
Abstract
The experimental realization of p-orbital systems is desirable because p-orbital lattices have been proposed theoretically to host strongly correlated electrons that exhibit exotic quantum phases. Here, we synthesize a two-dimensional Fe-coordinated bimolecular metal-organic framework which constitutes a honeycomb lattice of 1,4,5,8,9,12-hexaazatriphenylene molecules and a Kagome lattice of 5,15-di(4-pyridyl)-10,20-diphenylporphyrin molecules on a Au(111) substrate. Density-functional theory calculations show that the framework features multiple well-separated spin-polarized Kagome bands, namely Dirac cone bands and Chern flat bands, near the Fermi level. Using tight-binding modelling, we reveal that these bands are originated from two effects: the low-lying molecular orbitals that exhibit p-orbital characteristics and the honeycomb-Kagome lattice. This study demonstrates that p-orbital Kagome bands can be realized in metal-organic frameworks by using molecules with molecular orbitals of p-orbital like symmetry.
Similar content being viewed by others

Introduction
The orbital degree of freedom plays a central role in condensed matter physics. Orbital design in different lattice symmetries may lead to exotic quantum phases1,2,3,4,5,6. Notably, as the counterpart of pz-orbital graphene, the px/py honeycomb or other lattices can exhibit richer orbital-derived physics due to orbital degeneracy and spatial anisotropy of px/py orbitals7,8,9,10,11,12,13,14,15,16,17,18,19,20. The p-orbital lattices have been proposed theoretically to host strongly correlated phases, such as Wigner-crystal-like states7, itinerant flat band ferromagnetism15, non-trivial topological phases16, and Chern flat band, which holds the potential to realize high-temperature fractional quantum Hall states17,18,19,20. The experimental realization of p-orbital systems with exotic quantum phases has been demonstrated in optic lattice21,22, and lattices of coupled micropillars23,24. Another approach is to use a scanning tunneling microscope to manipulate CO molecules on an atomic clean surface to create artificial lattices25,26,27,28,29. Here, we demonstrate that a p-orbital honeycomb-Kagome lattice (HKL) model is materialized in a magnetic 2D metal-organic framework (2D MOF) system3,6.
2D MOFs are predicted to be a versatile platform for realizing exotic quantum phases, including the topological insulator30,31,32, Quantum anomalous Hall effect33, topological Chern insulator18, and superconductors34. The interplay of diffusive s and p electrons of molecular linkers with metal d-orbitals coupled with specific lattice symmetries provides rich opportunities to explore orbital design. 2D MOFs with various topologies, such as Kagome and honeycomb, have been synthesized, and their band structures are mostly constructed with molecular orbitals (MOs) with s-orbital nature. Experimental realization of 2D MOFs comprised of two sublattices made of two types of molecules to form a honeycomb-Kagome lattice has not been reported. In this work, we synthesize a 2D MOF using two types of molecular building blocks, 5, 15-di(4-pyridyl)-10, 20-diphenylporphyrin (DPyP) and 1,4,5,8,9,12-hexaazatriphenylene (HAT), coordinated with Fe atoms on a Au (111) substrate. As resolved using scanning tunneling microscopy (STM) and scanning tunneling spectroscopy (STS), this structure constitutes a honeycomb lattice of HAT and a Kagome lattice of Fe-metalated DPyP, interconnected via Fe-N coordination. We use density-functional theory (DFT) to analyze this structure and find a series of spin-polarized Dirac-cone bands and Chern flat bands near the Fermi level. The DFT-calculated band structure can be perfectly reproduced using a tight-binding (TB) p-orbitals model owing to the p-orbital characteristics of the low-lying molecular orbitals of DPyP and HAT. This work demonstrates that by choosing molecules with orbitals of p-orbital characteristics, one can use 2D MOFs as platforms to study p-orbital physics in myriad lattices. Considering the richness of molecular orbitals in terms of symmetry, parity, and geometry, this work opens new possibilities for designing band structures in 2D MOFs.
Results and discussion
Structural and electronic characteristics resolved by using STM and STS
After depositing DPyP and HAT molecules on an atomic flat and clean Au (111) surface held at room temperature under ultra-high vacuum conditions, two types of molecules segregate and form homo-molecular assemblies (Fig. S3). After dosing small amounts of Fe on this sample and a thermal annealing treatment at 170 °C, a porous 2D framework is formed on the surface, as shown in Fig. 1a. The framework extends over entire terraces of the Au(111) surface. Structural defects, such as domain boundaries and five-member, seven-member, and eight-member polygons (Fig. S4), are present in the network, presumably due to the flexible coordination interaction. Figure 1b is a high-resolution STM image of the hexagonal network. Two types of species are present in this structure: four-lobe squares and star-shaped triangles. In reference to the molecular appearance of DPyP and HAT shown in Fig. S3, we assign the squares to DPyP molecules and the triangles to HAT molecules. Each HAT molecule connects with three DPyP molecules and each DPyP molecule links to two HAT molecules via Fe atoms. A structural model is overlaid in Fig. 1b. In this structure, HAT molecules form a honeycomb lattice and DPyP molecules form a Kagome lattice, as represented with the red hexagonal and the blue David star, respectively, in Fig. 1b. The pore-to-pore distance is 4.61 ± 0.2 nm. Although Fe atoms are not resolved in the STM image, we propose the HAT-DPyP connection is established through a threefold Fe-N bonding motif in which a Fe atom coordinates with the pyridyl (py) ligand of DPyP and the bipyridine (bipy) ligand of HAT. A careful observation of Fig. 1b reveals that the majority of DPyP molecules feature a shallow valley in the middle of the saddle-shaped square, while the molecule pointed with the red arrow has a dip at the molecular center. This difference is attributed to the Fe metalation of central tetrapyrrole macrocycles of DPyP35,36, denoted as Fe-DPyP thereafter. The tunneling spectra recorded at the center of Fe-DPyP exhibit a symmetric U-shape around the zero bias with two steps at ±8.4 meV, as shown in the insert in Fig. 1c, whereas the metal-free DPyP molecules do not exhibit this feature (gray spectrum in the inset in Fig. 1c). This U-shape feature is in accordance with the spin-flip excitation of Fe37,38,39, indicating that the Fe atoms carry a magnetic moment. We term this structure as (HAT-Fe3)2 (Fe-DPyP)3.

a An overview STM image of monolayer \({({{{{{\rm{HAT}}}}}}-{{{{{{\rm{Fe}}}}}}}_{3})}_{2}{({{{{{\rm{Fe}}}}}}-{{{{{\rm{DPyP}}}}}})}_{3}\) coordination network grown on Au (111) substrate. b A high-resolution STM showing the network structure with molecular models and a honeycomb-Kagome lattice overlaid. C, N, H, and Fe atoms are gray, blue, white, and purple, respectively. c Site-specific tunneling spectra acquired at Fe-DPyP and HAT. Insert: a tunneling spectrum acquired at the center of Fe-DPyP, the gray spectrum is acquired at the center of a metal-free DPyP.
Figure 1c shows the site-specific tunneling spectra acquired at HAT and Fe-DPyP (the light gray spectrum is acquired at the bare Au (111) surface). Spectroscopically, HAT shows two step-like features below the Fermi level at −1.0 and −0.3 V, two shoulders at 0.1 and 0.5 V, and a broad peak at 1.0 V. Fe-DPyP shows two shoulders at −0.8 and 1.0 V and a dip at the Fermi level, which manifests the spin-flip signature of Fe. The metal-free DPyP features a salient peak at −0.5 V and a shoulder at 1.3 V (Fig. S5).
DFT+U calculated band structures and local density states
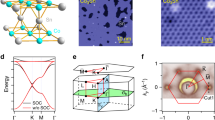
We perform DFT calculations with the U effect on Fe d-orbitals to analyze a free-standing structure of \({({{{{{\rm{HAT}}}}}}-{{{{{{\rm{Fe}}}}}}}_{3})}_{2}{({{{{{\rm{Fe}}}}}}-{{{{{\rm{DPyP}}}}}})}_{3}\). The relaxed structure is shown in Fig. 2a. The lattice constant of the optimized structure is 47 Å, which is in good agreement with the experimental data. In the threefold Fe-N coordination, the Fe-N(py) bond and the Fe-N(bipy) bonds are of the same bond length of 1.96 Å. The metalated Fe atom is bonded with four N atoms in the tetrapyrrole macrocycle core with a Fe-N bond length of 2.07 Å. The metalated Fe has a magnetic moment of 3.67 μB and the threefold coordinated Fe has a magnetic moment of 3.50 μB. We analyze several magnetic configurations, including colinear ferromagnetic state and ferrimagnetic state, as well as with spin–orbit coupling (SOC). All these cases exhibit similar band structures near the Fermi level (Fig. S6). Figure 2b shows the spin-polarized bands and project-density of states (PDOS) of \({({{{{{\rm{HAT}}}}}}-{{{{{{\rm{Fe}}}}}}}_{3})}_{2}{({{{{{\rm{Fe}}}}}}-{{{{{\rm{DPyP}}}}}})}_{3}\) with a ferromagnetic ground state. There are six sets of well-separated spin-polarized bands in the energy range from −0.5 eV to 0.3 eV. The PDOS shows that the Fe d- orbitals hybridize with the molecular orbitals. Thus, we argue that the Fe d electrons interact with the itinerant charges in the framework and lift the spin degeneracy of the electronic bands. In this work, we focus on the three sets of spin-down bands (colored in blue in Fig. 2b): a Dirac cone (denoted as D bands) around 0.4 eV below the Fermi level, a Dirac cone with one flat band (denoted as FD bands) near the Fermi level, and a Dirac cone sandwiched between two flat bands (denoted as FDF bands) around 0.1 eV above the Fermi level. Figure 2c shows the Gamma point wavefunctions at the band edges. The D bands are localized at HAT, the FD bands are at HAT and Fe-DPyP, and the FDF bands are at HAT at the low band edge and at HAT and Fe-DPyP at the high band edge, respectively.

a DFT-optimized free-standing structure of \({({{{{{\rm{HAT}}}}}}-{{{{{{\rm{Fe}}}}}}}_{3})}_{2}{({{{{{\rm{Fe}}}}}}-{{{{{\rm{DPyP}}}}}})}_{3}\). b Left: DFT-calculated spin-polarized band structure. Blue and red lines represent spin-down and spin-up channels. Right: spin-polarized DOS projected at Fe d-orbitals and molecular pz orbitals. c Gamma point wavefunctions at the band edges as indicated by the arrow lines. d DFT-calculated DOS projected at HAT and metalation Fe in Fe-DPyP.
Using a p-orbital tight-binding (TB) model to reconstruct the DFT-calculated band structure
To compare with the experimentally resolved local density of states (DOS) shown in Fig. 1c, we plot the DOS projected on HAT and the metalation Fe in Fe-DPyP in Fig. 2d. Both the metalated Fe (black curve) and HAT (purple curve) feature two sets of peaks at 1.1 and −1.0 eV. The two species differ in the low energy range: the metalation Fe shows a very weak PDOS signal, whereas HAT has two sets of pronounced peaks around −0.4 and 0.3 eV. Qualitatively, the PDOS captures the characteristics of the local DOS resolved in the site-specific STS (Fig. 1c) and STS maps (Fig. S7). The difference between the PDOS and the STS can be attributed to two effects: (1) the STS signal contains the contribution of the substrate states and (2) the framework is electronically coupled with the substrate, which broadens and shifts the intrinsic DOS of the free-standing framework.
As illustrated in Fig. 3a, we consider HAT and Fe-DPyP molecules as super-atoms represented by triangles and squares, respectively. Figure 3b shows the low-lying unoccupied molecular orbitals of HAT (LUMO and degenerated LUMO +1/+2) calculated using a Gaussian package. Figure 3c shows the LUMO +1 of Fe-DPyP. (Note: Fe-DPyP LUMO is localized at the central Fe atom, thus not being considered). One can find that the HAT LUMO features threefold rotation symmetry, and the degenerated LUMO + 1 and LUMO + 2 exhibit anti-phase symmetry along the vertical and horizontal central axis, respectively. The LUMO + 1 orbital of Fe-DPyP exhibits an anti-phase mirror symmetry along the central vertical axis. According to the symmetry and parity of these orbitals, we designate HAT LUMO as an s-orbital of the HAT super-atom (HAT-s), HAT LUMO + 1/+2 as HAT super-atom px and py orbitals (HAT-px/py), and Fe-DPyP LUMO + 1 as a rotated p-orbital of the DPyP super-atom (DPyP-p)40, as represented in Fig. 3b, c, respectively.

a The super-atom model of \({({{{{{\rm{HAT}}}}}}-{{{{{{\rm{Fe}}}}}}}_{3})}_{2}{({{{{{\rm{Fe-DPyP}}}}}})}_{3}\), where triangle/square represents HAT/Fe-DPyP. b Lowest unoccupied molecular orbital (LUMO), LUMO + 1/+2 of HAT. c LUMO + 1 of Fe-DPyP.
Based on the analysis above, we construct a TB model of nine superatomic orbitals, as shown in Fig. 4a, to reproduce the DFT band structure. The TB Hamiltonian is: \(H={\sum }_{i}{{\epsilon }}_{i}{c}_{i}^{+}{c}_{i}+{\sum }_{\langle i,j\rangle =\langle Hs,Dp\rangle }{t}_{sp\sigma }{c}_{i}^{+}{c}_{j}+{\sum } _{\langle i,j\rangle =\langle Dp,Hp\in \{H{p}_{x},H{p}_{y}\}\rangle }{t}_{pp\sigma }{c}_{i}^{+}{c}_{j}\) . We only consider the hoppings between nearest neighbors: where <i,j> represent the nearest-neighbor pair, and Hs, Dp, and Hp represent the HAT-s, Fe-DPyP-p, and HAT-px/py orbitals, respectively.

a The HKL with p-orbital on Kagome lattice (in blue) and s, px, py orbitals on honeycomb lattice (in red). b TB and DFT band structures. From the bottom up: the bands are constructed with HAT-s, Fe-DPyP-p, and HAT-px/py basis, respectively.
Setting the hopping terms tspσ = −0.04 eV and tppσ = −0.035 eV, the on-site energies \({{\epsilon }}_{{{{{{\rm{Dp}}}}}}}\) = 0.035 eV, \({{\epsilon }}_{{{{{{\rm{Hs}}}}}}}\) = −0.41 eV, \({{\epsilon }}_{{{{{{\rm{Hp}}}}}}}\) = 0.108 eV, the TB bands (dotted lines) are shown in Fig. 4b. From the bottom up, the D bands are contributed from the HAT-s orbital of the honeycomb lattice, the FD bands are contributed from the Fe-DPyP-p orbital of the Kagome lattice, and the FDF bands are contributed from the HAT-px/py orbitals of the honeycomb lattice. The TB-model bands (dashed lines) are overlaid on the DFT-calculated spin-down bands (solid lines) in Fig. 4b, which show perfect agreement between the two. Therefore, we demonstrate that the band structure near the Fermi energy of this MOF can be understood as an HKL with rotated p-MOs. In real materials, rotating the atomic orbitals is difficult to realize. Here we show that rotating orbitals can be effectively achieved in a MOF when the molecular orbitals exhibit p-orbital symmetry and parity. In this context, our work provides a design principle for obtaining Dirac and flat band structures in MOFs by choosing specific molecular orbitals.
Conclusion
In conclusion, the spin-down bands of the 2D MOF \({({{{{{\rm{HAT}}}}}}-{{{{{{\rm{Fe}}}}}}}_{3})}_{2}{({{{{{\rm{Fe}}}}}}-{{{{{\rm{DPyP}}}}}})}_{3}\) can be perfectly reconstructed using a p-orbital HKL TB model thank to that HAT and Fe-DPyP possess frontier molecular orbitals exhibiting p-orbital symmetry. This sheds light on a promising strategy of using spatial symmetry of molecular orbitals to design p-orbital lattices in 2D MOFs. The fully-spin-polarized band structure makes this system an appealing playground for studying Chern-bands-related quantum phenomena with broken time-reversal symmetry. More interestingly, a Chern flat band lies at the Fermi level, which provides opportunities for exploring both topological and strong correlated physics. Considering the topological bands of this structure are screened due to electronic coupling with the Au(111) substrate, future work is to synthesize this structure on an insulating substrate or an inert substrate which interacts weakly with the framework.
Methods
Sample preparation and measurement
All the experiments are performed in an ultra-high vacuum (UHV) system (omicron Nanotechnology) with a base pressure below 2.3 × 10−10 mbar. A single crystal Au (111) substrate is cleaned by Ar+ sputtering and annealing at 620 °C with several cycles. 5,15-di(4-pyridyl)-10,20-diphenylporphyrin (DPyP) is synthesized following the method described in ref. 41. 1,4,5,8,9,12-hexaazatriphenylene (HAT) is synthesized following the method described in SI (Figs. S1, S2). DPyP molecules are thermally evaporated by a molecular beam evaporator and deposited on the Au (111) substrate held at room temperature. After that, HAT molecules are deposited on the Au (111) surface using the same method. The evaporation temperatures for molecules DPyP and HAT are 330 and 165 °C, respectively. Finally, the Fe atoms are deposited on the surface using an e-beam evaporator, and the sample is annealed to 170 °C for 15 min. Without Fe, annealing at 170 °C desorbs HAT, while the remaining DPyP form close-packed molecular island. The STM measurement is performed at 4.8 K, and the bias voltage is applied to the sample plate. The standard lock-in technique with modulation of 5 mV (rms) and corresponding frequency 1.17 kHz is applied in differential tunneling spectra dI/dV measurement. The STS mapping is employed to image the electronic DOS distribution with the specific energies at different positions of the coordination network.
DFT calculations
The first-principles calculations are carried out using the Vienna ab initio simulation package (VASP)42. The Perdew–Burke–Ernzerhof (PBE) functional within generalized gradient approximation (GGA)43,44,45 framework is adopted to describe the exchange-correlation functionals. The interactions between electrons and ions are treated using the projector augmented wave (PAW) method46 with a plane-wave cutoff energy of 450 eV. The Brillouin zone is sampled with the Gamma-only point due to the large size of the unicell. A 20 Å vacuum layer is added to decouple the interactions between neighboring layers. All atoms are relaxed until the forces are smaller than 0.01 eV/Å. Due to the correlation effect of 3d electrons in Fe atoms, we employed the GGA+U scheme47. We calculate the energy of FM and FiM states under various U values, as shown in Table. S1. The magnetic ground states are FM for U = 2–5 eV, while FiM for U = 1 eV. As shown in the density of states under different U values in Fig. S8, variation of the U value only shifts the states/bands that are contributed from Fe d-orbitals and does not affect the states/bands that are mainly contributed from the molecules. Also, as shown in Fig. S6, the band structures of FM and FiM states share similar features near Fermi energy. Hence, we argue that the Kagome band structure is robust under a wide-range U value.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
Cui, B. et al. Creation of half-metallic f-orbital Dirac fermion with superlight elements in orbital-designed molecular lattice. Phys. Rev. B 96, 085134 (2017).
Gao, L. et al. Orbital design of topological insulators from two-dimensional semiconductors. Nanoscale 11, 22743–22747 (2019).
Jiang, W., Ni, X. & Liu, F. Exotic topological bands and quantum states in metal–organic and covalent–organic frameworks. Acc. Chem. Res. 54, 416–426 (2021).
Liu, H., Sethi, G., Meng, S. & Liu, F. Orbital design of flat bands in non-line-graph lattices via line-graph wave functions. Phys. Rev. B 105, 085128 (2022).
Wang, Z., Jin, K. H. & Liu, F. Computational design of two‐dimensional topological materials. Wiley Interdiscip. Rev. Comput. Mol. Sci. 7, e1304 (2017).
Liu, Z., Liu, F. & Wu, Y.-S. Exotic electronic states in the world of flat bands: from theory to material. Chin. Phys. B 23, 077308 (2014).
Wu, C., Bergman, D., Balents, L. & Sarma, S. D. Flat bands and Wigner crystallization in the honeycomb optical lattice. Phys. Rev. Lett. 99, 070401 (2007).
Wu, C. Orbital ordering and frustration of p-band Mott insulators. Phys. Rev. Lett. 100, 200406 (2008).
Wu, K. & Zhai, H. Theory of quantum antiferromagnetism of fermions in an optical lattice with a half-filled p band. Phys. Rev. B 77, 174431 (2008).
Li, X. & Liu, W. V. Physics of higher orbital bands in optical lattices: a review. Rep. Prog. Phys. 79, 116401 (2016).
Pinheiro, F., Bruun, G. M., Martikainen, J.-P. & Larson, J. XYZ Quantum Heisenberg models with p-orbital Bosons. Phys. Rev. Lett. 111, 205302 (2013).
Wu, C. Orbital analogue of the quantum anomalous Hall effect in p-band systems. Phys. Rev. Lett. 101, 186807 (2008).
Zhao, E. & Liu, W. V. Orbital order in Mott insulators of spinless p-band fermions. Phys. Rev. Lett. 100, 160403 (2008).
Wu, C. & Sarma, S. D. px,y-orbital counterpart of graphene: cold atoms in the honeycomb optical lattice. Phys. Rev. B 77, 235107 (2008).
Zhang, S., Hung, H.-h & Wu, C. Proposed realization of itinerant ferromagnetism in optical lattices. Phys. Rev. A 82, 053618 (2010).
Zhang, G.-F., Li, Y. & Wu, C. Honeycomb lattice with multiorbital structure: topological and quantum anomalous Hall insulators with large gaps. Phys. Rev. B 90, 075114 (2014).
Baidya, S., Kang, S., Kim, C. H. & Yu, J. Chern insulator with a nearly flat band in the metal-organic-framework-based kagome lattice. Sci. Rep. 9, 13807 (2019).
Liu, Z., Wang, Z.-F., Mei, J.-W., Wu, Y.-S. & Liu, F. Flat Chern band in a two-dimensional organometallic framework. Phys. Rev. Lett. 110, 106804 (2013).
Zhou, Y., Sethi, G., Zhang, C., Ni, X. & Liu, F. Giant intrinsic circular dichroism of enantiomorphic flat Chern bands and flatband devices. Phys. Rev. B 102, 125115 (2020).
Bergholtz, E. J. & Liu, Z. Topological flat band models and fractional Chern insulators. Int. J. Mod. Phys. B 27, 1330017 (2013).
Wirth, G., Ölschläger, M. & Hemmerich, A. Evidence for orbital superfluidity in the P-band of a bipartite optical square lattice. Nat. Phys. 7, 147–153 (2011).
Soltan-Panahi, P., Lühmann, D.-S., Struck, J., Windpassinger, P. & Sengstock, K. Quantum phase transition to unconventional multi-orbital superfluidity in optical lattices. Nat. Phys. 8, 71–75 (2012).
Jacqmin, T. et al. Direct observation of Dirac cones and a flatband in a honeycomb lattice for polaritons. Phys. Rev. Lett. 112, 116402 (2014).
Milićević, M. et al. Orbital edge states in a photonic honeycomb lattice. Phys. Rev. Lett. 118, 107403 (2017).
Gardenier, T. S. et al. p orbital flat band and Dirac cone in the electronic honeycomb lattice. ACS Nano 14, 13638–13644 (2020).
Qiu, W.-X., Ma, L., Lü, J.-T. & Gao, J.-H. Making an artificial px,y-orbital honeycomb electron lattice on a metal surface. Phys. Rev. B 104, 235404 (2021).
Slot, M. et al. p-Band engineering in artificial electronic lattices. Phys. Rev. X 9, 011009 (2019).
Drost, R., Ojanen, T., Harju, A. & Liljeroth, P. Topological states in engineered atomic lattices. Nat. Phys. 13, 668–671 (2017).
Paavilainen, S., Ropo, M., Nieminen, J., Akola, J. & Rasanen, E. Coexisting honeycomb and kagome characteristics in the electronic band structure of molecular graphene. Nano Lett. 16, 3519–3523 (2016).
Wang, Z., Liu, Z. & Liu, F. Organic topological insulators in organometallic lattices. Nat. Commun. 4, 1471 (2013).
Wang, Z., Su, N. & Liu, F. Prediction of a two-dimensional organic topological insulator. Nano Lett. 13, 2842–2845 (2013).
Zhang, L. et al. Intrinsic two-dimensional organic topological insulators in metal-dicyanoanthracene lattices. Nano Lett. 16, 2072–2075 (2016).
Wang, Z., Liu, Z. & Liu, F. Quantum anomalous Hall effect in 2D organic topological insulators. Phys. Rev. Lett. 110, 196801 (2013).
Zhang, X., Zhou, Y., Cui, B., Zhao, M. & Liu, F. Theoretical discovery of a superconducting two-dimensional metal–organic framework. Nano Lett. 17, 6166–6170 (2017).
Buchner, F., Schwald, V., Comanici, K., Steinrück, H. P. & Marbach, H. Microscopic evidence of the metalation of a free‐base porphyrin monolayer with iron. ChemPhysChem 8, 241–243 (2007).
Auwärter, W. et al. Controlled metalation of self‐assembled porphyrin nanoarrays in two dimensions. ChemPhysChem 8, 250–254 (2007).
Rubio-Verdú, C. et al. Orbital-selective spin excitation of a magnetic porphyrin. Commun. Phys. 1, 15 (2018).
Gopakumar, T. G., Tang, H., Morillo, J. & Berndt, R. Transfer of Cl ligands between adsorbed iron tetraphenylporphyrin molecules. J. Am. Chem. Soc. 134, 11844–11847 (2012).
Liu, B. et al. An iron-porphyrin complex with large easy-axis magnetic anisotropy on metal substrate. ACS Nano 11, 11402–11408 (2017).
Kim, D. & Liu, F. Realization of ideal flat band by rotated d-orbitals in Kagome metals. Preprint at arXiv:2212.09791 (2022).
Fleischer, E. B. & Shachter, A. M. Coordination oligomers and a coordination polymer of zinc tetraarylporphyrins. Inorg. Chem. 30, 3763 (1991).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Perdew, Burke, and Ernzerhof reply. Phys. Rev. Lett. 80, 891 (1998).
Perdew, J. P., Burke, K. & Ernzerhof, M. Errata: generalized gradient approximation made 521 simple. Phys. Rev. Lett. 78, 1396 (1997).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Liechtenstein, A. I., Anisimov, V. I. & Zaanen, J. Density-functional theory and strong interactions: orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 52, R5467 (1995).
Acknowledgements
This work is financially supported by Hong Kong RGC No. 16301820, 16301921, and N_HKUST615/21. B.X. and F.L. are supported by US DOE-BES (Grant No. DE-FG02-04ER46148).
Author information
Authors and Affiliations
Contributions
N.L. conceived the project; The molecules were synthesized by S.-Q.F. and J.-Y.C. under the supervision of P.-N.L. The sample preparation and characterization, and data analysis were carried out by X.-B.W. and C.-K.L. under the supervision of N.L. Theoretical analysis was completed by E.L., B.X., and D.K. under the supervision of F.L. Manuscript was prepared by X.-B.W., B.X., C.-K.L., and N.L.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, XB., Xia, B., Lyu, CK. et al. A p-orbital honeycomb-Kagome lattice realized in a two-dimensional metal-organic framework. Commun Chem 6, 73 (2023). https://doi.org/10.1038/s42004-023-00869-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-023-00869-7
This article is cited by
-
Crystal net catalog of model flat band materials
npj Computational Materials (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
