyl tetrahydrofuran (2-Me-THF), which represents a less hazardous and furthermore renewable solvent. A concentration of 0.5 M was found to be a good compromise between not using too much solvent and yet dissolving the monomers well enough to enable efficient stirring. The results of the polymerization reactions are shown in Table 3 and Supplementary Fig. 2, revealing that efficient formation of polymers with molecular weights >10 kDa takes place at the chosen conditions. Both solvents led to the formation of polymers (see Table 3, entries 1 and 2), with higher molecular weights being achieved in the case of the more sustainable 2-Me-THF.
To gain insight into the reaction taking place, control reactions were carried out in absence of either the diene monomer 13, the dithiol 20, UV irradiation or initiator. The results show that both monomers as well as irradiation with UV light are necessary for the formation of polymers. In absence of DMPA, oligomeric species with a molecular weight of Mn = 5.4 kDa were observed after two days of irradiation (see Table 3, entry 4), confirming that radical addition can also take place without an initiator. However, significantly higher molecular weights were achieved when adding DMPA to the reaction mixture. A reduction of the initiator concentration to 2.5 mol% also led to the formation of polymers with Mn > 10 kDa (see Table 3, entry 3). Yet, this molecular weight is lower than when using 5 mol% initiator.
The polymer could be precipitated from the crude mixture by dropping the solution into cold methanol. The 1H NMR spectrum of the precipitated polymer (see Supplementary Fig. 6) confirmed the presence of both the terpene moiety and the aliphatic chain of the dithiol within the material. Further, the IR spectrum of the precipitated polymer (see Supplementary Fig. 11) shows the presence of characteristic signals corresponding to the present urethane and thioether moieties. Together with the performed control reactions, this undermines the assumption of NIPU formation via thiol-ene polyaddition.
The formation of NIPUs from monomer 13 was monitored over time (see Supplementary Table 4 and Supplementary Fig. 1), showing that already after 1 h a molecular weight of 11.8 kDa was achieved. After 5 h, 14.1 kDa were observed, indicating that the polymerization can possibly be stopped earlier. Nevertheless, for a comparison between the different monomers, 24 h were kept as fixed reaction time to allow for slower reactions to still be observed.
After the first promising results, the determined reaction conditions were applied for the synthesis of linear NIPUs from all urethane monomers synthesized in this work. Besides the variation of the urethane monomer, a variation of the dithiol can be a possibility to achieve different properties. As renewable alternative to 20, dithiol 22 was synthesized from limonene. A summary of the used dithiols and of the results from the polymerization reactions is shown in Table 4 and Supplementary Table 5. SEC of the obtained polymeric materials was performed (chromatograms are shown in Supplementary Figs. 3–5) to determine Mn and Ð (see Table 4), revealing mostly high molecular weights and dispersities close to 2, as expected for a step-growth polymerization. In contrast to the results achieved with monomer 13, the use of monomer 14 (entry 4, Table 4) only led to the formation of oligomeric species, which can be attributed to the reduced reactivity of the double bond in α,β-position to the carbonyl group with respect to radical thiol-ene additions. The urethane monomers 15, 18 and 19 could successfully be used for the synthesis of NIPUs with Mn > 10 kDa. When compared to monomer 13, the polymer P6 derived from monomer 18 shows slightly higher molecular weight (entry 6, Table 4). The resulting polymer was precipitated and characterized (Supplementary Figs. 8 and 13). The observed Tg of 1 °C is significantly lower than that of polymer P1, which can be attributed to the longer alkyl chains and thus lower relative amount of urethane moieties. In the case of monomer 15, the molecular weight is limited (entry 5, Table 4), which could be related to an increased stiffness. The molecular weight was not sufficiently high for a successful precipitation; thus, the polymer could not be characterized for molecular and thermal analysis. Monomer 19 could also be successfully used for the synthesis of NIPUs. When using dithiol 20 (entry 8, Table 4), the polymer P8 with the highest molecular weight within this work of 31.2 kDa was obtained after precipitation. Its Tg of 23 °C is higher than that of P6, where the same amine was used for the carbonate opening. The higher Tg can be attributed to increased hydrogen bonding due to the presence of two urethane moieties per repeating unit. Structural characterization of P8 is found in Supplementary Figs. 9 and 14.
For a variation of the dithiol, the urethane monomer 13 was chosen as starting point. The use of dithiol 21 with a shorter chain length led to a polymer with lower molecular weight (entry 2, Table 4), which might be attributed to a less favorable structure in which the terpene units are relatively close. The resulting polymer P2 was purified and characterized (Supplementary Figs. 7 and 12), showing a Tg of 16 °C that is similar to that of P1. The renewable dithiol 22 in combination with monomer 13 did not yield a polymer with high molecular weights (entry 3, Table 4), supporting the previous assumption of close terpene moieties hampering the formation of longer polymer chains. For this reason, dithiol 22 was tested for a polymerization with monomers 18 and 19, which contain one and two additional decenyl spacers, respectively. Indeed, already the use of monomer 18 (entry 7, Table 4) with an elongated chain length compared to monomer 13 (entry 3, Table 4) led to higher molecular weight, yet not high enough for a precipitation of the polymer P7. Extending this to monomer 19 with an additional C10 chain led to the formation of NIPU P9 that could be precipitated and characterized (see Supplementary Figs. 10 and 15). As in the NMR spectrum of P9 the double bond signals are still visible as end groups, their integration can be used to calculate the molecular weight of P9 (see Supplementary Fig. 10 and Supplementary Eq. (1)). The calculated molecular weight of 11.8 kDa is in a similar range as the value from SEC measurements (see Table 4, entry 9). The observed Tg of 29 °C is higher than that of P8 with a linear dithiol, corresponding to a higher stiffness of the terpene unit.
Discussion
This work showed the application of thiourea catalysis for the functionalization of terpene-based carbonates towards urethane building blocks. The presence of a thiourea catalyst significantly improved the opening of the endocyclic carbonate groups by allylamine, whereas no activation was necessary in the case of the exocyclic carbonate structures. This enabled the access to AA monomers for the synthesis of linear NIPUs as potential application in polymer synthesis. By thiol-ene polyaddition with dithiols, NIPUs with molecular weights of up to 31 kDa were obtained, strongly depending on the structure of the respective monomers. By elongating the carbon chains within the urethane monomers, it was possible to achieve higher molecular weights and further implement a renewable dithiol from limonene. The Tg values, ranging from 1 to 29 °C, are slightly higher than those of literature-described terpene-containing NIPUs of similar molecular weight37,59,67, which can be attributed to additional OH groups59,67 or higher terpene content, respectively37.
This approach complements previous strategies of introducing urethane moieties into polymers via thiol-ene reaction59,68. It should be noted that the obtained materials contain additional thioether linkages as well as hydroxy groups in contrast to industrially used PUs. However, other works also include thioether linkages, e.g. for self-blowing NIPU foams69,70, showing the potential of such new structures. Further, this work brings forward the use of thiourea catalysis for NIPU production71, as potential strategy to activate more hindered cyclic carbonates. Although several examples have shown the potential of implementing terpene structures into polyurethanes22,36,37,38,67,72,73, their number remains limited.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and Supplementary Information. For experimental details and compound characterization data, see Supplementary Information. All other data are available from the corresponding author on reasonable request.
References
Statista. Market volume of polyurethane worldwide from 2015 to 2025, with a forecast for 2022 to 2029. https://www.statista.com/statistics/720341/global-polyurethane-market-size-forecast/ (2023).
Das, A. & Mahanwar, P. A brief discussion on advances in polyurethane applications. Adv. Ind. Eng. Polym. Res. 3, 93–101 (2020).
Slocombe, R. J., Hardy, E. E., Saunders, J. H. & Jenkins, R. L. Phosgene derivatives. the preparation of isocyanates, carbamyl chlorides and cyanuric acid. J. Am. Chem. Soc. 72, 1888–1891 (1950).
Karol, M. H. & Kramarik, J. A. Phenyl isocyanate is a potent chemical sensitizer. Toxicol. Lett. 89, 139–146 (1996).
Rokicki, G., Parzuchowski, P. G. & Mazurek, M. Non-isocyanate polyurethanes: synthesis, properties, and applications. Polym. Adv. Technol. 26, 707–761 (2015).
Maisonneuve, L., Lamarzelle, O., Rix, E., Grau, E. & Cramail, H. Isocyanate-free routes to polyurethanes and poly(hydroxy urethane)s. Chem. Rev. 115, 12407–12439 (2015).
Gomez-Lopez, A., Elizalde, F., Calvo, I. & Sardon, H. Trends in non-isocyanate polyurethane (NIPU) development. Chem. Commun. 57, 12254–12265 (2021).
Stachak, P., Łukaszewska, I., Hebda, E. & Pielichowski, K. Recent advances in fabrication of non-isocyanate polyurethane-based composite materials. Materials 14, 3497 (2021).
El Khezraji, S. et al. Recent progress of non-isocyanate polyurethane foam and their challenges. Polymers 15, 254 (2023).
Ghasemlou, M., Daver, F., Ivanova, E. P. & Adhikari, B. Bio-based routes to synthesize cyclic carbonates and polyamines precursors of non-isocyanate polyurethanes: a review. Eur. Polym. J. 118, 668–684 (2019).
Bobbink, F. D., van Muyden, A. P. & Dyson, P. J. En route to CO2-containing renewable materials: catalytic synthesis of polycarbonates and non-isocyanate polyhydroxyurethanes derived from cyclic carbonates. Chem. Commun. 55, 1360–1373 (2019).
Gandini, A. & Lacerda, T. M. From monomers to polymers from renewable resources: recent advances. Prog. Polym. Sci. 48, 1–39 (2015).
Zhu, Y., Romain, C. & Williams, C. K. Sustainable polymers from renewable resources. Nature 540, 354–362 (2016).
Tenorio-Alfonso, A., Sánchez, M. C. & Franco, J. M. A review of the sustainable approaches in the production of bio-based polyurethanes and their applications in the adhesive field. J. Polym. Environ. 28, 749–774 (2020).
Meier, M. A. R. Plant-oil-based polyamides and polyurethanes: toward sustainable nitrogen-containing thermoplastic materials. Macromol. Rapid Commun. 40, e1800524 (2019).
Sawpan, M. A. Polyurethanes from vegetable oils and applications: a review. J. Polym. Res. 25, 184 (2018).
Aristri, M. A. et al. Recent developments in lignin- and tannin-based non-isocyanate polyurethane resins for wood adhesives—a review. Appl. Sci. 11, 4242 (2021).
Winnacker, M. & Rieger, B. Recent progress in sustainable polymers obtained from cyclic terpenes: synthesis, properties, and application potential. ChemSusChem 8, 2455–2471 (2015).
Winnacker, M. In Advances in Polymer Science 1–30 (Springer Berlin Heidelberg, 2022).
Scheelje, F. C. M., Destaso, F. C., Cramail, H. & Meier, M. A. R. Nitrogen‐containing polymers derived from terpenes: possibilities and limitations. Macromol. Chem. Phys. 224, 2200403 (2023).
Hu, S., Chen, X. & Torkelson, J. M. Biobased reprocessable polyhydroxyurethane networks: full recovery of crosslink density with three concurrent dynamic chemistries. ACS Sustain. Chem. Eng. 7, 10025–10034 (2019).
Liu, X. et al. Fully bio-based polyhydroxyurethanes with a dynamic network from a terpene derivative and cyclic carbonate functional soybean oil. ACS Sustain. Chem. Eng. 9, 4175–4184 (2021).
Poussard, L. et al. Non-isocyanate polyurethanes from carbonated soybean oil using monomeric or oligomeric diamines to achieve thermosets or thermoplastics. Macromolecules 49, 2162–2171 (2016).
Paggiola, G. et al. Can bio-based chemicals meet demand? Global and regional case-study around citrus waste-derived limonene as a solvent for cleaning applications. Biofuels. Bioprod. Biorefin. 10, 686–698 (2016).
Pourbafrani, M., Forgács, G., Horváth, I. S., Niklasson, C. & Taherzadeh, M. J. Production of biofuels, limonene and pectin from citrus wastes. Bioresour. Technol. 101, 4246–4250 (2010).
Zebec, Ž., Poberžnik, M., Scrutton, N. S. & Lobnik, A. Enzymatic Hydrolysis of Textile and Cardboard Waste as a Glucose Source for the Production of Limonene in Escherichia coli. Life 12, 1423 (2022).
Schwab, W., Fuchs, C. & Huang, G.-C. Transformation of terpenes into fine chemicals. Eur. J. Lipid Sci. Technol. 115, 3–8 (2013).
Zielińska-Błajet, M., Pietrusiak, P. & Feder-Kubis, J. Selected monocyclic monoterpenes and their derivatives as effective anticancer therapeutic agents. Int. J. Mol. Sci. 22, 4763 (2021).
Della Monica, F. & Kleij, A. W. From terpenes to sustainable and functional polymers. Polym. Chem. 11, 5109–5127 (2020).
Louisy, E., Khodyrieva, V., Olivero, S., Michelet, V. & Mija, A. Use of limonene epoxides and derivatives as promising monomers for biobased polymers. ChemPlusChem 87, e202200190 (2022).
Palenzuela, M., Sánchez-Roa, D., Damián, J., Sessini, V. & Mosquera, M. E. In Advances in Organometallic Chemistry (Elsevier), Vol. 75 55–93 (Elsevier, 2021).
Sahu, P., Bhowmick, A. K. & Kali, G. Terpene based elastomers: synthesis, properties, and applications. Processes 8, 553 (2020).
Thomsett, M. R., Storr, T. E., Monaghan, O. R., Stockman, R. A. & Howdle, S. M. Progress in the synthesis of sustainable polymers from terpenes and terpenoids. Green Mater. 4, 115–134 (2016).
Wilbon, P. A., Chu, F. & Tang, C. Progress in renewable polymers from natural terpenes, terpenoids, and rosin. Macromol. Rapid Commun. 34, 8–37 (2013).
Winnacker, M. Pinenes: abundant and renewable building blocks for a variety of sustainable polymers. Angew. Chem. Int. Ed. 57, 14362–14371 (2018).
Bähr, M., Bitto, A. & Mülhaupt, R. Cyclic limonene dicarbonate as a new monomer for non-isocyanate oligo- and polyurethanes (NIPU) based upon terpenes. Green Chem. 14, 1447 (2012).
Schimpf, V., Ritter, B. S., Weis, P., Parison, K. & Mülhaupt, R. High purity limonene dicarbonate as versatile building block for sustainable non-isocyanate polyhydroxyurethane thermosets and thermoplastics. Macromolecules 50, 944–955 (2017).
Maquilón, C. et al. Renewable beta-elemene based cyclic carbonates for the preparation of oligo(hydroxyurethane)s. ChemSusChem 15, e202201123 (2022).
Lombardo, V. M. et al. Cooperative catalysis of cyclic carbonate ring opening: application towards non-isocyanate polyurethane materials. Eur. J. Org. Chem. 2015, 2791–2795 (2015).
Blain, M. et al. Rational investigations in the ring opening of cyclic carbonates by amines. Green Chem. 16, 4286–4291 (2014).
Alves, M. et al. DFT investigation of the reaction mechanism for the guanidine catalysed ring-opening of cyclic carbonates by aromatic and alkyl-amines. RSC Adv. 7, 18993–19001 (2017).
Lambeth, R. H. et al. Nonisocyanate polyurethanes from six-membered cyclic carbonates: catalysis and side reactions. J. Appl. Polym. Sci. 134, 44941 (2017).
Yu, A. Z., Setien, R. A., Sahouani, J. M., Docken, J. & Webster, D. C. Catalyzed non-isocyanate polyurethane (NIPU) coatings from bio-based poly(cyclic carbonates). J. Coat. Technol. Res. 16, 41–57 (2019).
Lambeth, R. H. & Henderson, T. J. Organocatalytic synthesis of (poly)hydroxyurethanes from cyclic carbonates and amines. Polymer 54, 5568–5573 (2013).
Blain, M. et al. Urea- and thiourea-catalyzed aminolysis of carbonates. ChemSusChem 9, 2269–2272 (2016).
Schreiner, P. R. & Wittkopp, A. H-bonding additives act like Lewis acid catalysts. Org. Lett. 4, 217–220 (2002).
Schreiner, P. R. Metal-free organocatalysis through explicit hydrogen bonding interactions. Chem. Soc. Rev. 32, 289–296 (2003).
Takemoto, Y. Recognition and activation by ureas and thioureas: stereoselective reactions using ureas and thioureas as hydrogen-bonding donors. Org. Biomol. Chem. 3, 4299–4306 (2005).
Wittkopp, A. & Schreiner, P. R. Metal‐free, noncovalent catalysis of Diels–Alder reactions by neutral hydrogen bond donors in organic solvents and in water. Chem. Eur. J. 9, 407–414 (2003).
Lippert, K. M. et al. Hydrogen-bonding thiourea organocatalysts: the privileged 3,5-bis(trifluoromethyl)phenyl group. Eur. J. Org. Chem. 2012, 5919–5927 (2012).
Robak, M. T., Trincado, M. & Ellman, J. A. Enantioselective aza-Henry reaction with an N-sulfinyl urea organocatalyst. J. Am. Chem. Soc. 129, 15110–15111 (2007).
Ban, S., Zhu, X., Zhang, Z., Xie, H. & Li, Q. Benzoylthiourea-pyrrolidine as another bifunctional organocatalyst: highly enantioselective michael addition of cyclohexanone to nitroolefins. Eur. J. Org. Chem. 2013, 2977–2980 (2013).
Fan, Y. & Kass, S. R. Electrostatically enhanced thioureas. Org. Lett. 18, 188–191 (2016).
Ganesh, M. & Seidel, D. Catalytic enantioselective additions of indoles to nitroalkenes. J. Am. Chem. Soc. 130, 16464–16465 (2008).
Nickisch, R., Gabrielsen, S. M. & Meier, M. A. R. Novel access to known and unknown thiourea catalyst via a multicomponent‐reaction approach. ChemistrySelect 5, 11915–11920 (2020).
Lipp, M., Dallacker, F. & Köcker, I. M. z. Über die addition von schwefel und selen an isonitrile. Mh. Chem. 90, 41–48 (1959).
Nguyen, T. B., Ermolenko, L. & Al-Mourabit, A. Three-component reaction between isocyanides, aliphatic amines and elemental sulfur: preparation of thioureas under mild conditions with complete atom economy. Synthesis 46, 3172–3179 (2014).
Kirchberg, A., Khabazian Esfahani, M., Röpert, M.-C., Wilhelm, M. & Meier, M. A. R. Sustainable synthesis of non‐isocyanate polyurethanes based on renewable 2,3‐butanediol. Macromol. Chem. Phys. 223, 2200010 (2022).
Filippi, L. & Meier, M. A. R. Fully renewable non-isocyanate polyurethanes via the lossen rearrangement. Macromol. Rapid Commun. 42, 2000440 (2021).
Kreye, O., Wald, S. & Meier, M. A. R. Introducing catalytic lossen rearrangements: sustainable access to carbamates and amines. Adv. Synth. Catal. 355, 81–86 (2013).
Hauenstein, O., Reiter, M., Agarwal, S., Rieger, B. & Greiner, A. Bio-based polycarbonate from limonene oxide and CO2 with high molecular weight, excellent thermal resistance, hardness and transparency. Green Chem. 18, 760–770 (2016).
Löser, P. S., Rauthe, P., Meier, M. A. R. & Llevot, A. Sustainable catalytic rearrangement of terpene-derived epoxides: towards bio-based biscarbonyl monomers. Philos. Trans. R. Soc. A 378, 20190267 (2020).
La Cruz-Martínez et al. Synthesis of bio-derived cyclic carbonates from renewable resources. ACS Sustain. Chem. Eng. 7, 20126–20138 (2019).
Dannecker, P.-K. & Meier, M. A. R. Facile and sustainable synthesis of erythritol bis(carbonate), a valuable monomer for non-isocyanate polyurethanes (NIPUs). Sci. Rep. 9, 9858 (2019).
Kreye, O., Kugele, D., Faust, L. & Meier, M. A. R. Divergent dendrimer synthesis via the Passerini three-component reaction and olefin cross-metathesis. Macromol. Rapid Commun. 35, 317–322 (2014).
Diakoumakos, C. D. & Kotzev, D. L. Non‐isocyanate‐based polyurethanes derived upon the reaction of amines with cyclocarbonate resins. Macromol. Symp. 216, 37–46 (2004).
Firdaus, M. & Meier, M. A. R. Renewable polyamides and polyurethanes derived from limonene. Green Chem. 15, 370–380 (2013).
Merckle, D., Constant, E. & Weems, A. C. Linalool derivatives for natural product-based 4D printing resins. ACS Sustain. Chem. Eng. 9, 12213–12222 (2021).
Purwanto, N. S., Chen, Y. & Torkelson, J. M. Reprocessable, bio-based, self-blowing non-isocyanate polyurethane network foams from cashew nutshell liquid. ACS Appl. Polym. Mater. 5, 6651–6661 (2023).
Purwanto, N. S., Chen, Y., Wang, T. & Torkelson, J. M. Rapidly synthesized, self-blowing, non-isocyanate polyurethane network foams with reprocessing to bulk networks via hydroxyurethane dynamic chemistry. Polymer 272, 125858 (2023).
Cornille, A. et al. Room temperature flexible isocyanate-free polyurethane foams. Eur. Polym. J. 84, 873–888 (2016).
Blattmann, H. & Mülhaupt, R. Multifunctional β-amino alcohols as bio-based amine curing agents for the isocyanate- and phosgene-free synthesis of 100% bio-based polyhydroxyurethane thermosets. Green Chem. 18, 2406–2415 (2016).
Gupta, R. K., Ionescu, M., Radojcic, D., Wan, X. & Petrovic, Z. S. Novel renewable polyols based on limonene for rigid polyurethane foams. J. Polym. Environ. 22, 304–309 (2014).
Acknowledgements
The authors want to acknowledge Michelle Karsten and Elsa Brudy for synthetic support and the analytical department of the Institute of Organic Chemistry (IOC) at KIT and Dr. Andreas Rapp, Despina Savvidou, Tanja Ohmer-Scherrer, and Lara Hirsch for their support with the NMR and ESI measurements.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
F.C.M.S. and M.A.R.M. conceived and designed the project. F.C.M.S. is the lead author of the manuscript and carried out and directed the experiments and synthesis of the compounds. M.A.R.M. directed the work and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Malte Winnacker, Di Cai, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Scheelje, F.C.M., Meier, M.A.R. Non-isocyanate polyurethanes synthesized from terpenes using thiourea organocatalysis and thiol-ene-chemistry. Commun Chem 6, 239 (2023). https://doi.org/10.1038/s42004-023-01041-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-023-01041-x
This article is cited by
-
Organomediated polymerization
Communications Chemistry (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
