
Abstract
Technetium-101 (101Tc) has been poorly studied in comparison with other Tc isotopes, although it was first identified over ~80 years ago shortly after the discovery of the element Tc itself. Its workable half-life and array of production modes, i.e., light/heavy particle reactions, fission, fusion-evaporation, etc., allow it to be produced and isolated using an equally diverse selection of chemical separation pathways. The inherent nuclear properties of 101Tc make it important for research and applications related to radioanalytical tracer studies, as a fission signature, fusion materials, fission reactor fuels, and potentially as a radioisotope for nuclear medicine. In this review, an aggregation of the known literature concerning the chemical, nuclear, and physical properties of 101Tc and some its applications are presented. This work aims at providing an up-to-date and first-of-its-kind overview of 101Tc that could be of importance for further development of the fundamental and applied nuclear and radiochemistry of 101Tc.
Similar content being viewed by others

Introduction
The element technetium, with atomic number (Z) Z = 43, was discovered in 1937 through the transnational collaboration between American particle physicist E. O. Lawrence and Italian radiochemists E. Segrè and C. Perrier1. It was serendipitously synthesised by Lawrence during the prolonged bombardment of a molybdenum (Mo) plate with deuteron particles. Segrè and Perrier, responsible for the subsequent radiochemical workup of the activated Mo plate, isolated a new material with chemical and nuclear properties unlike any of the known elements at the time. Determining that they had isolated a new element, Segrè and Perrier were credited with the discovery and later in 1947 coined the novel element technetium (Tc), derived from the Greek word for artificial, τεχνητoζ2,3. With the help of B. Cacciapuoti, the first isotopes of Tc to be identified from this initial experiment were 95mTc (t1/2 = 61 d) and 97mTc (t1/2 = 91 d)4,5.
Today, there are ~35 isotopes of Tc that are known with atomic masses (A) spanning from A = 85 to 120. None of the isotopes of Tc are stable, making it the lightest inherently radioactive element on the periodic table6. On Earth, 99gTc (t1/2 = 2.11 × 105 y) is the predominant naturally occurring Tc isotope where it is found in ultra-trace quantities due to the spontaneous fission of 238U (uranium), induced fission of 235U by neutron capture in U-bearing ores, and capture of cosmic-ray neutrons in Mo-ores7,8. However, the dominant sources of Tc are anthropogenic in origin and are artificially generated through nuclear transmutation reactions.
The most prevalent Tc isotope today is 99gTc, which is primarily sourced from fission of both 235U (with a fission yield ~6.2%) and 239Pu in nuclear reactors and nuclear weapons detonations. Because of its high fission yield and long t1/2, 99gTc is generated in appreciable quantities in the spent fuel where it accumulates9. Although current waste strategies focus on retention and controlled disposal of Tc10,11, legacy mitigation policies were less stringent and discharge of Tc-carrying effluent streams into the environment was a common practice12. Thus, the introduction of bulk Tc into the biosphere can be attributed to a combination of these various practices and actions13.
99gTc is also formed via 99mTc (t1/2 = 6.01 h), which serves as the backbone of the radiopharmaceutical industry, pertaining to diagnostic imaging, and presently constitutes for tens of millions of procedures worldwide annually14. From an applications perspective, 99mTc is the most frequently used Tc isotope for commercial, industrial, and/or economic purposes. Despite Tc’s broad capacity in regards to coordination chemistry, catalysis, superconductivity, corrosion resistance, etc., the innate radioactivity associated with its longer-lived isotopes makes its handling and widespread use complicated9; thus, the use of its shorter-lived Tc isotopes (i.e., 94mTc (t1/2 = 52.0 min), 94gTc (t1/2 = 293 min), 95mTc, 95gTc (t1/2 = 20.0 h), 96mTc (t1/2 = 51.5 min), 96gTc (t1/2 = 4.28 d), 99mTc, and 101Tc (t1/2 = 14.2 min) in radioanalytical or medical tracer studies and similar applications are the most commonly employed.
Of the aforementioned isotopes, 101Tc is comparatively one of the lesser discussed in the literature. However, its unique nuclear properties and varying routes of production make it an interesting Tc isotope nonetheless. Moreover, it is present in an array of applications, where its shorter t1/2 is exploited. The motivation behind this review was our recent work highlighting 101Tc production and isolation using a fusion-based compact accelerator neutron source15. Thus, a collection of some of the existing literature on 101Tc pertaining to its discovery, nuclear properties, synthesis, and application is provided in order to allow a better understanding of the isotope. With rapid developments being made in the areas of nuclear safeguards, medical isotopes, fusion energy materials, and novel fission technologies, it is plausible that 101Tc and the A = 101 isobar may see a mirrored growth in interest from the scientific community.
Discovery of 101Tc
Nystrom and Thoennessen provide a comprehensive list of the known Tc isotopes and a brief description of their discoveries, including 101Tc16. They state that the discovery of 101Tc, was simultaneous amongst two groups in 1941: (1) Maurer and Ramm17, and (2) Hahn and Strassmann18,19. The credit for the discovery of 101Tc was given to Maurer and Ramm as well as concession by Hahn and Strassmann for identifying the puzzle of the near-identical half-lives of 101Mo and 101Tc. However, prior publications in the literature, which are actually alluded to in the work of Hahn and Strassmann19, point to evidence of the production and identification of 101Tc, contradicting this account.
Japanese researchers Sagane et al. reported in two publications from 1940 a new isotope of masurium (Ma), which was an unofficial name of Tc predating its current title, with a mass of A = 10120,21. Samples of Mo were activated with slow neutrons, and using an undisclosed, rapid separation the new Tc (Ma) isotope was isolated from its parent radionuclide 101Mo. Both half-lives were measured: for 101Mo, a t1/2 value of 19 ± 1 min was determined, whereas for 101Tc (101Ma) the value was 9 ± 1 min with a corresponding β− energy of 1.14 MeV20. Likewise, Sagane et al. were able to determine that 101Tc (101Ma) was effectively produced by exposing Mo in a slow neutron field, but not a fast one whereas 99Ma (i.e, 99mTc) was, which is consistent with what is known about these isotopes today (e.g., 100Mo(n, 2n)99Mo; 99Mo → 99mTc)22. Therefore, although the works of both Maurer and Ramm and Hahn and Strassmann provided a more accurate measure of 101Tc according to its t1/2, Sagane et al. were the initial group to show isolation and preliminary data that assisted the field in its identification. It should be mentioned that the suggestion of a hypothetical 101Ma by Hahn and Strassmann in the late 1930s prior to its correct identification23 did aid in explaining the process of fission by Meitner and Frisch24.
Interestingly, Sagane et al. point out the recently discovered 99Ma (t1/2 = 6.6 h; i.e., 99mTc) by Segré and Seaborg25 and make a comparative note about developing chemistry of the element using these newly discovered isotopes. They state the use of 101Tc (101Ma) would be more convenient for uncovering its chemistry due to its ease of production with slow neutrons for a short irradiation time (i.e., ~24 min) and its energetic β− emission20. However, in retrospect, 99mTc and 99gTc became much more crucial in the development of Tc chemistry than 101Tc due to other technological advances as discussed earlier9.
Nuclear properties
The isotope 101Tc is situated in the neutron-rich region of known Tc isotopes with 58 neutrons (N) for its 43 protons. Considering its mass, located in the A ~100 transitional region of nuclei shapes from spherical to well-deformed, the ground-state nuclei of the neutron-rich 101Tc is reported to have a higher degree of deformation in comparison to the lighter proton-rich ones, e.g., spherical, semi-magic 93Tc (N = 50)26,27, and lesser than the heavier ones, such as 103Tc (N = 60)28. For example, in the positive and negative parity bands of odd-Z Tc isotopes (A = 95–101), a systematic decrease was observed in level energies with the same spin according to increasing neutron number. Likewise, the level order inversion for the low-spin states in the π5/2+ state, following this same trend, reduces across 95,97,99,101Tc, finally disappearing for 103Tc. These phenomena were attributed to the increasing deformation parameter and decreasing Coriolis interaction consistent with the increase in neutron number29. Accompanying the increase in neutron number from N = 50 to 60, a transition from a near-spherical to a triaxial-deformed nuclei shape along with an increase in the quadrupole deformation was determined using Routhian surface (RTS) calculations. Triaxiality is predicted to initiate in 97Tc and gradually evolve throughout 99,101,103Tc29. It was determined that the shape transition of the 101Tc nucleus is similar to that of of 101Mo, where shape coexistence of the nucleus occurs. Due to the proximity of Z = 43 to the Z = 40 sub-shell closure, the strength of proton-neutron interactions can alter the effects of this sub-shell, thereby manifesting shape changes and coexistence30.
In the A = 101 isobar chain, 101Tc is directly preceded by its parent isotope 101Mo (t1/2 = 14.61 min) and is formed through β− decay (Eq. 1)31,32,33. The decay occurs through the Jπ = +1/2 ground-state transition of 101Mo to the Jπ = +9/2 ground-state of 101Tc. Wiles conducted a study on low-level decays of 101Mo and 101Tc, and was unable to identify a p1/2 isomeric state for 101Tc34; however, an isomeric state of 760 ± 50 µs at Eγ = 192.0 ± 0.2 keV was induced via the (γ, p) photonuclear reaction with 26 MeV LINAC Bremsstrahlung on a ruthenium (Ru) target35, which was also observed by Bartsch et al.36. Meanwhile, high-spin structures of 101Tc have been studied up to Jπ = +31/2 via its production as a fission fragment from the bombardment of 176Yb with 28Si at 145 MeV; backbending in 101Tc was shown to occur similar to that of 102Ru with the same rotational frequency and excitation energy37.
As shown in Fig. 1, 101Tc decays by β− emission, where it generates the stable end-member of the A = 101 isobar, 101Ru, with a transition from the Jπ = +9/2 to the Jπ = +5/2 state33. The t1/2 determination of 101Tc has been the topic of many studies as shown in Table 138,39,40. The presently accepted value for the t1/2 of 101Tc is 14.22(1) min, which most closely resembles that determined by Abouzi et al., i.e., 14.224 ± 0.008 min41. However, more recent measurements have highlighted discrepancies in this value, and the measured t1/2 of 101Tc, which was free from 101Mo during the measurement, was 14.02 ± 0.01 min42. It is noted that in another recent experiment, da Silva et al. conducted measurements on 22 samples and resolved the t1/2 of 101Tc from the weighted results to be 13.725(13) min; the significantly smaller value compared to the tabulated one was attributed to the larger t1/2 measured for 101Mo, i.e., 14.893(13) min, being an interference43. The work of Hamida represents a much longer t1/2 in comparison to others, although no indication was provided for this discrepancy44. The most recent value of 16.0 min comes from Steinetz et al. in 2017, where 101Tc was generated via photon irradiation of deuterated lanthanide-Mo samples. Gamma-ray (γ) spectroscopy was performed using HPGe detectors, although no separation of parent-daughter isotopes was executed prior to measurements45. Considering all values collected here, the average t1/2 of 101Tc was determined to be 14.42 ± 1.92 min, or 1.39% difference from the reported value of 14.22 min.

Image generated via Evaluated Nuclear Structure Data File (ENDSF) database33.
The overall Q-value associated with the decay of 101Tc is roughly 1613 keV (Fig. 1). Table 2 shows the β− emissions associated with 101Tc decay and their respective properties. The average Eβ is ~473.6 keV and is made up of several distinct β− emissions occurring at 225 keV (0.85%), 263 keV (1.92%), 385 keV (6.44 %), and 487 keV (90.3%). Its βmax of ~1320 keV corresponds to its highest energy β− decay as shown in Table 233. As previously stated, 101Tc decays into stable 101Ru, the end-member of the A = 101 isobar.
There are a host of characteristic γ-rays emitted during the decay of 101Tc to 101Ru as shown in Fig. 1 and Supplementary Table S1. Martin et al. was the first to study the γ-ray transitions associated with this occurrence, establishing 22 γ-rays46, and has since been the subject of multiple studies41,47,48,49,50,51,52,53. A more recent study by Hammed et al. catalogues 30 characteristic γ-ray peaks that were consistent with those reported in the literature, although not all of the ones reported previously were present in their investigation31. The dominant γ-rays associated with the decay of 101Tc and their respective absolute intensities per 100 decays are: 127.22 keV (2.63%), 184.12 keV (1.60%), 306.83 (89%), and 545.05 (5.96%).
Production
Table 3 summarises the production routes for 101Tc addressed in this section.
Neutron-driven reactions
Neutron capture reactions provide the production of 101Tc through both direct and indirect processes depending on the target material being irradiated. To clarify, a direct route would be the formation of 101Tc directly from the interaction of the neutron field with the target material, whereas indirect would generate an intermediate radionuclide that in turn yields 101Tc from its decay. The most likely known pathway for producing 101Tc is indirectly via 101Mo. For example, neutron capture on 100Mo generates the parent isotope 101Mo, i.e., 100Mo(n, γ)101Mo, which subsequently decays to 101Tc as shown in Eq. 1. This indirect production route was first explored in 1940 by Sagane et al.20,21 as previously described.
Shown in Supplementary Fig. S1 is the ENDF/B-VIII calculated neutron capture cross-section for 100Mo up to En = 20 MeV. The thermal neutron capture-cross section of 100Mo is σc = 0.199 ± 0.003 b. For epithermal, intermediate, and resonance energy neutrons, there is an increased probability of interaction on 100Mo, and the resonance integral cross-section (Ic) of 100Mo is approximately Ic = 3.76 ± 0.15 b54,55,56. The epithermal to thermal neutron capture cross section ratio is ~18.8.
For fast neutrons, the probability of neutron capture on 100Mo drastically decreases after En ~1 MeV, and the likeliness of other neutron-driven reactions, such as (n, 2n), (n, p), (n, n’), etc. begin to dominate57,58. In fact, Sagane et al. observed this behaviour by using lithium-deuterium (Li-D) fast neutrons to irradiate a 100Mo-containing source, which was enclosed in a cadmium (Cd) box filled with boric acid in order to remove the slow neutron component. They ascertained that the lack of measurable presence of a t1/2 similar to that identified for 101Ma (i.e., 101Tc), suggested that its formation was predominated by (n, γ) reactions on 100Mo59. Similarly, Mausolf et al. demonstrated the production of 99mTc and 101Tc via (n, γ) reactions using a D-D neutron generator outputting 2 × 1010 n·s−1 2.45 MeV fast neutrons that were scattered down into the epithermal and thermal regions in an aqueous ammonium heptamolybdate (AHM, (NH4)6Mo7O24·4H2O) target15. In general, when using the neutron capture route for 99Mo/99mTc production, whether it be an accelerator, reactor, or another neutron source, the co-production of 101Mo/101Tc will occur when using targets that are of natural isotopic composition or contain fractions of 100Mo. Those responsible for commercial irraditations in the 99Mo/99mTc industry generally view 101Mo/101Tc as non-problematic impurities, as they typically decay away within a relatively short period post-irradiation60.
Steintez et al. observed the production of 99mTc and 101Tc from their respective Mo parent radionuclides via low-energy (~2 MeV) photon irradiation of deuterated lanthanide elements erbium (Er) and hafnium (Hf) mixed with Mo, which was used as a “witness material” for tracking initiated nuclear reactions, in a deuterated paraffin matrix, e.g., ErD2.8 + C36D74 + Mo and HfD2 + C36D74 + Mo. The irradiations produced neutrons with thermal to epithermal energies, neutrons in the 1.4–2.5 MeV fast range, and those with energies >10 MeV were also detected. Irradiations performed under similar conditions of the hydrogenated counterparts as control samples yielded no induced radioactivity in the Er, Hf, or Mo materials45.
Daly et al. presented the simultaneous production of 101Tc using secondary neutrons generated during the proton irradiation of an Mo target in a cyclotron in order to determine the contribution 101Tc formation either from (n, γ) or (p, γ) reactions on 100Mo in the target itself. For this, a secondary Mo target was placed outside of the incoming beam of the cyclotron and was irradiated with neutrons emitted from the primary target to determine the contribution arising from the (n, γ) pathway alone61. Similarly, Mayordomo et al. demonstrated the simultaneous production of 101Tc and 99mTc during routine 18F[FDG] production on a low-energy biomedical cyclotron. Secondary, natural Mo targets were situated adjacent to the primary 18F target, where anisotropic neutrons are generated as a byproduct via the 18O(p, n)18F reaction specifically, and irradiated within the resulting neutron field62.
Production routes using alternative target elements other than Mo have been investigated, such as rhodium (Rh) and Ru. For Rh (Supplementary Table S2), the first report of the 103Rh(n, 3He)101Tc reaction was from Fervert in 196563. Shortly after, Gray et al., using a Crockcoft-Walton accelerator with a D2+ beam to generate 14.7 MeV deuterium-tritium (D-T) neutrons with fluxes of 1 × 109 to 5 × 1010 n·s−1, irradiated samples of Rh(OH)3 and RhCl364. The Q-value of the reaction was determined to be −8.7 MeV and the measured cross-section was < 400 nb. The reported value by Csikai in the same year was similar for 14.7 MeV neutrons at σ = 1.3 ± 0.6 µb65. Husain et al. explored the 103Rh(n, 3He)101Tc reaction using 14.8 MeV neutrons with a yield of 1010–1011 n·s−1 generated in a 400 kV Cockcroft-Walton accelerator via the T(d, n)4He fusion reaction. The experimentally determined cross-section was 2.0 ± 0.6 µb at En = 14.8 MeV66. Using a Rh foil enriched in 103Rh and a Cockcroft-Walton D-T accelerator, Diksic et al. obtained the highest cross-section value at 14.6 MeV of 16 ± 7 nb67.
With fast neutrons 101Tc can be generated through both direct and indirect routes using Ru targets. For the direct method, the reaction 101Ru(n, p)101Tc was first investigated by Paul and Clarke in 1953, however, unintentionally. The nuclide they intended to generate was 101Ru via neutron bombardment on an Ru target, although it is now known the identified ~15 min t1/2 is attributed to 101Tc and 101Ru is stable68. Luo et al. later studied this reaction and the measured cross-sections were: 15.7 ± 2.0, 18.4 ± 2.7 and 22.0 ± 2.4 mb at 13.5 ± 0.2, 14.1 ± 0.2 and 14.8 ± 0.2 MeV incident neutron energies, respectively69. Taking into account other potential reaction pathways from Ru targets as presented in Table 3, the authors were able to more accurately assign contributions arising from each route to be 0.316σ(102Ru(n, d*)101Tc) + 0.17σ(101Ru(n, p)101Tc) reaction cross-sections. These values were consistent with those reported previously70,71 as shown in Supplementary Fig. S2, although it is noted that both the calculated cross-sections from ENDF/B-VIII and JEFF-3.3 tend to overestimate these values, especially JEFF-3.3 in the lower energy regime between ~6 and 15 MeV.
The reaction 102Ru(n, d*)101Tc is another direct route72 with a reported Q-value of −6.119 MeV. Using fast neutrons, the reaction cross-sections were determined to be ~0.63 ± 0.07 mb, 1.23 ± 0.10 mb, and 2.77 ± 0.1 mb at 13.5 ± 0.2 MeV, 14.1 ± 0.2 MeV, and 14.8 ± 0.2 MeV, respectively69,73. The experimental values measured for this reaction are underestimated by ENDF/B-VIII calculations and slightly overestimated for JENDL-5, although the JENDL-5 trends better in general (Supplementary Fig. S3).
For the indirect method, the 104Ru(n, α)101Mo reaction proceeds with a Q-value ~2.05 MeV64. The most recent experimentally derived cross-sections were determined to be 4.6 ± 0.5 mb at En ~13.5 MeV and 6.4 ± 0.2 mb at 14.8 MeV74. For the latter data point, it trends better with those from earlier reports, i.e., Paul et al. and Gray et al., than those reported by Kasugai et al. in a similar energy regime (Supplementary Fig. S4)75.
Fission-based reactions
Concerning the library of knowledge on fission reactions and the formation of fission products, it is not the intention of this review to fully encompass all studies that have been performed. The majority of these are comprised of the fission of actinide elements that either occurs spontaneously or is induced by a bombarding particle, such as n, p, d, α or γ. Instead, the intent here is to showcase a handful of examples that demonstrate the production of 101Tc through fission-based means. Interestingly, the mass region in which A = 101 is encompassed, i.e., A ~ 96–106, exhibits enhanced fission yields of primary fission fragments, containing 82 neutrons or 50 protons, and their respective secondary fission fragments arising from reactions such as 235U(n, f), 238U(γ, f), 235U(d, f), 238U(d, f), 233U(n, f), and spontaneous fission of 242Cm (curium), due to fine structure associated with closed nuclear shells76.
The first report of the fission-derived A = 101 isobar 101Mo and/or 101Tc was Hahn and Strassmann, identifying it post-U fission18,19. Fission of U in a thermal or fast neutron flux forming these isotopes of interest has been the subject of several studies77,78. The neutron-induced fission yields from 235U of A = 101 are relatively abundant with ~5.17% and ~5.24% for thermal and fast neutrons, respectively; however, for 14 MeV neutrons the yield is slightly less (Table 4)79. For the A = 101 decay chain, 101Mo is preceded by 101Nb, but due to its shorter t1/2, i.e., (t1/2 = 7.1 s), it is typically not observed unless monitoring the reaction in real-time. It is also noted that the independent fission yield for 101Tc from 235U(n, f) is rather low, i.e., < 2.0 × 10−4%80, therefore in these types of experiments, contribution from this pathway is very little in comparison to the A = 101 isobar.
In another example, Srivastava et al. evaluated the yields of the heavier, neutron-rich isotopes of Tc including 101Tc from the thermal neutron-induced fission of 239Pu (plutonium); yields were compared with those obtained from the neutron-induced fission of 235U and the spontaneous fission of 252Cf (californium)81. The reported fission yields of 239Pu for thermal and fast neutrons for A = 101 are ~6.18% and 6.63%, respectively. In comparison to the photon-induced fission of 240Pu, the respective cumulative yield is relatively higher than those previously mentioned for 239Pu, where for photofission 101Mo and 101Tc account for the highest fission product yields amongst the lighter fission fragments82.
Rattan and colleagues demonstrated that 209Bi (bismuth) could be fissioned with high-energy α-particles generated in a variable energy cyclotron (VEC) to generate 101Mo83. The measured fractional cumulative yields of 101Mo were 0.655 ± 01.34% and 0.688 ± 0.114% at 55.7 and 58.5 MeV, respectively. The σfission values of the two different energies were comparable to each other, e.g., 3790 ± 242 µb at 55.7 MeV and 3640 ± 522 µb at 58.5 MeV.
Light ion reactions (p, d, t, and α beams)
Several light ion reactions have been established for generating 101Tc using various particle accelerators. These include accelerators employing incident beams of p, d, t, and α. Typically, reactions of this nature are performed using high-energy cyclotrons, although alternative means of production have been identified. Under these circumstances, both routes, direct and indirect, for production of 101Tc are attainable depending on the target isotopics, beam type and beam energy employed.
Daly et al. irradiated 100Mo with 5-20 MeV protons and identified the 100Mo(p, γ)101Tc pathway61, which has been the subject of several studies. This pathway is of particular interest as it is a side reaction that occurs during the production of 99mTc via (p, 2n) on 100Mo84. The calculated Q-value assigned to this reaction is +7.441 MeV, although it is reported that this reaction is rare for medium mass targets. The measurements performed by Daly et al. were consistent with nuclear calculations by Qaim et al. with a σmax (~1 mb) at Ep ~14 MeV. However, more recent data from Lamere et al. and Gagon et al. show consistent, yet relatively lower σmax than previously reported as shown in Supplementary Fig. S585,86.
Alternatively, direct production of 101Tc can be accomplished via the (d, n) reaction utilising high-energy deuterons, i.e., ~13 MeV, on 100Mo with a corresponding Q-value of 5.2 MeV. Shown in Supplementary Fig. S6 is the experimentally measured cross-sections for this reaction between Ed = 3.9 and 11.3 MeV, where σmax (~300 mb) occurs at Ed ~7.3 MeV. For Ed = 11.7 MeV, the thick target yield (Y) and production rate (R) were determined to be 10,378 µCi·µA−1 h−1 and 3493 µCi·µA−1, respectively87.
For the indirect production of 101Tc via 101Mo, Randa et al. measured the excitation functions for the 100Mo(d, p)101Mo reaction with deuteron beams < 13 MeV88. The resulting cross-section data for this reaction is shown in Supplementary Fig. S7, where the corresponding Y and R values were 6495 µCi·µA−1 h−1 and 2280 µCi·µA−1, respectively. In comparison to this experimental dataset, calculations via TENDL-2019 tend to underestimate reaction cross-sections with increasing energies ≥ 6 MeV. With the purpose of studying the nuclear structure of Mo isotopes such as 101Mo, Hjorth et al. have also reported the irradiation of Mo targets with 15 MeV deuterons89. Habib et al. performed irradiations of 100Mo with both a 12 MeV deuteron and a 14 MeV triton beam in order to target the 100Mo(d, p)101Mo and 100Mo(t, d)101Mo reactions, respectively, with the intention of studying the nuclear structure and spectroscopic states of 101Mo90.
Shown in Supplementary Fig. S8 is the experimental cross-section data for the 98Mo(α, p)101Tc reaction measured by Levkovski91. Under the conditions tested, the σmax was measured at Eα ~24 MeV with a value of ~14.5 mb. In comparison to the TENDL-2019 calculations, the experimental cross-section data is notably higher for the corresponding energies than those provided by the calculation.
Heavy ion fusion-evaporation reactions
Heavy ion fusion-evaporation reactions are another potential route for the production of either 101Tc or 101Mo that are usually performed at high-energies situated at or slightly above the Coulomb barrier for a particular heavy ion beam on a given target. These can be further characterised by complete and incomplete fusion reactions, depending on the degree/capacity for breakup fragments to fuse with the target. In general, incomplete fusion (ICF) or massive-transfer (MT) reactions proceed through a compound-nucleus-like de-excitement that yields an array of products by evaporation or γ-ray decay, similarly to that of the process of fission.
Dejbakhsh et al. studied high-spin states of 101Tc via the 100Mo(7Li, α2nγ)101Tc reaction30. Experimentally, this was completed by bombarding a metallic target of Mo with a 49-MeV 7Li beam in a high-energy cyclotron, and successively assessing the Z = 1 and 2 exit channels, i.e., 101-104Ru and 101-103Tc, with 102Ru and 101Tc yielding the highest statistics for the two channels, respectively. Agarwal et al. also reported the formation of 101Tc during the ICF dynamics of the high-energy bombardment of 18O on a 93Nb (niobium) target through the 93Nb(18O, 2α2p)101Tc reaction pathway92. Once again, 101Tc is one of many residues that was formed under these circumstances. The excitation functions of this particular pathway are shown in Supplementary Fig. S9.
Photonuclear reactions
Perlman et al. studied various photonuclear reactions on different elemental targets including Ru. For example, Ru metal was irradiated with photons produced using a betatron via the high-energy electron bombardment (i.e., 50 and 100 MeV) of a tungsten plate, and the formation of 101Tc was ascribed to the (γ, p) reaction on 102Ru38. As mentioned previously, both groups Uyttenhove et al. and Bartsch et al. employed high-energy linear electron accelerators (i.e., 32 and 65 MeV, respectively) to induce the 102Ru(γ, p)101Tc reaction35,36.
Although not a direct result of the photonuclear reaction itself, both 101Mo and 101Tc were reported during the photonuclear production of 99Mo in enriched 100Mo targets. It is likely that secondary neutrons liberated within the Mo target via the 100Mo(γ, n)99Mo reaction were captured within itself, in turn forming this radionuclidic pair93. For example, calculations corresponding to yields at the end of bombardment (EOB), the two most prominent radioisotopes aside from 99Mo were 101Mo and 101Tc using a 20.2 MeV, 45 µA beam over an 82-min irradiation94. A similar process was described by Tsechanski et al. when employing a one-stage approach for the photonuclear production of 99Mo using natural Mo targets. Under this scenario, 99Mo would be generated from two different nuclear transformation pathways: (1) the primary photonuclear reaction on 100Mo, and (2) successive neutron-capture on 98Mo from secondary neutrons generated as described above95.
Separation
There exists a myriad of separation techniques for removing Tc from simple or complex mixtures of neighbouring host elements, such as Mo, Ru, Rh, etc.9. However, due to the short-half life of 101Tc, as well as its parent isotope 101Mo when not generated directly, it is necessary that the separation protocol implemented must be fairly rapid, i.e., approximately equivalent to one t1/2 of 101Tc96. Therefore, not all of the known options for separating longer-lived Tc isotopes from a host target material may be feasible for 101Tc. In this regard, there have been a number of viable separation platforms for isolating 101Tc that have been reported in the literature, including liquid-liquid extraction, chromatographic/solid-phase extraction, precipitation, and volatilisation (Table 5). Each of these methods will be expounded upon in the following section.
Liquid–liquid extraction
Liquid–liquid extraction is an efficient methodology for partitioning a mixture of elements where differences in charge and/or polarity of elements or complexes in solution can be exploited, for example, the use of polar aqueous and non-polar organic systems, which are immiscible or only moderately miscible with each other. When only small volumes of liquid are required to be processed and sufficient distribution coefficients (Kd) for a system can be achieved, then liquid-liquid-based separations can be completed rapidly and on a continuous or near-continuous basis.
A predominant oxidation state of Tc is Tc(VII), where the hallmark compound under oxidising conditions is [TcO4]−, a chaotropic, low charge density complex that has the tendency to uptake into/onto non-polar solvents/mediums97. This behaviour makes it ideal for its recovery into non-polar solvents, which has been the key principle behind the low-specific activity 99Mo generator using methyl ethyl ketone (MEK) to remove [99mTcO4]− from aqueous solutions of 99Mo-containing targets98.
Most literature using liquid–liquid extraction for isolating 101Tc reported employing CHCl3 as an organic solvent in conjunction with bulky coordinating cations, such as [(C6H5)4As]+ and [C25H46N]+, for removing the corresponding [TcO4]-salt from a caustic aqueous solution50,51. For example, while exploiting the varying chemistries between Ru/Rh and Tc, Gray et al. were able to perform quick separations on the order of ~12 min to isolate 101Tc from the corresponding target material. A 99mTc/99gTc tracer was used to track separation efficiencies, which varied between 50 and 80%, where 101Tc was extracted into an organic phase of CHCl3 from an aqueous solution of the dissolved Ru/Rh trichlorides targets containing 1% (C6H5)4AsCl. Subsequent analyses of 101Tc were performed on the isolated organic phase, in which Tc is found as the complexed form (C6H5)4As[TcO4]64.
Using comparable extraction conditions, e.g., (C6H5)4AsCl in CHCl3 and KBrO3/HNO3, Hild et al. employed a chemistry device called a MicroSISAK for isolating short-lived Tc isotopes, 101Tc and 104Tc99. The MicroSISAK is a micro-membrane extractor that was constructed for the purpose of continuously isolating short-lived, heavy elements such as bohrium (Bh). The Tc isotopes were generated online during the neutron-induced fission of 235U in a TRIGA reactor and transported with He/KCl gas jet from the target to the device. The device contains a micro-mixer element that intimately contacts the organic and aqueous phases prior to phase separation with a hydrophobic Teflon membrane. The extraction yield at 40 °C was 76 ± 1% for 104Tc, which was comparable for similar experiments performed with 99mTc with 83 ± 3%99.
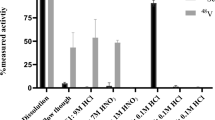
You et al. demonstrated the isolation of 101Tc from an irradiated aqueous AHM target using liquid–liquid extraction42. The irradiated solution was acidified with HNO3, after which the bulk Mo target material was removed using a solution of α-benzoic oxime-ethyl acetate as the organic phase. The Tc daughter remained in the acidified aqueous phase and was extracted again with the organic phase to remove any remaining Mo (i.e., 93Mo, 99Mo, and 101Mo) prior to performing subsequent measurements to ensure only 101Tc was present. The experimental design was aimed at maximising 101Tc yield and purity, so a quick irradiation in combination with a rapid separation protocol was adopted.
Experiments by Groening and Harbottle on “hot atom chemistry,” or the recovery of recoil products via radioactive decay or activation using a nuclear reactor or accelerator, investigated the separation 101Mo and 99Mo from a host matrix of Mo(CO)6 post-neutron bombardment100. Retention of 101Mo in the host phase was upwards of ~60% post-irradiation, however, effects of isochronal annealing (i.e., 10 min) of the target material prior to liquid-liquid extraction showed correlative increases in 101Mo retention upwards of ~85% at an annealing temperature of 120 °C. The separation of 101Tc/99mTc from an irradiated Mo(CO)6 matrix containing 101Mo/99Mo was also performed. Tc isotopes were removed in the aqueous solution, where no stable partition between the organic and aqueous phases could be established. This phenomenon, which was more appreciably observed for 101Tc, was attributed to likely instability, such as hydrolysis, of a Tc-CO and/or mixed Tc–Mo-CO species in the presence of an aqueous HClO4 environment. For 99mTc, the data showed that ~59% of it present post-irradiation was in a non-volatile form, which also indicates potential for separation of Tc from Mo(CO)6 via volatilisation100.
Ion exchange (IE)/extraction chromatography
The use of IE or extraction chromatography can be a robust and rapid technique for isolating a single elemental fraction from a heterogeneous mixture of elements in solution. This technique is widely used for isolating short-lived radioisotopes in heavy element chemistry, radiopharmaceutical applications including positron emission tomography (PET), and for front or backend refinement in neutron activation analysis (NAA). In regards to 101Tc there are several accounts on the use of IE/extraction chromatography for its isolation.
For example, Brodskaya reported irradiating a sample of Mo with neutrons, forming both 99mTc and 101Tc. A rapid extraction of these Tc isotopes from an aqueous Mo solution was completed using column chromatography. From the record, it is not certain what the stationary phase was composed of, although it is suspected that HDEHP on an inert support matrix was used101. Under these conditions, HDEHP is selective for Mo, and it is likely the resulting Tc is eluted from the column as in a standard radionuclide generator. Mausolf et al. reported the isolation of 101Tc and 99mTc from low-specific activity 101Mo and 99Mo, respectively, using activated carbon (AC) to remove carrier-free Tc from an aqueous, acidic solution of AHM15. The separation was performed immediately after a ~15 min neutron irradiation by passing the AHM solution containing 101Mo/101Tc over a small column of AC, which was subsequently washed and isolated for successive characterisation. Several physicochemical mechanisms behind the uptake of Tc onto AC have been reported, such as electrostatic interactions, physisorption, chemisorption/ion-exchange, etc. For example, the protonation of AC surface functional groups at pHs below the point of zero charge (pzc), where carboxylic, carbonyl, laconic, and phenolic groups provide positively charged R–C=O and R–C–OH moieties, helps facilitate the AC sequestration of Tc as [TcO4]–.
Precipitation
The use of radiochemical separations via precipitation is a fairly common, straightforward procedure where the insolubility of one element or compound is exploited over another. However, one drawback to this technique is that a carrier-agent is typically added to promote precipitation and to ensure full recovery of the radioisotope, which typically exists in very minute quantities according to mass. When a stable isotope of the chemical element of interest is available, then this is suggested for use, but if there is no stable isotope, such as for Tc, then a homologous element is considered, i.e., rhenium (Re), or one with similar chemistry may be an alternate option. If it can be handled in appreciable quantities, then a long-lived isotope such as 99gTc is also technically feasible.
Daly et al. reported the use of precipitation as a means of separating 101Tc from a proton-irradiated sample of 100Mo via the (p, γ) reaction61. A short irradiation was performed, i.e., 15 min, on enriched MoO3 (96.9% 100Mo) and the resulting target material was dissolved in a mixture of NaOH and Na2S2O8. Following dissolution, the liberated 101Tc was recaptured via precipitation with a Re-carrier and (C6H5)4AsCl in the form of (C6H5)4As[ReO4]. Isolated samples with 95% yields were produced within one t1/2 of 101Tc.
Volatilisation
The elements Tc and Mo, as well as some of their neighbouring elements Ru and Rh, are able to form various volatile compounds according to the chemical and physical environment. By exploiting the differences in vapour pressures of elements or compounds of interest, selective volatilisation can potentially be employed for separating them. In particular, oxide, halide, oxyhalide, and carbonyl complexes of Mo and Tc are well-known for their volatile nature9.
Götz et al. have demonstrated the atom-at-a-time production and separation of the A = 101 chain member 101Mo via the neutron-induced fission of 235U and spontaneous fission of 248Cm. The application of flowing N2/CO(g) at elevated temperatures and pressures over the fission-based targets allowed generation of isotopes of Mo as the volatile, mononuclear Mo(CO)6 complex and their continuous separation102. The total efficiency (εtot) of 101Mo(CO)6 was determined to be 49%. Because Tc does not form a labile mononuclear CO complex, but rather a binuclear one Tc2CO10103, it is typically disregarded for atom-at-a-time production and separation104.
In general, the breadth of known separation methods for 101Tc is underwhelming in comparison to those that have been applied for other Tc isotopes, in particular 99mTc and 99gTc. Because of the commercial application of 99mTc and the implications of 99gTc removal from spent nuclear fuel, most of the separations knowledgebase of for Tc have focused on its isolation from low-specific activity Mo targets for 99mTc and fissioned uranium (U)-based materials for 99gTc105. For the latter, recovery of Tc from fission-generated feedstocks can be performed using a miscellany of techniques such as ion exchange resins (i.e., Purolite®-A520E/A530E/A532E, AmberliteTM-IRA-400, TEVA®, SuperLig®−639, ABEC, and ReillexTM HPQ), cationic inorganic frameworks (layered double hydroxides (LDH), etc.), or metal organic frameworks (MOFs) to name a few, none of which have yet been applied for 101Tc105,106,107. However, the viabilities of these separation methods will be dictated on the ability to streamline processing of a fission-based target and reduce competition and co-extraction of other fission products. Thus far, selective and real-time volatilisation of Mo or Tc as recoiled fission products from the fission source has proven most effective for isolating minute quantities of 101Mo/101Tc102.
Of the separation pathways presented here, the majority are focused on the use of Mo targets as the source of transmuted 101Tc. A variety of techniques have been explored for isolating Tc from Mo targets under the circumstances for 99mTc radiopharmaceuticals108,109. These include many of the same presented here for 101Tc, such as liquid–liquid extraction, column chromatography, volatilization, etc. Interestingly though, it was not apparent that many of those that have been thoroughly investigated for 99mTc have also been tested for 101Tc. For example, liquid-liquid extraction using MEK as the organic extractant for [TcO4]–110; chromatography using high sorption capacity resins for Mo (meso- or nano-based inorganic supports such as Al2O3, TiO2, CeO2, etc.)111; chromatography based on Tc sorption resins (i.e., ABEC-2000, Analig® Tc02, Chemmatrix, Dowex-1×8, etc.)112; molybdate gel generators113; and thermal chromatographic volatilization114. Exceptions are the use of HDEHP, AC, and variations on the use of Mo(CO)6. Separation methods such as carrier-added precipitations, which are typically not used for 99mTc production, are still a commonly employed radiochemical technique. However, the depth of work that has been reported for recovery of 101Tc via precipitation is not obviously extensive, and other bulky precipitating agents for [MO4]– (M = Tc, Re) that could provide similar results include tetraphenylpyridinium (TPPy), tetraphenylphosphonium (TPP), tetrabutylammonium (TBA), etc., or exploiting the limited solubilities of reduced valent states of Tc/Re, such as for MO2 and MS2 (M = Tc, Re), and other precipitating or redox agents, i.e., Fe(OH)29,115.
For alternative production routes via Ru or Rh targets, only liquid-liquid extraction was experimented for the isolation of 101Tc. Separations of fission-derived Tc, Ru, and Rh from each other are well-studied under the context of fuel reprocessing as well as is separating transmuted Ru from bulk Tc targets for waste processing; however, it appears that the study of separating trace Tc from bulk Ru or Rh is less known. The account from Gray et al. reported here demonstrates the ability to exploit the varying chemistries of the elements, i.e, [TcO4]– versus M3+ (M = Ru, Rh), which could also be leveraged for other separation techniques64.
Applications
Neutron activation analysis (NAA) of Mo samples
NAA has been a technique heavily employed for the determination of trace quantities of Mo in samples of inorganic or organic origin. In this context, the use of the 101Mo/101Tc radionuclide pair can be an alternative to the 99Mo/99mTc pair for NAA. In 1958, Fukai and Meinke introduced the use of this radionuclidic pair for NAA for determination of Mo content in marine samples via the separation and measurement of 101Tc. Separations were performed using (C6H5)4AsCl and detection of the 307 keV characteristic γ-ray was used116,117. For example, Diksic et al. also demonstrated that the shorter-lived 101Mo/101Tc chain could be utilised to measure the Mo content in bovine liver and orchard leaves118. NAA for the analysis of Mo in plant material was performed by van Zanten et al. via 99Mo and 101Mo. For 101Mo, samples (dried clover, ~0.5 g) were irradiated in a thermal neutron flux of 1.4 × 101 n·cm−2 s−1 for 20 min, dissolved in a sulfuric acid medium, and the 101Mo was recovered with a Mo carrier using liquid-liquid extraction in tri-n-octylamine in kerosene over a 15 min period. The entire process required ~1 h and the detection limit for Mo using 101Mo was 0.1 µg of Mo with a standard error of ~35%, which was qualified using values determined by 99Mo NAA and spectrophotometry119. Other biological materials that have been investigated for Mo content are human hair120, wool120, and urine121.
Sun et al. applying the 101Mo/101Tc pair in combination with pre-concentration via co-precipitation with thionalide–ammonium pyrrolidinedithiocarbamate from high pH solutions, were able to determine the concentration of Mo in the form of molybdate [MoO4]2− in natural waters by NAA122. The detection limit of [MoO4]2− was determined to be about 1 ng·L−1 with ppb to sub-ppb detection limits for natural water samples. The stated advantages were less interferences in the spectra or yield calculations in comparison to the 99Mo/99mTc system (i.e., 59Fe, Eγ = 143 keV; 235U(n, f)99Mo;102Ru(n, α)99Mo), shorter irradiation/measurement times, higher sensitivities with lower background, and safer handling due to less activation in the samples.
Hetherington proposed and investigated the use of NAA to determine the enrichment of Mo targets for 99Mo zirconium molybdate gel generators123. The intent was to determine concentrations of 98Mo, 100Mo, and 92Mo via their activation products and corresponding radionuclidic daughters when applicable, i.e., 99Mo/99mTc, 101Mo/101Tc, and 93Mo (t1/2 = 4000 y). Simulated samples were synthesised by mixing weighted quantities of enriched 98MoO3 (96.8% 98Mo) with natural isotopic MoO3 in various ratios, and the samples were irradiated for durations of minutes to hours with similar cooling periods depending on the radionuclidic pair to be analysed. This technique was shown to be effective with an accuracy of ±2% for determining the isotopic abundances of 98Mo and 100Mo in enriched samples, whereas the resulting activation was insufficient to collect appreciable data on 93Mo.
Fission monitor in nuclear reactors and materials interrogation
Because of its association with fission-based processes, the 101Mo/101Tc radionuclidic pair and A = 101 isobar end-member 101Ru can serve as unique signatures and dialogistic tools78,124,125. For instance, Tohmay et al. were able to measure and differentiate fission product yield ratios of short-lived fission products to certify the enrichment of a natural U sample via thermal neutron fission of 235U and fast neutron fission of 238U; an Am-Be neutron source was used as a source of neutrons coupled with delayed γ-ray measurements on a HPGe detector for analysing fission products. Using this method, variations in fissile and fertile compositions (i.e., enrichment and depletion) could be measured, while specific fission product radionuclides including 101Tc, along with 88Kr, 91Sr, 92Sr, 92Y, and 105Ru, were determined to be prominent signatures of enrichment124. The authors suggested that a small, portable neutron source, such as the Am-Be source used in the study or one similar, could be implemented for nuclear materials interrogation by applying these techniques.
In the nuclear reactor environment, the use of γ-ray analysis of select fission products as a complementary method with delayed neutron measurements for monitoring potential fuel failure was performed in the 30 MW HANARO reactor in Korea. Several Tc isotopes originating from fission were detected in the coolant water: 99mTc, 101Tc, and 104Tc with concentrations of 4.73 × 103 Bq·L−1, 1.95 × 104 Bq·L−1, and 8.35 × 103 Bq·L−1, respectively. Considering t1/2, decay scheme, peak area, and potential interferences, 101Tc was chosen for its 307 keV γ-peak, amongst other fission radionuclides, which could be measured with relatively small uncertainties despite the presence of a large Compton continuum located throughout the energy regime in the area of interest. The methodology proved to be beneficial as it was able to identify a very small fuel defect during normal reactor operation that would typically go undetected by the fuel failure detection (FFD) system based on delayed neutrons126.
A radioanalytical method that has been employed for determining the origin of atmospheric releases in fission-based accidents is measuring the isotopic ratio of non-naturally occurring Ru isotope pairs. For instance, sources of Ru generated from fission are typically depleted in the lighter Ru isotopes (i.e., 96Ru, 98Ru, 99Ru, and 100Ru), due to formation of stable Mo or long-lived Tc within these isobars, and enriched in heavier ones (i.e., 101Ru, 102Ru, and 103Ru), which are preceded by isotopes with relatively short half-lives. It was shown that ratios of 100Ru/101Ru and 102Ru/101Ru were higher for material originating from civilian power reactors due to contributions arising from fission of 239Pu in-grown in later fuel cycles, whereas these ratios were lower in Hanford groundwater where 235U fission was dominant over 239Pu127.
The use of the A = 101 isobar in combination with other isotopic markers has been key for developing an understanding of geological transformations and geochemical behaviours of different isotopes and radioisotopes that were formed in the Oklo natural fission reactor in Gabon. For example, one study on the micro-metallic and ε-phase aggregates, including the fission-derived elements Ru, Rh, Pd, Te, Pb, As, S, and Bi, were collected and the isotopic ratios of different isotopic systems, i.e., 235U/238U, 90Zr/91Zr, 95Mo/97Mo, and 99Ru/101Ru, were analysed128. Whereas the 90Zr/91Zr and 95Mo/97Mo ratios in the metallic aggregates varied little in composition, likely due to constant separate mixing of fissiogenic and non-fissiogenic components, the 99Ru/101Ru ratios exhibited large variations, which could not be described by such processes. On one hand, it was suggested that chemical fractionation between Tc and Ru during reactor criticality had occurred, where long-lived 99gTc and quickly formed 101Ru would have behaved dissimilarly under dynamic redox conditions arising from the radiolysis of water. It is suggested that fractionation would have occurred over the timeframe of the t1/2 of 99gTc or ~2.11 × 105 y, where the leaching behaviour of Tc, said to be 103–104 times that of Ru, would have been a strong driver of fractionation between the two elements. Likewise, the chemical fraction of Tc and Ru could have taken place during the incorporation of each into the metallic aggregate128. On the other hand, Groopman et al. attributed enrichments of the 99Ru/101Ru ratio to the propensity of the 99gTc(n, γ)100Tc reaction to proceed, which would have occurred under the high neutron flux of the reactor ~1.2 × 1021 n·cm−2 s−1 , post-Tc sequestration129. Under the scenario of Tc mobility within the uraninite or in other phases, a more even distribution of 100Ru throughout the particles would have been observed, which was not the case. Thus, it was suggested that if chemical fraction via leaching of Tc from the ε-phase had occurred, then it was likely not significant in extent and would have occurred when most Tc had already decayed to Ru.
Uranium-molybdenum (U-Mo) fission reactor fuels
Low enriched uranium (LEU)-Mo alloy dispersion and monolith fuels, i.e., LEU-10%Mo, have been implemented as an alternative to high enriched uranium (HEU) fuels for fission reactors. The desirable refractory behaviour of Mo combined with its high solid solubility in the gamma (γ) bcc phase of U makes it a viable candidate for generating a robust, high-temperature, higher-density alloy130,131. The isotopics of the Mo used in the fuel, however, are non-negligible, where the average neutron capture cross-section is σc = 2.57 b for natural Mo, a value 13-times larger than that of zirconium (Zr), a common structural material used in the core of thermal reactors. Therefore, in order to increase efficiencies in fuel burn-up, geometry, composition, and overall neutronics, the use of enriched Mo fractions has been proposed. The isotope 95Mo constitutes the second most naturally abundant Mo isotope (i.e., 15.9%), and it has the highest neutron cross-sections for both thermal and epithermal neutrons, i.e., σc = 13.6 b and Ic = 117 b. The use of either light or heavy enriched fractions of Mo depleted in 95Mo has been proposed, where the average absorption cross-section is reduced by a factor of 2 when using the heavy fraction (i.e., 96-100Mo)132. Centrifugal enrichment of 98Mo and 100Mo for the commercial production of medical isotopes is an ongoing process; thus, this could be a potential source for providing enriched Mo for U-Mo fuel fabrication133.
In relation to 101Mo/101Tc, it is noted that whether using natural Mo or a heavy-enriched fraction in U-Mo fuels, the formation of this mother-daughter isotopic pair will occur due to neutron capture on the Mo component of the fuel. As previously discussed, the A = 101 isobar is also a product of fissioning actinides, and in the context of scenarios where fission-rich processes take place, this will become another source of 101Mo/101Tc. On one hand, this radionuclidic pair could become a potential interference if the A = 101 isobar is used a fission signature, whereas on the other hand, if the neutronics and the production rate of 101Mo/101Tc through neutron capture on Mo are well-known under the circumstances of interest, then the added Mo could serve as a secondary internal reference for comparison of the two processes.
Mo-containing materials for fusion reactor components
Due to its robust structural and physical properties, the use of Mo or Mo-alloys (i.e., titanium-zirconium molybdenum (TZM)) in fusion reactors, for example, in the structural components of the reactor such as the divertor or blanket, has been considered. However, because of the potential activation of Mo under the high, fast neutron fluxes when using either D-D or D-T fusion fuels, considerable dose rates from the induced activity and the subsequently generated waste from these materials can be problematic during operation, decommissioning, and in the case of a potential accident134,135,136. Hence, specific materials have been chosen that will reduce the induced radioactivity, mostly those based on carbon (C), silicon (Si), titanium (Ti), iron (Fe), chromium (Cr), and vanadium (V), which include materials such as ferritic-martensitic steels (e.g., EUROFER97, F28H, HT9, etc.), and sintered aluminium. The exchange of Mo for W, its heavier chemical homolog, in component materials has been a particular design strategy to reduce induced radioactivity, although the presence of Mo at limited concentrations is considered acceptable; this has been the basis of reduced activation ferritic-martensitic steels (RAFMs)137,138. Alternatively, the use of Mo enriched in specific Mo isotopes that do not significantly activate, i.e., 96Mo and 97Mo, has been proposed; however, because of enrichment costs, better economic options will need to be developed for commercial applications139.
Iida et al. calculated that short-lived radionuclides and their daughters, such as 91Mo, 99Mo/99mTc, and 101Mo/101Tc, would contribute to the rapid rise in induced radioactivity during the beginning of reactor operation and throughout its operating lifetime. After two years of operation, the radioactivity of Mo used as a structural material in the blanket of the JAERI tokamak D-T fusion reactor (JFDR) would be nearly two orders of magnitude more than the stainless steel reflector, much of which is attributed to decaying 99Mo/99mTc. Seven days post-shutdown, the corresponding dose rate inside of the Mo shield would be around 100 mrem·h−1, however, due to adequate thickness of the proposed shield the dose outside would significantly less at about 10 mrem·h−1 134. The activation profiles for Mo in ferritic steels such as HT9 were investigated by Youssef and Conn for the SATYR D-D and WITAMIR-I D-T blankets140. They showed for the D-T system that 101Mo produced in the reactor exhibited a high short-lived level or isotopic radioactive index (IRIA). The “softer” spectrum for the D-D system is predicted to enhance (n, γ) reactions, thus 101Mo would have a slightly higher contribution to total activity at shutdown. The activity of 101Mo/101Tc in the ferritic steel SATYR blanket as a function operating time indicated that, although there is a slight decrease, the activity of the two radioisotopes remains relatively constant throughout the 30 years operation period141.
Despite the fact that W is a leading high-Z material for surface components in the divertor and first wall of a fusion reactor, there are concerns about potential performance issues and the strategic uncertainty of relying on one specific material for construction. Brooks et al. surveyed other possible refractory metals, such as Zr, Nb, Mo, Hf, and Ta, and calculated their performance under the ARIES-ACT-1 fusion reactor operating conditions142. For example, the U.S. waste disposal rating (WDR) was used to categorise the contribution of these elements after 3.8 MWy·m−2 of divertor neutron irradiation, where for a W-based divertor this qualified as low-level waste (WDR < 1 Ci·m−3 at 100 y after replacement), while Mo was considered HLW (WDR > 1) even when the armour and divertor were measured together, which reduced the total value. However, the dose rate exposure for remote handling equipment necessary to recycle the divertor are nearly similar for W and Mo after a day or two, in such that recycling for either system could be initiated immediately after shutdown. Similarly to the activity profile, the heat load for Mo (~5 × 105 W·m−3) decreases significantly after one day, which also improves possibilities associated with post-processing142. Cepraga et al. presented calculations demonstrating similarities when considering TZM alloy as a divertor material for the Next European Torus (NET)143. For TZM (99.369 wt% Mo), both 101Mo and 101Tc along with 91Mo (t1/2 = 15.49 min) were the first isotopes to decay in less than a day, which also correlated to a diminishment in the total associated radioactivity and decay heat.
Potential use in nuclear medicine for diagnostics, therapeutics, and/or theranostics
It was not apparent from the literature that 101Tc has never been utilised for nuclear medicine purposes in a laboratory or clinical setting. As previously described here in, there is the redundant affiliation of 101Tc in nuclear medicine as an impurity generated alongside 99mTc during production. However, there are several accounts that have studied its decay properties in the context as a hypothetical diagnostic, therapeutic, and/or theranostic agent, where the works of our group, i.e., Mausolf et al. and Mayordomo et al., constitute the most detailed explanations to date15,62. In these works, the logistical model for fusion-based neutron generator, or an alternative compact accelerator neutron source, production and application of 101Tc was compared to the combination of emerging short-lived radiotherapeutic isotopes, such as 226Th (t1/2 = 30.57 min) and 214Pb (t1/2 = 27.06 min), and accelerator-produced PET radioisotopes, such as 15O (t1/2 = 122.24 s), 13N (t1/2 = 9.97 min), and 11C (t1/2 = 20.36 min), which are generated and distributed in relative proximity to the end-user. Although the use of shorter-lived radioisotopes generally goes against the guidelines concerning the t1/2 of an ideal therapeutic as outlined by Qaim144, where half-lives on the order of several hours to a week are considered the most efficient, it is acknowledged that a part of this justification is associated with the ability to conveniently ship, store, and stockpile these radioisotopes as most are typically produced in a nuclear reactor or require a high-power accelerator isolated an appreciable distance from the patient145. However, the possibility of real-time, onsite production using a compact accelerator source, similarly to how radionuclide generators or PET isotopes are utilised, circumvents many of the supply logistics and constraints associated with the use of a short-lived radioisotope.
Furthermore, an assessment of the fundamental decay properties (i.e., β– and γ-ray emissions) of 101Tc were compared to the therapeutic properties of 186Re (t1/2 = 3.72 d; βmax = 1.072 MeV, Iβ = 71.0%), 188Re (t1/2 = 17.01 h; βmax = 2.120 MeV, Iβ = 70.7%), and 89Sr (t1/2 = 50.563 d; βmax = 1.495 MeV, Iβ = 70.7%), and the diagnostic ones of 131I (t1/2 = 8.025 d; Eγ = 364.48 keV, Iγ = 99.9%) for single-photon emission computed tomography (SPECT) imaging15. The clinical application of these isotopes include palliative care, synovectomy, endovascular irradiation, treatment of osseous metastases, neuroendocrine tumours, prostate cancer and thyroid ablation to name a few. Research published by Manjunatha presented dosimetry calculations to determine the effects of various β–emitting nuclides in human bone, in particular on parameters regarding yield, intensity, and β– bremsstrahlung dose in human skeletal tissues146. The bremsstrahlung dose (Gy·Bq−1) as a function of distance from a 101Tc source (βmax = 1.613 MeV) in cortical bone is shown in Supplementary Fig. S10. The calculated bremsstrahlung exposure rates (mR·h−1) for 101Tc in cortical bone, red marrow, yellow marrow, spongiosa, and cartilage are 78.661, 41.272, 40.642, 55.352, and 49.663 mR·h−1, respectively. Other β-emitting radionuclides with βmax energies comparable to 101Tc that were presented include 32P (t1/2 = 14.268 d; βmax = 1.710 MeV) and 89Sr (Supplementary Fig. S10), both which have historically been utilised in the clinical setting for treating bone diseases147,148,149.
Because of the extensive knowledge-base behind 99mTc diagnostics, it is likely that similar applications and chemistry will be translational for 101Tc, although experimental testing will be necessary to demonstrate this. In this regard, 99mTc chemistry related to cold-kits and tagging is well-studied and an array of formulations are produced for routine clinical use. However, limitations related to the time required for synthesis and delivery of the drug will make those which are less time intensive necessary in order to maximize ease of use. For example, pertechnetate [TcO4]– is the chemical form eluted from commercial 99Mo/99mTc generators, which is both the starting material for successive radiopharmaceutical compounding and an imaging agent in itself. Cold kits that require minimal manipulations and synthesis time (i.e., ≤ 1 t1/2) which could theoretically be applied for 101Tc include but are not limited to: methylene diphosphonate (MDP), hydroxydiphosphonate (HDP), pyrophosphate (PYP), ethylenediamine pentaacetic acid (DTPA), glucoheptonate (GH), N-(2,6-diethylacetanilido) iminodiacetic acid (EHIDA), phytate, sulfur colloid, human serum albumin (HSA) (nano)colloid, HSA microspheres, d,l-hexamethylpropylene amine oxime (d,l-HMPAO), methoxyisobutylisonitrile (MIBI), and tetrofosmin150. New kit formulations specifically designed for 101Tc, or the development of rapid, automated techniques that streamline compounding, such as microwave-assisted syntheses or microfluidic chip systems, could provide alternative solutions151,152. Similarly, there are drug delivery routes, such as Technegas® (e.g., inhalation) and the use of catheters (e.g., intra-arterial or intracavitary infusion brachytherapy, convection-enhanced delivery (CED), etc.) that allow for the accelerated introduction of radiopharmaceuticals to points of critical concern, which could be leveraged for this shorter-lived isotope153,154.
Conclusion and outlook
From the shadows of its isotopic brethren 99mTc and 99gTc, the short-lived 101Tc emerges as an equally interesting and potentially useful isotope. The known literature summarised here showcases its array of unique nuclear properties, production and isolation routes, and applications in various areas of research and industry. Improved understanding of its fundamental nuclear structure as a heavier, neutron-rich Tc isotope gives further clarity to the relationship amongst all Tc isotopes and insight into the inherent radioactive nature of Tc. The straightforward production of 101Tc through (n, γ) reactions on natural or enriched 100Mo-containing samples, even with lower yielding neutron fluxes, is quite promising considering ongoing advances in compact accelerator neutron sources (CANS) technology. Meanwhile, its shorter t1/2 makes it manageable to work with and its stable isobar end-member 101Ru negates the need for radioactive waste management or the possibility of long-term contamination scenarios. As the element Tc, an array of oxidation states and rich coordination chemistry are accessible, and it will be interesting to see what similar chemistries for other Tc isotopes will be applicable for 101Tc.
A better understanding of the potential of fission-derived and neutron-activated 101Tc, either paired together or as unique signatures, for applications related to on- or offline nuclear fuel monitoring may play a role in determining fuel composition, enrichment, burnup, etc., and may aid in the context of nuclear proliferation and materials interrogation that has only been previously touched upon. This function will be particularly important with the geopolitical and technological shift from HEU-fueled reactors to LEU ones, where high-density U-Mo compositions have become of primary interest. There are also implications for improvements in neutron economy and the potential for reducing radiotoxicity and/or long-lived waste generation in primary and ancillary nuclear components depending on the Mo isotopics. With the recent efforts and focus on the development and deployment of commercial fusion energy, it will be key to utilise materials that are not only structurally and functionally robust, but will also minimise problematic radioactive wastes; thus, tailoring Mo isotopics could be beneficial where short-lived radioactive isotopes, e.g., 101Mo/101Tc, or neutron poisons, e.g., 95Mo, are dominant.
Stable isotope enrichment is another area of scientific, commercial, and industrial interest. The large-scale use of stable isotopes, however, is significantly impeded by the small number of enrichment facilities worldwide and the lack of cost/resource-effective processes that can be employed, especially for bulk quantities. As discussed in this review, enrichment of particular Mo isotopes fractions, i.e., 98Mo and 100Mo, is crucial for some applications, such as radioisotope production, and would greatly benefit others where sourcing appreciable amounts is not quite yet an option, i.e., nuclear fuels and materials. However, not discussed but relevant to the topic of isotope enrichment, is the potential for generating an isotopically pure stream of 101Ru from 101Tc. When using a Mo target and a neutron source, the Mo (natural or enriched in 100Mo) starting material is simultaneously transmuted and upgraded from a minor metal of relative fiscal value through 101Mo/101Tc to Ru, a platinum group metal (PGM) worth several orders of magnitude more. Mining PGMs, such as Ru, from spent nuclear fuel has been proposed155; whether appreciable amounts of Ru could be generated through non-fission-based means and whether it would be financially worthwhile has yet to be determined.
Within the field of nuclear medicine, the complexities related to just-in-time production and distribution of radioisotopes, especially ones that require unique infrastructure like a high-flux nuclear reactor or high-energy accelerator, make logistics of production and supply a challenge156,157. The authors believe that providing an alternate pipeline of medical radioisotopes that can be produced in a distributed fashion closer to the point of use may help to alleviate and possibly avoid some supply chain issues. 101Tc may be a likely candidate for this model, however, significant research and clinical testing will first be necessary to determine its effectiveness and whether there is potential for it as a medical radioisotope.
Although many pathways for its production have been established, the knowledge of isolation and separation methodologies as well as known applications for 101Tc are limited. In general, the gaps in the known literature concerning 101Tc, for example, in comparison to its Tc isotope brethren 99mTc and 99gTc, highlight the apparent need for further investigative work in order to better elucidate trends in nuclear and chemical behaviour across the range of Tc isotopes. The authors hope that this will be the first of many reviews on this unique and interesting Tc isotope.
Data availability
Data is contained within the article and Supplementary Information.
References
Hackney, J. C. Technetium − Element 43. J. Chem. Educ. 28, 186–190 (1951).
Perrier, C. & Segrè, E. Some chemical properties of element 43. J. Chem. Phys. 5, 712–716 (1937). First isolation of Tc and a compilation of its preliminary chemical properties, which was a precedent for subsequent chemistry related to isolating different Tc isotopes.
Perrier, C. & Segrè, E. Technetium: the element of atomic number 43. Nature 159, 24 (1949).
Cacciapuoti, B. N. & Segrè, E. Radioactive isotopes of element 43. Phys. Rev. 52, 1252–1253 (1937). First measurements of the half-lives of different Tc isotopes, which helped to establish the list of known Tc isotopes.
Cacciapuoti, B. N. Radioactive isotopes of element 43. Phys. Rev. 55, 110 (1939).
Johnstone, E. V., Yates, M. A., Poineau, F., Sattelberger, A. P. & Czerwinski, K. R. Technetium: the first radioelement on the periodic table. J. Chem. Edu. 94, 320–326 (2017). Presents a combined theoretical and empirical-based explanation behind the innate radioactivity of Tc and its isotopes, which provides context behind the radioactivity of Tc-101 and its neighbouring Tc isotopes.
Boyd, G. E. & Larson, Q. V. Report on the occurrence of technetium on the Earth’s crust. J. Phys. Chem. 60, 707–715 (1956).
Kenna, B. T. & Kuroda, P. K. Technetium in nature. J. Inorg. Nucl. Chem. 26, 403–499 (1964).
Schwochau, K. In Technetium: Chemistry and Radiopharmaceutical Applications (Wiley-VCH, 2000). A review that provides a thorough overview of fundamental Tc chemistry and compilation of Tc radiopharmaceuticals.
Luksic, S. A., Riley, B. J., Schweiger, M. & Hrma, P. Incorporating technetium in minerals and other solids: a review. J. Nuc. Mater. 466, 526–528 (2015).
Poineau, F., Mausolf, E. J., Jarvinen, G. D., Sattelberger, A. P. & Czerwinski, K. R. Technetium chemistry in the fuel cycle: combining basic and applied studies. Inorg. Chem. 52, 3573–3578 (2013).
Luykx, F. In Technetium Discharges into the Environment. 21–27 (Springer, 1986). https://doi.org/10.1007/978-94-009-4189-2_2
Schulte, E. H. & Scoppa, P. Sources and behaviour of technetium in the environment. Sci. Tot. Environ. 64, 163–179 (1987).
Blower, P. J. A nuclear chocolate box: the periodic table of nuclear medicine. Dalton Trans. 44, 4819–4844 (2015). Review of different elements and their corresponding radioisotopes and coordination complexes that are employed in the field of nuclear medicine, which includes a discussion on Group 7 elements, such as Tc.
Mausolf, E. J. et al. Fusion-based neutron generator production of Tc-99m and Tc-101: a prospective avenue to technetium theranostics. Pharmaceuticals 14, 875 (2021). Work from our group on fusion-based production of Tc-101 and its isolation and its proposed use as a medical radioisotope, which served as the motivation behind this review.
Nystrom, A. & Thoennessen, M. Discovery of yttrium, zirconium, niobium, technetium and ruthenium isotopes. At. Dat. Nucl. Dat. Tab. 98, 95–119 (2012).
Maurer, W. & Ramm, W. Untersuchung über das „19-Minuten“-Isotop von Molybdän und das daraus entstehende Isotop von Element 43. Naturwissenschaften. 29, 368 (1941).
Hahn, O. & Strassmann, F. Über die bei der Uranspaltung auftretenden Molybdän-Isotope. Naturwissenschaften. 29, 369 (1941).
Hahn, O. & Strassmann, F. Über die bei der Uranspaltung auftretenden Molybdän-Isotope. Z. Phys. 117, 789 (1941). Identification of Tc-101 arising from fission-based processes and first detailed measurement of half-life.
Sagane, R., Kojima, S., Miyamoto, G. & Ikawa, M. A new radioactive isotope of massurium, 43Ma101. Phys. Rev. 57, 70 (1940). First report on the production and discovery of Tc-101, which describes its ease of production with a neutron source.
Sagane, R., Kojima, S., Miyamoto, G. & Ikawa, M. Artificial radioactivity induced in Zr and Mo. Phys. Rev. 57, 1179 (1940).
Nagai, Y. & Hatsukawa, Y. Production of 99Mo for nuclear medicine by 100Mo(n, 2n)99Mo. J. Phys. Soc. Jpn. 78, 033201 (2009).
Hahn, O. & Strassmann, F. Über den Nachweis und das Verhalten der bei der Bestrahlung des Urans mittels Neutronen entstehenden Erdalkalimetalle. Naturwissenschaften 27, 11–15 (1939).
Meitner, L. & Frisch, O. R. Disintegration of uranium by neutrons: a new type of nuclear reaction. Nature 143, 239–240 (1939). First report of the theoretical presence of Ma-101 (Tc-101) formed during the proposed process of fission.
Seaborg, G. T. & Segrè, E. Nuclear isomerism in element 43. Phys. Rev. 55, 808 (1939).
Li, H. J. et al. Collective band structures in the Tc-99 nucleus. Phys. Rev. C 91, 054314 (2015).
Shen, S. F., Wang, F. G., Fang, K. M. & Xu, F. R. Study of nuclear level structure in Tc isotopes with mass A~100. High. Ener. Phys. Nucl. Phys. 31, 543–547 (2007).
Zeghib, S. 103Tc nuclear structure and systematic evolution of g9/2 parentage in odd-A 95,97,99,101,103Tc isotopes. Can. J. Phys. 93, 862–870 (2015). Decription of nuclear structure shape and systematic change across odd-A Tc isotopes, which gives context to the Tc-101 nucleus and how it compares with other odd-A Tc isotopes.
Crowe, B. et al. 99Tc produced by the (3He, pnγ) reaction. Phys. Rev. C 57, 590 (1998).
Dejbakhsh, H., Mouchaty, G. & Schmitt, R. P. Level structure of 101Tc investigated by means of massive transfer reaction. Phys. Rev. C 44, 120–127 (1991).
Hammed, M. A., Mahon, Mac, Naboulsi, T. D. & Decay, A. H. scheme data for 101Mo and 101Tc. Nuc. Instrum. Meth. A 334, 484–494 (1993). Most recent report on the decay scheme and inherent gamma-rays accompanying the Mo-101 to Tc-101 transition.
O’Kelley, G. D., Larson, Q. V. & Boyd, G. E. Decay chain 101Mo–101Tc. Bull. Am. Phys. Soc. 2, 24 (1957).
Kinsey, R. R. Plots produced using the code ENSDAT, National Nuclear Data Center, Brookhaven National Laboratory, Upton, NY, U.S.A (1995).
Wiles, D. R. Search for the p1/2 isomeric state in 101Tc and identification of energy levels. Phys. Rev. 93, 181 (1954).
Uyttenhove, J., Demuynck, J., Dorikens, M. & Dorikens-Vanpraet, L. Experimental study of isomeric states 101mRu, 103mRu, and 101Tc. Z. Phys. 238, 90–98 (1970). First identification and measurement of an isomeric state of Tc-101 originating from photonuclear-induced reactions on Ru targets.
Bartsch, H., Günther, W., Huber, K., Kneissl, U. & Krieger, H. Systematic trends in the analysis of photo nuclear cross section ratios. Z. Phys. A 285, 71–75 (1978).
Hoellinger, F. et al. High-spin structures observed in the 101Tc fission fragment. Eur. Phys. J. A 4, 319–321 (1999).
Perlman, M. L. & Friedlander, G. Relative yields of some X-ray induced nuclear reactions. Phys. Rev. 74, 442–448 (1948).
Mock, D. L., Waddell, B. C., Fagg, L. W. & Tobin, R. A. Photo-induced reactions at 20 MeV. J. Phys. Rev. 74, 1536 (1948).
Kumabe, I., Poularikas, A. D., Preiss, I. L., Gardner, D. G. & Fink, R. W. (n, 3He) Reactions of medium weight nuclei induced by 14.8-MeV neutrons. J. Phys. Rev. 117, 1568 (1960).
Abzouzi, A., Antony, M., Ndongué, V. & Oster, D. Redetermination of several half-lives. J. Radioanal. Nucl. Chem. 145, 361–364 (1990).
You, X., Shengdong, Z., Zhihong, Y., Ding, Y. & Cui, A. Preparation and measurement of the half-life of 101Tc. J. Radioanal. Nucl. Chem. 287, 267–272 (2011). High-accuracy half-life measurement of Tc-101 isolated from Mo-101.
da Silva, A. C. O., Genezini, F. A., Zamboni, C. B., Zahn, G. S. & da Cruz, M. T. F. Half-life of 101Mo and 101Tc β–-decay. AIP Conf. Proceed 1139, 185 (2009).
Hamida, E. Quantifying the nuclear s-process via neutron activation. Uni. Surrey Master’s thesis (2010).
Steinetz, B. M. et al. Experimental observations of nuclear activity in deuterated materials subjected to a low-energy photon beam. NASA/TM—2017-218963 http://ntrs.nasa.gov (2017).
Martin, D. W., Bursons, S. B. & Cork, J. M. Decay of 101Mo (14.6 min). Bull. Am. Phys. Soc. 1, 329 (1956).
Cook, W. B. & Johns, M. W. Decay of 101Mo and 101Tc. Can. J. Phys. 50, 1957 (1972).
Siwamogsatham, B. & Easterday, H. T. Note on the 101Tc–101Ru decay. Nucl. Phys. A162, 42 (1972).
Svensson, L. G., Backlin, A., Solhed, H. & Lindskog, J. Transition probabilities in odd-A Tc isotopes. Proc. Intern. Conf. Nuc. Struct. Spectrosc. 1, 132 (1974).
Abecasis, S. M., Civitarese, O. & Krmpotic, F. Analysis of odd-mass technetium isotopes with the Alaga model. Z. Phys. A278, 309 (1976).
Svensson, L. G., Backlin, A., Solhed, H. & Lindskog, J. Transition rates between the positive parity states in 101Tc. Uppsala Uni. Report UUIP—873 (1976).
Wright, J. A. Five-particle, shell-model calculation using spin-dependent potential as applied to 101Tc and the nuclear decays of 101Mo, 101Tc, 142Xe, and 142Cs. Iowa State University Ph.D. thesis. (1974).
Aras, N. K., Fettweis, P., Chilosi, G. & O’kelley, G. D. Levels in 101Ru populated by the decay of 101Tc. Nucl. Phys. A 169, 209–224 (1971).
Cabell, M. J. The thermal neutron capture cross-section and resonance capture integral of 100Mo. J. Nuc. Ener. A 12, 172–176 (1960). Neutron capture measurements for thermal and resonance energy neutrons, which provides an understanding of neutron-driven production of Mo-101 from Mo-100.
Weigmann, H., Raman, S., Harvey, J. A., Macklin, R. L. & Slaughter, G. G. Neutron resonances in 100Mo and valence neutron capture. Phys. Rev. C 20, 115 (1979).
Remley, K. E. et al. Transmission measurements and resonance parameter analysis for Mo-98 and Mo-100. Ann. Nucl. Ener. 122, 23–36 (2018).
Amemiya, S., Ishibashi, K. & Katoh, T. Neutron activation cross-section of molybdenum isotopes at 14.8 MeV. J. Nucl. Sci. Tech. 19, 781–788 (1982).
Parashari, S. et al. Systematic analysis of the neutron-induced reaction cross sections for natMo isotopes within 10–20 MeV. Phys. Rev. C 99, 044602 (2019).
Sagane, R., Kojima, S., Miyamoto, G. & Ikawa, M. Radioactive isotopes of molybdenum and their daughter products. Proc. Phys. Math. Soc. Jpn. 3rd 24, 499–509 (1941).
Toth, J. J. et al. Production of Molybdenum-99 Using Neutron Capture Methods; Technical report; PNNL-19895, RPT-59331-01 Rev 0; Pacific Northwest National Laboratory (PNNL): Richmond, WA, USA, (2011). Report of co-production of Mo-101 / Tc-101 as a short-lived contaminant during Mo-99 / Tc-99m production using low-specific activity Mo targets in a nuclear reactor.
Daly, P. J., Seppelt, B. M. & Shaw, P. F. D. Radiative capture cross sections in medium-weight and heavy nuclei. Nucl. Phys. A 119, 673–690 (1968). First report of the 100Mo(p, γ)101Tc reaction on a cyclotron.
Mayordomo, N. et al. CANS production of Tc-99m and Tc-101. IAEA Conf.: Accelerators for Research and Sustainable Development: From Good Practices Towards Socioeconomic Impact, Vienna, Austria, 23-27 May, 2022. Report on co-production of Mo-101 / Tc-101 in a secondary target irradiated with secondary neutrons generated during routine 18F[FDG] production in a low-energy biomedical cyclotron.
Fervert, E. (n,3He) cross-section measurements on 55Mn, 59Co, 75As and 103Rh for 14.8 neutrons. Acta. Phys. Austriaca. 100, 202 (1965).
Gray, P. R., Zander, A. R. & Ebery, T. G. Activation cross sections for reactions of Rh and Ru with 14.7 MeV neutrons. Nuc. Phys. 75, 215–225 (1966). Direct production of Tc-101 using a fast neutron source with either Ru or Rh targets.
Csikai, J. Investigation of 103Rh(n, 3He)101Tc reaction. Act. Phys. Aca. Sci. Hung. 21, 229–233 (1966).
Husain, L., Bari, A. & Kuroda, P. K. 14.8 MeV Neutron cross-section for (n,3He) and other rare nuclear reactions on 103Rh. J. Inorg. Nucl. Chem. 30, 3145–3149 (1968).
Diksic, M., Strohal, P. & Slaus, I. (n, 3He) and (n, t) reaction cross-sections at 14 MeV. J. Inorg. Nucl. Chem. 36, 477–485 (1974).
Paul, E. B. & Clarke, R. L. Cross section measurements of reactions induced by neutrons of the 14.5 MeV energy. Can. J. Phys. 31, 267 (1953).
Luo, J. et al. Measurements of activation cross-sections for the 101Ru(n, p)101Tc reaction for neutrons with energies between 13 and 15 MeV. J. Radioanal. Nucl. Chem. 296, 1225–1230 (2013).
Kasugi, Y., Tokushima, T., Kawade, K., Yamamoto, H. & Katoh, T. Measurement of activation cross section of short-lived nuclei produced by 14 MeV neutrons - Ru, Pd, Cd, Sn. Proc. Symp. on Nuc. Dat.; Tokyo, Jap. JAERI-M—92-027, 268–277 (1991).
Kielan, D., Marcinkowski, A. & Garuska, U. Isotopic effect in (n, p) reaction on ruthenium. Nucl. Phys. A 559, 333–346 (1993).
Sakane, H. et al. Measurement of activation cross-sections of (n, np+d) reactions producing short-lived nuclei in the energy range between 13.4 and 14.9 MeV using an intense neutron source OKTAVIAN. Ann. Nucl. Ener. 29, 53–66 (2002).
Kasugai, Y. et al. Conf. Report, JAERI-M Reports. 92, 027:268 (1992).
Luo, J. et al. Activation cross sections for reactions induced by 14 MeV neutrons on natural ruthenium. Phys. Rev. C 76, 057601 (2007).
Kasugi, Y., Yamamoto, H., Kawade, K. & Iida, T. Measurement of (n, α) cross-sections for short-lived products by 13.4–14.9 MeV neutrons. Ann. Nucl. Ener. 25, 1485–1502 (1998).
Wiles, D. R. & Coryell, C. D. Fission yield fine structure in the mass region 99–106. Phys. Rev. 96, 696 (1954). Provides a description of fission yields relevant for A = 101 including Mo-101 / Tc-101 from different fission processes.
Kiso, Y., Matsushita, R., Takada, J., Takemi, H. & Tamai, T. Quick separation of fission production molybdenum and gamma-rays of Mo-102. J. Nucl. Sci. Tech. 13, 141–143 (1976).
Elmaghraby, E. K., Tohamy, M. & Comsan, M. N. H. Multiscale time-bin analysis of delayed gamma-ray spectra of fission products. Phys. Scr. 96, 125318 (2021).
Joint Evaluated Fission and Fusion File (JEFF) 3.11, accessed Mar 2022.
Chu, S. Y. F., Ekström, L. E. & Firestone, R. The Lund/LBNL nuclear data search. http://nucleardata.nuclear.lu.se/toi/ (1999).
Srivastava, A. et al. Fragment shell effect in low energy fission: Independent yields of technetium isotopes in the thermal-neutron-induced fission of 239Pu. Phys. Rev. C 33, 969 (1986).
Silano, J. et al. Validating the Bohr hypothesis: comparing fission yields from photon-induced fission of 240Pu and neutron-induced fission of 239Pu. EPJ Web Conf. 239, 03004 (2002).
Rattan, S. S., Ramaswami, A., Singh, R. J. & Prakash, S. Alpha particle induced fission of 209Bi at 55.7 and 58.6 MeV. Radiochim. Acta 55, 169–172 (1991).
Gagon, K. et al. Cyclotron production of 99mTc: experimental measurement of the 100Mo(p, x)99Mo, 99mTc and 99gTc excitation functions from 8 to 18 MeV. Nucl. Med. Biol. 38, 907–916 (2011). Report on cyclotron production of Tc-99m and corresponding excitation functions, which includes the co-contaminant reaction of the formation of Tc-101 from Mo-100.
Gagnon, K., Wilson, J. S. & McQuarrie, S. A. Experimental cross section measurements for the 100Mo(p, x)101Tc, 96Nb, 97Nb reactions in the energy range of 10 to 18 MeV. Nucl. Med. Biol. 39, 923–925 (2012).
Lamere, E. et al. Proton-induced reactions on molybdenum. Phys. Rev. C 100, 034614 (2019).
Randa, Z. & Svodoba, K. Excitation functions and yields of (d, n) and (d, 2n) reactions on natural molybdenum. J. Inorg. Nucl. Chem. 38, 2289–2295 (1976).
Randa, Z. & Svoboda, K. Excitation functions and yields of the (d, p) reactions on natural molybdenum for deuteron energies less than 13 MeV. J. Inorg. Nucl. Chem. 39, 2121–2123 (1977).
Hjorth, S. A. & Cohen, B. L. Nuclear structure studies in the molybdenum isotopes with (d, p) and (d, t) reactions. Phys. Rev. 138, B920–B933 (1964).
Habib, E. E., Cameron, J. A., Din, G. U., Janzen, V. & Schubank, R. Low-lying states in 97Mo and 101Mo by (t,d) and (d,p) reactions on the neighbouring molybdenum isotopes. Can. J. Phys. 68, 1322–1329 (1990).
Levkovski, V. N. Cross-Section of Medium Mass Nuclide Activation (A = 40–100) by Medium Energy Protons and Alpha-Particles (E = 10–50 MeV); Inter-Vesi: Moscow, USSR (1991).
Agarwal, A. et al. Effect of neutron excess in the entrance channel on the 18O + 93Nb system: an experimental study relevant to incomplete-fusion dynamics. Phys. Rev. C 103, 034602 (2021).
Sabel’nikov, A. V., Maslov, O. D., Molokanova, L. G., Gustova, M. V. & Dmitriev, S. N. Preparation of 99Mo and 99mTc by 100Mo(γ, n) photonuclear reaction on an electron accelerator, MT-25 Microtron. Radiochem 48, 191–194 (2006).
Kelsey IV, C. et al. MCNPX-CINDR’90 simulation of photonuclear Mo-99 production experiments. Tenth International Topical Meeting on Nuclear Applications of Accelerators, 2011-04-03/2011-04-07, Knoxville (2011). Formation of Mo-101 / Tc-101 during the photonuclear production of Tc-99m with an electron linear accelerator and Mo-targets.
Tsechanski, A., Bielajew, A. F., Archambault, J. P. & Mainegra-Hing, E. Electron accelerator-based production of molybdenum-99: Bremsstrahlung and photoneutron generation from molybdenum vs. tungsten. Nucl. Instru. Meth. Phys. Res. Sect. B 366, 124–139 (2016).
Herrmann, G. & Trautmann, N. Rapid chemical methods for identification and study of short-lived nuclides. Ann. Rev. Nucl. Part. Sci. 32, 117–147 (1982).
Ravi, A. et al. Finding a receptor design for selective recognition of perrhenate and pertechnetate: hydrogen vs. halogen bonding. Chem. Comm. 54, 4826–4829 (2018).
Narasimhan, D. & Mani, R. Chemical and radiochemical evaluation of the purity of 99mTc extracted by MEK. J. Radioanal. Nucl. Chem. 33, 81–100 (1976).
Hild, D. et al. MicroSISAK: continuous liquid–liquid extractions of radionuclides at ≥ 0.2 mL/min. Radiochim. Acta 101, 681–689 (2013).
Groening, H. R. & Harbottle, G. Recoil and annealing studies in neutron-irradiated crystalline molybdenum hexacarbonyl. Radiochim. Acta 14, 109–111 (1970).
Brodskaya, G. A. Method of fast radiochemical separation of technetium-99m and technetium-101 from molybdenum irradiated by neutrons. AN Uzbekskoj SSR, Tashkent. Inst. Yadernoj Fiziki (1983). First report of the use of column chromatography for the separation and isolation of Tc-101 from Mo-101.
Götz, M. et al. Gas phase synthesis of 4d transition metal carbonyl complexes with thermalized fission fragments in single-atom reactions. Radiochim. Acta 109, 153–165 (2021). Report on the use of volatilization as a separation pathway for recovery of Mo-101 / Tc-101 fission fragments from a thermalized fission source.
Calderazzo, F. et al. Reduction of ammonium pertechnetate and ammonium perrhenate with CO: synthesis of M2(CO)10 (M: Tc, Re) and crystal and molecular structure of the trinuclear cyano-bridged derivative Re3(CN)3(CO)12. Gazz. Chim. Ital., Soc. Chim. Ital. 119, 241–247 (1989).
Even, J. et al. In situ synthesis of volatile carbonyl complexes with short-lived nuclides. J. Radioanal. Nucl. Chem. 303, 2457–2466 (2014).
Xiao, C., Khayambashi, A. & Wang, S. Separation and remediation of 99TcO4– from aqueous solutions. Chem. Mater. 31, 3863–3877 (2019).
Banerjee, D., Kim, D., Schweiger, M. J., Kruger, A. A. & Thallapally, P. K. Removal of TcO4− ions from solution: materials and future outlook. Chem. Soc. Rev. 45, 2724–2739 (2016).
Jarvinen, G. D. et al. Separation of pertechnetate from uranium in a simulated UREX processing solution using anion exchange extraction chromatography. Solvent Extr. Ion-. Exc 31, 416–429 (2013).
Dash, A., Knapp, F. R. Jr & Pillai, M. R. A. 99Mo/99mTc separation: an assessment of technology options. Nucl. Med. Biol. 40, 167–176 (2013).
Hasan, S. & Prelas, M. A. Molybdenum-99 production pathways and the sorbents for 99Mo/99mTc generator systems using (n,γ) 99Mo: a review. SN Appl. Sci. 2, 1–28 (2020).
Martini, P. et al. Highly efficient micro-scale liquid-liquid in-flow extraction of 99mTc from molybdenum. Molecules 26, 5699 (2021).
Nawar, M. F. & Türler, A. New strategies for a sustainable 99mTc supply to meet increasing medical demands: promising solutions for current problems. Front. Chem. 10 (2022).
Gumiela, M. Cyclotron production of 99mTc: comparison of known separation technologies for isolation of 99mTc from molybdenum targets. Nucl. Med. Biol. 58, 33–41 (2018).
Le, V. S. Generator development: up-to-date recovery technologies for increasing the effectiveness of utilisation. Sci. Tech. Nucl. Install. 2014 (2014).
Nagai, Y. et al. High thermo-separation efficiency of Tc-99m from molten (MoO3)-Mo-100 samples by repeated milking tests. J. Phys. Soc. Jap. 83 (2014).
Mausolf, E., Droessler, J., Poineau, F., Hartmann, T. & Czerwinski, K. R. Tetraphenylpyridinium pertechnetate: a promising salt for the immobilization of technetium. Radiochim. Acta 100, 325–328 (2012).
Maddock, R. S. & Meinke, W. W. Activation analysis; nuclear chemical research; radiochemical separations. Univ. of Michigan Prog. Rep. 1–124 (1958-1959). Presents the use of Mo-101 / Tc-101 for neutron activation analysis for trace analysis of Mo in natural substances.
Fukai, R. & Meinke, W. W. Activation analysis of vanadium, arsenic, molybdenum, tungsten, rhenium, and gold in marine organisms. Limnol. Oceanogr. 7, 186–200 (1962).
Diksic, M. & Cole, T. F. Fast determination of molybdenum and tellurium by neutron activation analysis. Analy. Chim. Acta 93, 261–266 (1977).
van Zanten, B., Decat, D. & Leliar, G. Activation analysis of molybdenum in plant material. Tantala 9, 213–218 (1962).
Healy, W. B. & Bate, L. C. Determination of molybdenum in hair and wool by neutron activation analysis. Anal. Chim. Acta 33, 443–448 (1965).
Cornelis, R., Versieck, J., Desmet, A., Mees, L. & Vanballenberghe, L. Neutron activation analysis of the trace element molybdenum in urine of healthy persons. Bull. Soc. Chim. Belg. 90, 289–295 (1981).
Sun, Y. C., Yang, J. Y. & Tzeng, S. R. Rapid determination of molybdate in natural waters by coprecipitation and neutron activation analysis. Analyst 124, 421–424 (1999).
Hetherington, E. L. R. Evaluation of neutron activation analysis for the measurement of isotopic abundances in molybdenum-98 enriched molybdenum. AAEC Report E644. 1–11 (1987). Describes using neutron activation via Mo-101 / Tc-101 for determining the degree of enrichment and purity of Mo-98-containing samples.
Tohamy, M., Elmaghraby, E. K., El-hakim, E. H. & Comsan, M. N. H. Thermal/fast fission yield ratio signature for neutron interrogation of nuclear materials. Phys. Part. Nucl. Lett. 19, 152–161 (2022). Describes the use of tracking different radioisotopes including Mo-101 / Tc-101 arising from different fission processes / enriched material to determine enrichment of fissionable materials.
Chiba, G., Tsuji, M., Narabayashi, T., Ohoka, Y. & Ushio, T. Important fission product nuclides identification method for simplified burnup chain construction: Physor 2014. J. Nucl. Sci. Tech. 52, 953–960 (2015).
Kim, M., Kim, H., Ahn, G., Lee, C. & Lim, I. Investigation of possibility for fuel defect detection by analysis of radionuclide in primary coolant of HANARO. Conference: RRFM 2009: 13. International topical meeting on Research Reactor Fuel Management (RRFM), Vienna, (2009).
Hopp, T., Zog, D., Kleine, T. & Steinhauser, G. Non-natural ruthenium isotope ratios of the undeclared 2017 atmospheric release consistent with civilian nuclear activities. Nat. Comm. 11, 2744 (2020). Reports the use of Ru-101 in context with the A = 101 isobar for identifying origins of material from an undeclared atmospheric release.
Kikuchi, M., Hidaka, H. & Gauthier-Lafaye, F. Formation and geochemical significance of micrometallic aggregates including fissiogenic platinum group elements in the Oklo natural reactor, Gabon. Geochim. Cosmochim. Acta 74, 4709–4722 (2010).
Groopman, E. E., Nittler, L. R., Willingham, D. G., Meshik, A. P. & Pravdivtseva, O. V. Long-term retention and chemical fractionation of fissionogenic Cs and Tc in Oklo natural nuclear reactor fuel. Appl. Geochem. 131, 105047 (2021).
Prabhakaran, R. U-Mo monolithic fuel for nuclear research and test reactors. JOM 69, 2529–2531 (2017).
van de Berghe, S. & Lemoine, P. Review of 15 years of high-density low-enriched UMo dispersion fuel development for research reactors in Europe. Nucl. Eng. Tech. 46, 125–146 (2014).
Bakker, K. & Wijtsma, F. Using molybdenum depleted in 95Mo in UMo fuel. Int. Meeting of Reduced Enrichment for Research and Test Reactors (RERTR). Bariloche (2002). Describes the importance of Mo isotopics in regard to the implementation of LEU U-Mo fuels in a nuclear reactor.
Shmelev, A. N. & Kozhahmet, B. K. Use of molybdenum as a structural material of fuel elements for improving the safety of nuclear reactors. IOP Conf. Ser.: J. Phys. Conf. Ser. 781, 012022 (2017).
Iida, H., Seki, Y. & Ide, T. Induced activity and dose rate in a fusion reactor with molybdenum blanket structure. J. Nucl. Sci. Tech. 14, 836–838 (1977). Describes the activation of Mo isotopes which includes Mo-101 / Tc-101 in the context for materials to be used in an operating fusion reactor, which can impact actions regarding handling and waste disposal.
Youssef, M. Z. & Corn, R. W. Induced radioactivity and influence of materials selection in deuterium-deuterium and deuterium-tritium fusion reactors. Nucl. Tech. Fusion 3, 361–384 (1983).
Piet, S. J., Kazimi, M. S. & Lidsky, L. M. Relative public health effects from accidental release of fusion structural radioactivity. Nucl. Tech. Fusion 4, 533–538 (1983).
Qiu, G.-X., Zhan, D.-P., Cao, L. & Jiang, Z.-Z. Review on development of reduced activated ferritic/martensitic steel for fusion reactor. J. Iron Steel Res. Int 367, 142–146 (2022).
Lyakishev, N. P. et al. Prospect of development and manufacturing of low activation metallic materials for fusion reactor. J. Nuc. Mater. 233, 1516–1522 (1996).
Gilbert, M. R., Packer, L. W. & Stainer, T. Experimental validation of inventory simulations on molybdenum and its isotopes for fusion applications. Nucl. Fusion 60, 106022 (2020).
Youssef, M. Z. & Conn, R. W. On isotopic tailoring for fusion reactor radioactivity reduction. Nucl. Tech. Fusion 4, 1177–1182 (1983).
Youssef, M. Z. & Conn, R. W. Induced radioactivity and influence of materials selection in deuterium-deuterium and deuterium-tritium fusion reactors. Nucl. Tech. Fusion. 3, 361–384 (1983).
Brooks, J. N., El-Guebaly, L., Hassanein, A. & Sizyuk, T. Plasma facing material alternatives to tungsten. Nucl. Fusion 55, 043002 (2014).
Cepraga, D. G. et al. Activation of TZM and stainless steel divertor materials in the NET fusion machine. J. Nuc. Mater. 212, 644–648 (1994).
Qaim, S. M. Therapeutic nuclides and nuclear data. Radiochim. Acta 89, 297–302 (2001).
Gudkov, S. V., Shilyagina, N. Y., Vodeneev, V. A. & Zvyagin, A. V. Targeted radionuclide therapy of human tumors. Int. J. Mol. Sci. 17, 33 (2015).
Manjunatha, H. C. A dosimetric study of Beta induced bremsstrahlung in bone. Appl. Rad. Iso 94, 282–293 (2014). Reports dosimetric calculations for bremsstrahlung radiation arising from different beta sources, including Tc-101, in human bone.
Storaasli, J. The role of radiotherapy and radioactive phosphorus (32P). JAMA 210, 1077–1078 (1969).
Hoskin, P. J. Radiotherapy in the management of bone pain. Clin. Orthop. Relat. Res 312, 105–119 (1995).
Montebello, J. F. & Hartson-Eaton, M. The palliation of osseous metastasis with 32P or 89Sr compared with external beam and hemibody irradiation: a historical perspective. Cancer Investig. 7, 139–160 (1989).
IAEA. Technetium-99m Radiopharmaceuticals: Manufacture of Kits. Technical report series no. 466. https://www.iaea.org/publications/7867/technetium-99m-radiopharmaceuticals-manufacture-of-kits (2008).
Kimura, H. et al. Microwave-assisted synthesis of organometallic complexes of 99mTc(CO)3 and Re(CO)3: its application to radiopharmaceuticals. Chem. Pharm. Bull. 60, 79–85 (2012).
Wang, J., Chao, P. H. & van Dam, R. M. Ultra-compact, automated microdroplet radiosynthesizer. Lab a Chip 19, 2415–2424 (2019).
Lemb, M., Oei, T. H., Eifert, H. & Günther, B. Technegas: a study of particle structure, size and distribution. Eur. J. Nucl. Med. 20, 576–579 (1993).
Kickingereder, P. et al. Intracavitary brachytherapy using stereotactically applied phosphorus-32 colloid for treatment of cystic craniopharyngiomas in 53 patients. J. Neurooncol. 109, 365–374 (2012).
Bourg, S. & Poinssot, C. Could spent nuclear fuel be considered as a non-conventional mine of critical raw materials? Prog. Nucl. Energ. 94, 222–228 (2017).
Vogel, W. V., van der Marck, S. C. & Versleijen, M. W. J. Challenges and future options for the production of lutetium-177. Eur. J. Nucl. Med. Mol. Imaging 48, 2329–2335 (2021).
Bodei, L., Herrmann, K., Schöder, H., Scott, A. M., Lewis, J. S. Radiotheranostics in oncology: current challenges and emerging opportunities. Nat. Rev. Clin. Oncol. https://doi.org/10.1038/s41571-022-00652-y (2022).
Acknowledgements
N.M. acknowledges the support from the TecRad (02NUK072) project funded by the German Federal Ministry of Education and Research (BMWF).
Author information
Authors and Affiliations
Contributions
Conceptualization, E.V.J., E.J.M. and N.M.; investigation, E.V.J., E.J.M. and N.M.; writing—original draft—E.V.J., E.J.M. and N.M.; writing—review and editing, E.V.J., E.J.M. and N.M.; funding acquisition, E.V.J., E.J.M. and N.M.
Corresponding author
Ethics declarations
Competing interests
E.J.M. and E.V.J. are developing patent-pending technology related to 101Tc production and isolation entitled: (US/International Patent) Direct, Continuous Transmutation of Molybdenum (Mo) for the Production and Recovery of Technetium (Tc) and Ruthenium (Ru) using a Neutron Source, submitted 2018. IFS, LLC consults on topics related to radioisotope production, use, and disposal. N.M. declares no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work. Peer Review reports available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Johnstone, E.V., Mayordomo, N. & Mausolf, E.J. Discovery, nuclear properties, synthesis and applications of technetium-101. Commun Chem 5, 131 (2022). https://doi.org/10.1038/s42004-022-00746-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-022-00746-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
