
Abstract
Morpholines and morpholinones are important building blocks in organic synthesis and pharmacophores in medicinal chemistry, however, C3-disubstituted morpholines/morpholinones are extremely difficult to access. Here we show the ZnCl2-catalyzed cyclizative 1,2-rearrangement for the efficient synthesis of morpholinones bearing aza-quaternary stereocenters. A series of structurally diverse C3-disubstituted morpholin-2-ones which are difficultly accessible by existing methods were efficiently constructed from readily available two achiral linear compounds. Notably, mechanistic studies reveal that this reaction proceeds via an unusual sequence of direct formal [4 + 2] heteroannulation regioselectively delivering specific α-iminium/imine hemiacetals followed by a 1,2-esters or amides shift process, which is different from the reported mechanism of the aza-benzilic ester rearrangements.
Similar content being viewed by others
Introduction
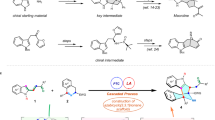
C-substituted morpholines and morpholinones represent important building blocks1,2 in organic synthesis and pharmacophores in medicinal chemistry, such as commercially available drugs of Finafloxacin3, Plk-2 inhibitor4 and Aprepitant5 (Fig. 1). Consequently, efficient access to these structural targets have been in great demand. Thus far, there have been a number of methods for the synthesis of C-functionalized morpholines or morpholinones6,7,8 benefiting from metal-catalyzed cyclization reactions9,10,11,12,13,14, allylic alkylation15, hydrogenations16,17 or other multi-step transformations18,19,20. On the contrary, approaches for the catalytic preparation of C3-disubstituted morpholines/morpholin-2-ones are far less developed, which is due probably to the steric hindrance effect of the incoming aza-quaternary carbon and availability of the corresponding branched starting materials21,22,23. Furthermore, asymmetric synthesis of such compounds was envisioned to be even more challenging, which must overcome the formidable challenges in both reaction reactivity and stereoselectivity. In this regard, an elegant work by Carreira and coworkers dealing with ring expansion of 3-oxetanone-derived spirocycles under In(III) catalysis is noteworthy (Fig. 2A)24. Mechanistically, the newly formed aza-quaternary carbon results from the Strecker reaction25 of oxazolidine with trimethylsilyl cyanide, and a subsequent stereoselective desymmetrizing 6-exo-tet cyclization finally affords morpholine in high dr value. To the best of our knowledge, so far, this is the only example of asymmetric synthesis of morpholine derivative with an aza-quaternary carbon center which is in situ formed rather than originates from a substrate. Thus, the highly useful and efficient approaches for the de novo construction of structurally diverse C3-disubstituted morpholines/morpholin-2-ones from readily available linear compounds are highly desirable.

Chiral morpholines bearing aza-tertiary stereocenters or aza-quaternary stereocenters.

A In(III)-catalyzed synthesis of morpholine containing an aza-quaternary center by Carreira. B Aza-benzilic ester rearrangement for the construction of aza-tertiary center of morpholinone. C Construction of morpholinone bearing aza-quaternary center by ZnCl2 catalysis (this work).
Condensation of arylglyoxals with vicinal amino alcohols delivering 2-acyloxazolidines which further isomerize to morpholinones is well-known26,27,28,29,30. Recently, this reaction has been applied to the diastereoselective synthesis of morpholinones by Walczak group31. Very recently, we demonstrated an enantioselective aza-benzilic ester rearrangement for the synthesis of enantioenriched morpholinones by chiral Brønsted acid (Fig. 2B)32. In both cases, however, only morpholinones bearing aza-tertiary stereocenters have been successfully synthesized while all efforts aimed at the construction of C3-disubstituted morpholin-2-ones afford either oxazolidine intermediates or degradation at higher temperature31,32. To the best of our knowledge, 1,2-rearrangement for the construction of morpholinones bearing aza-quaternary stereocenters has not been achieved in spite of the significant synthetic importance of the resulting compounds33,34. In connection with our ongoing research interest in developing catalytic asymmetric cyclizative rearrangement (CACR)32,35, we herein report the successful realization of this endeavor, which delivers a wide range of C3-disubstituted morpholin-2-ones 3 from readily available linear 2,3-diketoesters 1 and vicinal amino alcohols 2 under the catalysis of ZnCl2 complex (Fig. 2C). Moreover, a highly diastereoselective version of such transformation has also been achieved by employing chiral vicinal amino alcohols as reaction partners. More importantly, rather than previously reported formal [4 + 1] oxazolidination/ring-expansive hemiacetalization/1,2-aryl or alkyl shift cascade (Fig. 2B)32,35, mechanistic investigations indicated that this transformation proceeded via a completely different reaction pathway consisting of an initial direct [4 + 2] cyclization (Int) followed by a 1,2-ester/amide shift process (Fig. 2C)36.
Results
Reaction development
With our on-going efforts to further examine its reactivity profile35,36, the multi-functionalizable vicinal tricarbonyl compounds37,38,39,40,41,42,43,44,45,46,47 were chosen as the reaction partner with aminoethanols. Our studies commenced with a mixture of ethyl 2,3-dioxo-3-phenylpropanoate and its hydrate (1a, 1.0 equiv), 2-((4-methoxyphenyl)amino)ethan-1-ol 2a (1.2 equiv) in 1,2-dichloroethane (c 0.1 M) at 80 °C under argon. Initially, screening of various strong Brønsted acids, such as trifluoroacetic acid, trifluoromethanesulfonic acid, and p-toluenesulfonic acid, failed to give the rearranged products (entries 1-3, Table 1). Additionally, even aqueous hydrochloric acid could not give the desired product. As typical bifunctional catalysts, chiral phosphoric acids (CPA)48,49 could not give the rearranged product 3a as well. Boron trifluoride-diethyl etherate (BF3 ∙ Et2O) was also ineffective (entry 4). Screening of a series of strong Lewis acids, such as Mg(OTf)2, InCl3, and Zn(II) complexes (entries 5-10), in the presence of tricarbonyl compound 1a and amino alcohol 2a at 80 °C resulted in the formation of the desired C3-disubstituted morpholin-2-one 3a, and ZnCl2 gave the best result (55% yield). Using ZnCl2 as catalyst, the reaction conditions were further optimized by varying the solvent (entries 11–13), catalyst loading (entry 14), and reaction temperature (entries 15–16). Overall, the best conditions consisted of performing the reaction in DCE (c 0.1 M) at 100 °C in the presence of ZnCl2 (0.2 equiv). Under these conditions, C3-disubstituted morpholin-2-one 3a was isolated in 61% yield (entry 15).
Substrate scope exploration
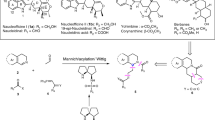
With the optimized conditions in hand, this ZnCl2-catalyzed cyclizative 1,2-rearrangement was applicable to a wide range of substrates (Fig. 3). Regarding the aryl group of 3-aryl substituted 2,3-diketoesters, the presence of electron-neutral (H, Ph), electron-donating (MeO) groups and electron-withdrawing (F, CF3, CO2Et) at para and meta positions of the phenyl ring were well tolerated (3a-3j). Unfortunately, an aryl group bearing an ortho substituent failed to give the desired product which is due probably to the steric hinderance effect. The structure of compound 3a was confirmed by X-ray crystallographic analysis (see Supplementary Data 1). In the presence catalytic amount of ZnCl2, (S)-(+)-Ibuprofen derived 2,3-diketoester could give the corresponding C3-disubstituted morpholin-2-one 3g in moderate yield. Moreover, arylcoupled Aspirin could be smoothly functionalized with the present methodology (3h). 3,4-Dichlorophenyl, 2-naphthyl, and even heterocycles, such as benzo[d][1,3]dioxole, 3-thiophene and 2-thiophene, were transferred without event (3k-3o). To our delight, with aliphatic 2,3-diketoesters (1p-1s), these rearrangement reactions efficiently afforded the corresponding product 3p-3s as well. Interestingly, instead of vicinal tricarbonyl compounds, 1,2-diketone bearing CF3 group (1t)50 could also give the desired rearranged product (3t) with decreased yield. Furthermore, 2,3-diketoesters derived from different alcohols, such as methyl, isopropyl, and benzyl, were all well-accepted substrates (3u-3w). Gratefully, 2,3-diketoamides participated in this ZnCl2-catalyzed cyclizative 1,2-rearrangement to provide the corresponding rearranged products 3x and 3y in high yields. Performing the gram scale reactions of 1y (6.78 mmol) and 2a (8.14 mmol) in the presence of ZnCl2 (20 mol%) afforded 3y without obvious erosion of yield (75%). L-(-)-Menthol derived 2,3-diketoesters gave the corresponding product 3z in good yield as well. Alternatively, other N-substituted amino alcohols bearing phenyl, 2-methylphenyl, methyl and benzyl groups were also examined, affording the corresponding products 3aa-3ad without event. Notably, amino alcohol bearing electron-withdrawing protecting group on nitrogen, such as Ts and Ns, failed to afford the desired product 3. Furthermore, N-unsubstituted amino alcohols could give the corresponding rearranged products (3ae-3af) smoothly.

[a] Conditions: 1 (0.10 mmol), 2 (0.12 mmol), ZnCl2 (0.02 mmol), DCE (c 0.1 M), sealed tube, argon, 12 h. [b] Yield of gram scale reaction.
Strategy extension
Asymmetric catalytic construction of multi-functionalized morpholin-2-ones bearing aza-quaternary stereocenters represents a formidable challenge. To further exploit the structural diversity of the products, we divert our attention to the diastereoselective synthesis of C3-disubstituted morpholin-2-ones by employing optically pure N-substituted 2-aminoethan-1-ols. Gratefully, without any modifications to the reaction conditions, this protocol was applicable to a wide range of chiral amino alcohols delivering structurally diverse products bearing multi-stereocenters in high stereoselectivities (Fig. 4). Various N-substituted 2-aminoethan-1-ol, such as (S)-2-aminopropan-1-ol, (R)-2-amino-3-phenylpropan-1-ol and (R)-2-amino-3-methylbutan-1-ol, underwent this rearrangement reaction smoothly affording the desired products 3ag-3ai in high diastereoselectivities. 2,3-Diketoamides 1aj and 1ak were also well tolerated leading to the formation of the products 3aj and 3ak in good yields with excellent stereoselectivities (>20/1 d.r.). Moreover, C2-phenyl substituted amino alcohol 1al afforded the product 3al in excellent d.r. value as well. N-benzyl substituted amino alcohol 1am delivered decreased stereoselectivity (6/1 d.r.). Subsequently, Other more challenging chiral amino alcohols were further examined. Interestingly, even N-PMP substituted (S)-1-aminopropan-2-ol 1an where the original stereocenter was far away from the newly formed aza-quaternary carbon could also afford the product 3an in good diastereoselectivity (4/1 d.r.). Substrates 1ao and 1ap derived from cyclic amino alcohols underwent the cyclizative 1,2-rearrangement without event, which furnished the corresponding products (3ao-3ap) bearing three stereocenters in excellent and good diastereoselectivities. The absolute configurations of the products 3ag, 3am, 3an and 3ao were determined to be (3S,5S), (3R,5R), (3S,6S), and (3 R,4aR,8aR), respectively (see Supplementary Data 2–5). Consequently, the absolute configurations for other morpholinones (3ah-3al and 3ap) were assigned accordingly (Fig. 4). Unfortunately, all efforts aimed at developing a catalytic enantioselective version of this rearrangement gave almost racemic products (see Supplementary Table S1 for details).

[a] Conditions: 1 (0.10 mmol), 2 (0.12 mmol), ZnCl2 (0.02 mmol), DCE (c 0.1 M), sealed tube, argon, 12 h. [b] Yield of 1.0 mmol scale.
Mechanistic studies
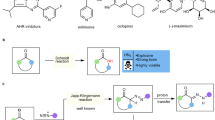
A series of control experiments were performed to gain insight into the reaction mechanism. In the presence of p-toluenesulfonic acid (p-TSA), a formal [3 + 2] reaction of 1a with 2a occurred at 80 °C affording an oxazolidine compound 4a in moderate yield (Fig. 5a). Further treating intermediate 4a with catalytic amount of p-TSA (20 mol%) gave trace of the product 3a (Fig. 5a). To our surprise, however, resubmitting 4a to the standard reaction conditions also afforded only trace of the rearranged product 3a with concurrent formation of a linear compound 5 in 43% yield and some decomposition species (Fig. 5b). As 2-aminoethan-1-ol derivatives 2 are capable of serving as bidentate ligands to Zn(II)51, we further examine the reactivity of (2a)∙ZnCl2 complex. Under the catalysis of the in situ formed (2a)∙ZnCl2 complex, the reaction of the intermediate 4a gave product 3a in very low yield (10%) and a linear compound 5 was formed as a main product (Fig. 5c). Alternatively, a set of experiments were performed with 2,3-diketoester 1a and amino alcohol 2a under the standard conditions and the yields of both 3a and 4a were detected at different time. As it is seen clearly from the diagram shown in Fig. 5d, the yield of 3a increased steadily as the reaction proceeded with the concurrent formation of the low yield of 4a. These results revealed that, in this case, oxazolidine intermediate 4a resulted from formal [4 + 1] condensation of 1a and 2a is not a major reservoir for product 3a, which is different from the reported Brønsted acid catalyzed aza-benzilic ester rearrangements31,32. Notably, during the screening of the reaction conditions, another benzilic ester rearrangement product 636 was observed in the presence of catalytic amount of Zn(OTf)2 (Fig. 6a). However, ZnCl2 complex only gave a trace of compound 6 (Fig. 6b). Furthermore, resubmitting 6 to our standard reaction conditions failed to give the desired product 3a (Fig. 6c). These results indicate that an intramolecular aza-cyclization to the final product 3a via a possible in situ formed carbocation intermediate 6’ derived from dehydration of 6 could be excluded. Subsequently, we realized that, at the first step of this tandem reaction, a direct formal [4 + 2] reaction might be competitive to the [4 + 1] cyclization of 1a and 2a. All our efforts aimed at trapping the key intermediate of the rearrangement by employing various excessive nucleophilic reagents failed to give the corresponding products. To our delight, when the reaction of 1a and N-unsubstituted amino alcohol 2ae was performed at lower temperature (60 °C), a unique α-imine hemiacetal intermediate 7 was successfully isolated in good yield (Fig. 6d). Surprisingly, the imine moiety was formed from the condensation of primary amine with the carbonyl group linked to phenyl rather than with the middle one, which is also different from the previous reports31,32,35. Notably, no oxazolidine intermediate 4ae was detected throughout the whole reaction process26,27,28,29,30. Interestingly, a predictable regioisomer 8 resulted from the formal [4 + 2] reaction was actually not observed as well, which is due probably to the relatively high thermodynamic stability of the α-imine hemiacetal 7. Importantly, under the standard condition, the six-membered ring compound 7 underwent the rearrangement reaction smoothly, which delivered the corresponding product 3ae in high yield (Fig. 6e). In the absence of ZnCl2 catalyst, intermediate 7 failed to give 3ae. The reaction of 1a with another branched amino alcohol 2af gave similar results (Fig. 6f, g). The structure of the important α-imine hemiacetal 9 was further confirmed by X-ray single crystal diffraction technique (see Supplementary Data 6). These results clearly suggested that a six-membered ring intermediate resulting from a direct formal [4 + 2] cyclization should be the real reservoir for product 3. Moreover, treating compound 9 with catalytic amount of either p-TSA or (R)-TRIP-CPA at 100 °C failed to yield the product 3af, which not only underlined the great importance of ZnCl2 catalyst in triggering the key 1,2-ester shift step but also highlighted the difference in the reaction mechanism between current rearrangement and Brønsted acid-catalyzed benzilic ester-type rearrangement (Fig. 6h)31,32,35. Finally, the reaction of diphenyl-substituted 1,2-diketone 10 with the amino alcohol 2a gave neither the rearranged product nor any intermediates (Fig. 6i), which indicated that the extra ester group is of vital importance for this rearrangement. For all NMR spectra of the new compounds, see Supplementary Data 7.

a–c Conversion of 4a catalyzed by p-TSA, ZnCl2 and (2a)·ZnCl2, respectively. d Kinetic curve for the formation of 3a and 4a.

a, b Reaction of 1a and 2a catalyzed by Zn(OTf)2 and ZnCl2, respectively. c Conversion of 6 catalyzed by ZnCl2. d, e Reaction of 1a and 2ae. f–h Reaction of 1a and 2af. i Reaction of 10 and 2a catalyzed by ZnCl2.
As the conversion of 1 and 2 to the five-membered oxazolidine intermediates 4 is extremely low, and the transformation of 4 to the products 3 is also highly difficult (see Fig. 5b–d), the compound 4 should be not the preferred intermediates under the conditions. Moreover, the isolation of the only [4 + 2] cyclization products 7 and 9 which could be smoothly transformed into the rearranged products 3ae and 3af clearly indicates the real reaction pathway as well. Therefore, on the basis of the mechanistic experiments, this rearrangement reaction is proposed to proceed via an unprecedent direct [4 + 2] cyclization/1,2-ester or amide migration sequence (Fig. 7a). Initially, the 2,3-diketoester 1a and amino alcohol 2a predominately undergo a formal [4 + 2] heteroannulation reaction to generate a cyclic α-iminium hemiacetal A. Subsequently, the key intermediate A then participate in a 1,2-ester shift reaction under the catalysis of in situ formed (2a)∙ZnCl2 complex affording the final C3-disubstituted morpholin-2-one 3a. The in situ formed a small amount of 4a is more inclined to generate compound 5 rather than the rearrangement product 3a, which is due probably to the driving force differences to these two compounds, respectively. The side product 5 might result from the carbon-carbon cleavage of intermediate B to produce a carbene species which is quenched by water and a potential hydride transfer reagent 2a52,53.

a The reaction of 1a and 2a was employed to demonstrate the reaction mechanism. b The stereochemical pathway for the formation of 3ah was shown.
A plausible pathway leading to the observed stereochemical outcome is also illustrated in Fig. 7b. Because the ester moiety of the intermediate A1 is remote from benzyl group, the Re face of the iminium moiety is open to the ester group 1,2-migration, thus favorably affording 3ah. In contrast, the Si face of the iminium moiety is shielded by benzyl group in A2, thus making the 1,2-ester shift disfavored and resulting in the formation of the enantiomer ent-3ah in a minor amount. This scenario is in accordance with the configuration of the products as established by X-ray crystal structure analysis of four representative morpholines (3ag, 3am, 3an and 3ao, see Fig. 4).
In conclusion, we have developed the first examples of ZnCl2-catalyzed cyclizative 1,2-rearrangement for the asymmetric synthesis of morpholinones bearing aza-quaternary stereocenters which are far more challenging to obtain efficiently by existing methods. A series of structurally diverse C3-disubstituted morpholin-2-ones were efficiently constructed from readily available two achiral linear compounds. Importantly, on the basis of mechanistic studies, this rearrangement proceeds via an unprecedent sequence of direct formal [4 + 2] heteroannulation followed by a 1,2-ester or amide migration process, which is highly distinct from the reported reaction mechanism of aza-benzilic acid-type rearrangements.
Methods (Supplementary Methods in the SI)
General procedure for the synthesis of 3
To a mixture of 1 (0.1 mmol, 1.0 equiv), 2 (0.12 mmol, 1.2 equiv), and ZnCl2 (0.02 mmol, 0.2 equiv) was added 1,2-dichloroethane (1.0 mL) under argon. The reaction mixture was stirred at 100 °C for indicated hours. After completion of the reaction (monitored by TLC), the solvent was removed under vacuum. The residue was purified by column chromatography on silica gel, eluting with petroleum ether/ethyl acetate to afford the products 3 in moderate to high yields.
Data availability
Cif. files (see Supplementary Data 1–6); all NMR spectra of new compounds (see Supplementary Data 7). All data generated and analyzed during this study are included in this article, its Supplementary Information, and also available from the authors upon reasonable request. The X-ray crystallographic coordinates for structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 2225996, 2246927, 2246928, 2246926, 2246925 and 2266070 (see Supplementary Notes and Supplementary Tables S2-43 for details). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data request/cif.
References
Enders, D., Meyer, O., Raabe, G. & Runsink, J. (S,S)-3,5-Dimethylmorpholine, a novel C2-Symmetric auxiliary. First application in [4+2]-Cycloadditions leading to 4-Oxohexahydropyridazine derivatives. Synthesis 1994, 66–72 (1994).
Baldoli, C. et al. The first chiral racemic Fischer-type aminocarbene complex bearing a C2 symmetry amine: stereochemical characteristics and application to Michael addition reactions. J. Organomet. Chem. 486, 279–282 (1995).
Flick, A. C. et al. Synthetic approaches to the 2014 new drugs. Bioorg. Med. Chem. 24, 1937–1980 (2016).
Aubele, D. L. et al. Selective and brain-permeable polo-like Kinase-2 (Plk-2) inhibitors that reduce a-Synuclein phosphorylation in rat brain. ChemMedChem 8, 1295–1313 (2013).
Nelson, T. D. Synthesis of Aprepitant. In Strategies and Tactics in Organic Synthesis; Harmata, M., Ed.; Academic Press, 2005; Vol. 6, pp 321–351.
Wijtmans, R. et al. Biological relevance and synthesis of C-Substituted morpholine derivatives. Synthesis 2004, 641–662 (2004).
Trstenjak, U., Ilaš, J. & Kikelj, D. Advances in the synthesis of Morpholin-3-ones and morpholin-2-ones. Synthesis 44, 3551–3578 (2012).
Yamada, R., Sakata, K. & Yamada, T. Electrochemical synthesis of substituted morpholines via a decarboxylative intramolecular etherification. Org. Lett. 24, 1837–1841 (2022).
Uozumi, Y., Tanahashi, A. & Hayashi, T. Catalytic asymmetric construction of morpholines and piperazines by palladium-catalyzed tandem allylic substitution reactions. J. Org. Chem. 58, 6826–6832 (1993).
Wilkinson, M. C. Asymmetric synthesis of an aminomethyl morpholine via double allylic substitution. Tetrahedron Lett. 46, 4773–4775 (2005).
Bandini, M., Monari, M., Romaniello, A. & Tragni, M. Gold catalyzed direct activation of allylic alcohols in the stereoselective synthesis of functionalized 2-Vinyl-Morpholines. Chem. Eur. J. 16, 14272–14277 (2010).
Wang, Y., Zhang, W.-Y. & You, S.-L. Ketones and aldehydes as O-Nucleophiles in iridium-catalyzed intramolecular asymmetric allylic substitution reaction. J. Am. Chem. Soc. 141, 2228–2232 (2019).
Zhai, H., Borzenko, A., Lau, Y. Y., Ahn, S. H. & Schafer, L. L. Catalytic asymmetric synthesis of substituted morpholines and piperazines. Angew. Chem., Int. Ed. 51, 12219–12223 (2012).
Luescher, M. U. & Bode, J. W. Catalytic synthesis of N-unprotected piperazines, morpholines, and thiomorpholines from aldehydes and SnAP reagents. Angew. Chem., Int. Ed. 54, 10884–10888 (2015).
Numajiri, Y., Jiménez-Osés, G., Wang, B., Houk, K. N. & Stoltz, B. M. Enantioselective synthesis of dialkylated N-heterocycles by palladium-catalyzed allylic alkylation. Org. Lett. 17, 1082–1085 (2015).
Kuwano, R., Karube, D. & Ito, Y. Catalytic asymmetric hydrogenation of 1-aza-2-cycloalkene-2-carboxylates catalyzed by a trans-chelating chiral diphosphine PhTRAP-rhodium complex. Tetrahedron Lett. 40, 9045–9049 (1999).
Li, M. et al. Asymmetric hydrogenation for the synthesis of 2-substituted chiral morpholines. Chem. Sci. 12, 15061–15066 (2021).
Lau, Y. Y., Zhai, H. & Schafer, L. L. Catalytic asymmetric synthesis of morpholines. Using mechanistic insights to realize the enantioselective synthesis of piperazines. J. Org. Chem. 81, 8696–8709 (2016).
Leathen, M. L., Rosen, B. R. & Wolfe, J. P. New strategy for the synthesis of substituted morpholines. J. Org. Chem. 74, 5107–5110 (2009).
Palchykov, V. A. & Chebanov, V. A. Recent progress in the synthesis of morpholines. Chem. Heterocycl. Compd. 55, 324–332 (2019).
Siau, W.-Y. & Bode, J. W. One-step synthesis of saturated spirocyclic N-heterocycles with stannyl amine protocol (SnAP) reagents and ketones. J. Am. Chem. Soc. 136, 17726–17729 (2014).
Narczyk, A. & Stecko, S. An entry to non-racemic β-tertiary-β-amino alcohols, building blocks for the synthesis of aziridine, piperazine, and morpholine scaffolds. Org. Biomol. Chem. 18, 5972–5981 (2020). The structural diversity of the substrates derived from natural amino acids is extremely limited, especially for those bearing aza-quaternary stereocenters.
Brands, K. M. J. et al. Efficient synthesis of NK1 receptor antagonist aprepitant using a crystallization-induced diastereoselective transformation.J. Am. Chem. Soc. 125, 2129–2135 (2003).
Ruider, S. A., Müller, S. & Carreira, E. M. Ring expansion of 3-Oxetanone-derived spirocycles: facile synthesis of saturated nitrogen heterocycles. Angew. Chem., Int. Ed. 52, 11908–11911 (2013).
Strecker, A. Ueber die künstliche Bildung der Milchsäure und einen neuen, dem Glycocoll homologen Körper. Ann. Chem. Pharm. 75, 27–45 (1850).
Ukaji, Y., Yamamoto, K., Fukui, M. & Fujisawa, T. Stereodivergent synthesis of optically active tertiary alcohols via addition reaction of Chiral 2-Acyl oxazolidine with organometallics. Tetrahedron Lett. 32, 2919–2922 (1991).
Agami, C., Couty, F. & Lequesne, C. N-Boc 2-Acyloxazolidines: useful precursors to enantiopure 1,2-Diols via highly diastereoselective nucleophilic additions. Tetrahedron 51, 4043–4056 (1995).
Polyak, F., Dorofeeva, T., Zelchans, G. & Shustov, G. Regio- and stereoselectivity of the formation of 1,3-Oxazolidines in the reaction of L-Ephedrine with phenylglyoxal. unexpected rearrangement of 2-Benzoyl-3,4-dimethyl-5-phenyl-l,3-oxazolidine to 4,5-Dimethyl-3,6- diphenylmorpholin-2-one. Tetrahedron Lett. 37, 8223–8226 (1996).
Mlostón, G., Wróblewska, A., Linden, A. & Heimgartner, H. Studies on the reaction of Aryl Glyoxals with L-Prolinol: unexpected formation of Morpholin-2-one derivatives and stereoselective trifluoromethylation of the bicyclic system. Asian J. Org. Chem. 4, 770–777 (2015).
Zhao, M. M. et al. Practical asymmetric synthesis of aprepitant, a potent human NK-1 receptor antagonist, via a stereoselective Lewis acid catalyzed trans acetalization reaction. J. Org. Chem. 67, 6743–6747 (2002).
Powell, W. C. & Walczak, M. A. Asymmetric synthesis of chiral 1,2-Amino alcohols and Morpholin-2-ones from arylglyoxals. J. Org. Chem. 83, 10487–10500 (2018).
He, Y., Wu, H., Wang, Q. & Zhu, J. Catalytic enantioselective synthesis of morpholinones enabled by aza-benzilic ester rearrangement. J. Am. Chem. Soc. 143, 7320–7325 (2021).
Wang, B. & Tu, Y.-Q. Stereoselective construction of quaternary carbon stereocenters via a semipinacol rearrangement strategy. Acc. Chem. Res. 44, 1207–1222 (2011).
Wu, H., Wang, Q. & Zhu, J. Recent advances in catalytic enantioselective rearrangement. Eur. J. Org. Chem. 2019, 1964–1980 (2019).
He, Y., Quan, R., Li, X., Zhu, J. & Wu, H. Asymmetric construction of α,α-disubstituted piperazinones enabled by benzilic amide rearrangement. Angew. Chem., Int. Ed. 62, e202217954 (2023).
Wu, H., Wang, Q. & Zhu, J. Catalytic enantioselective benzilic ester rearrangement. Angew. Chem. Int. Ed. 59, 7261–7265 (2020).
Rubin, M. B. Chemistry of vicinal polyketones. Chem. Rev. 75, 177–202 (1975).
Wasserman, H. H. & Han, W. T. Vicinal Tricarbonyl products from singlet oxygen reactions.: application to the synthesis of carbacephams. Tetrahedron Lett. 25, 3743–3746 (1984).
Wasserman, H. H. & Han, W. T. A synthesis of antibiotic (±)-PS-5. Tetrahedron Lett. 25, 3747–3750 (1984).
Wasserman, H. H. & Han, W. T. Penem synthesis through C3-N ring closure of a β-lactam precursor. J. Am. Chem. Soc. 107, 1444–1446 (1985).
Wasserman, H. H. et al. The chemistry of vicinal tricarbonyls. A stable vinyl tricarbonyl hydrate as a di- and trielectrophile. J. Am. Chem. Soc. 111, 371–372 (1989).
Wasserman, H. H. & Blum, C. A. The chemistry of vicinal tricarbonyls. Use of vinyl and acetylenic derivatives in the synthesis of substituted indoles. Tetrahedron Lett. 35, 9787–9790. (1994).
Rubin, M. B. & Gleiter, R. The chemistry of vicinal polycarbonyl compounds. Chem. Rev. 100, 1121–1164 (2000).
Wasserman, H. H. & Parr, J. The chemistry of vicinal tricarbonyls and related systems. Acc. Chem. Res. 37, 687–701 (2004).
Truong, P. M., Zavalij, P. Y. & Doyle, M. P. Highly enantioselective carbonyl-ene reactions of 2,3-Diketoesters: efficient and atom-economical process to functionalized chiral α-Hydroxy-β-Ketoesters. Angew. Chem. Int. Ed. 53, 6468–6472 (2014).
Wang, L., Zhong, J. & Lin, X. Atroposelective phosphoric acid catalyzed three-component cascade reaction: enantioselective synthesis of axially chiral N-Arylindoles. Angew. Chem. Int. Ed. 58, 15824–15828 (2019).
Chen, Z. et al. Organocatalytic enantioselective synthesis of axially chiral N,N’-Bisindoles. Angew. Chem. Int. Ed. 62, e202300419 (2023).
Akiyama, T., Itoh, J., Yokota, K. & Fuchibe, K. Enantioselective mannich-type reaction catalyzed by a chiral brønsted acid. Angew. Chem. Int. Ed. 43, 1566–1568 (2004).
Uraguchi, D. & Terada, M. Chiral brønsted acid-catalyzed direct mannich reactions via electrophilic activation. J. Am. Chem. Soc. 126, 5356–5357 (2004).
Wang, P. et al. Asymmetric 1,2-Perfluoroalkyl migration: easy access to enantioenriched α-Hydroxy-α-perfluoroalkyl esters. J. Am. Chem. Soc. 137, 4626–4629 (2015).
Liu, R. et al. Lewis-acid-catalyzed asymmetric alkynylation of alkynyl 1,2-Diketones: controllable formation of 3(2H)‑furanones and α‑Hydroxy ketones. Org. Lett. 22, 6948–6953 (2020).
Peng, B. & Maulide, N. The redox-neutral approach to C-H functionalization. Chem. Eur. J. 19, 13274–13287 (2013).
F. G. West, B. N. Naidu, Applications of Stevens [1,2]-shifts of cyclic ammonium ylides. A Route to Morpholin-2-ones, J. Org. Chem. 1994, 59, 6051–6056.
Acknowledgements
Financial support for this work was provided by the National Natural Science Foundation of China (Grant No. 22101172, 22371183), and startup funding from Shanghai Jiao Tong University (SJTU). Dr. Y.-P. He thanks the China Postdoctoral Science Foundation (grant no. 2022M710094).
Author information
Authors and Affiliations
Contributions
X.L. Y.H. and H.W. conceived the work, designed the experiments and analyzed the data. X.L. optimized the reaction conditions and performed the experiments. H.W. conceived and directed the project and wrote the paper. All the authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, XZ., He, YP. & Wu, H. Zinc chloride-catalyzed cyclizative 1,2-rearrangement enables facile access to morpholinones bearing aza-quaternary carbons. Commun Chem 6, 216 (2023). https://doi.org/10.1038/s42004-023-01016-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-023-01016-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
