
Abstract
Energy storage devices using electrochemical reactions have become an integral part of our daily lives, and further improvement of their performance is highly demanded. An important task for this purpose is to thoroughly understand the electrochemical processes governing their chemistry. Here we develop a method based on Kelvin probe force microscopy that enables dynamic visualization of changes in the internal potential distribution in an operating electrochemical device and use it to characterize an all-solid-state lithium ion battery. Observation of the cathode composite regions during a cyclic voltammetry operation reveals differences between the behavior of local electrochemical reactions in the charge and discharge processes. Based on careful inspection of the results, we show that the difference arises from a change in the state of an electronic conductive path network in the composite electrode. Our method provides new insights into the local electrochemical reactions during electrochemical operation of devices.
Similar content being viewed by others


Introduction
Rechargeable battery technologies, for example, Li ion batteries (LIBs), have attracted considerable attention as demand for their use in portable electronics, electric vehicles, and so on has increased1,2,3. To further improve device performance, a deeper understanding of their working mechanisms is essential to gain valuable insights into design guidelines. A battery is generally operated by controlling electrochemical reactions via a voltage applied to the outer electrodes, and the battery is evaluated by electrochemical measurements, including charge/discharge tests and cyclic voltammetry (CV)4. These macroscopic characterizations are the first necessary steps for grasping battery performance; however, to understand the fundamental working principles of a device, additional direct microscopic analyses are desired to clarify how local electrochemical reactions proceed during electrochemical measurements. One of the most effective techniques for this purpose is dynamic observations of the internal potential distributions generated as a result of controlling the outer electrode potential, because the local potential is a key physical quantity that governs local electrochemical reactions. However, there have been few reports of such observation owing to the lack of characterization techniques applicable to a wide range of electrochemical devices5. To date, most works have been limited to static characterizations, where changes in the potential distributions were observed only before and after electrochemical operations6,7,8,9,10,11,12.
In this work, we develop a method for dynamically imaging the internal potential distribution in a solid-state electrochemical device under the device working conditions (the so-called operando technique) by incorporating an electrochemical measurement system into the previously developed in situ Kelvin probe force microscopy (KPFM) system9. Using this method, we characterize the cathode composite regions of an all-solid-state LIB (ASS-LIB)—a potential next-generation energy storage device13,14,15. Our results clarify that electrochemical reactions proceed nonuniformly from the outer electrode side in the charge process, whereas those in the discharge process progress uniformly over the composite electrode. By directly comparing the variation of the internal potential distribution with the CV behavior, we demonstrate that this difference can be explained by a change in the state of the electronic conductive path network in the composite electrode arising from a change in the electronic conductivity of the cathode active material due to charge/discharge reactions (Li\({}^{+}\) ion extraction/insertion). Our methods provide valuable information about local electrochemical reactions that cannot be obtained by analyzing the macroscopic electrochemical characteristics or from static measurements of the potential distributions only before and after electrochemical operations, and thus opens a new opportunity for understanding macroscopic properties of electrochemical devices from a microscopic point of view.
Results
Experimental setup of operando KPFM measurement
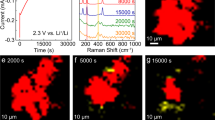
Experiments were conducted on the cross section of the ASS-LIB in a KPFM setup equipped with an electrochemical measurement system, as illustrated in Fig. 1a (see Methods for details). Note that KPFM is an atomic force microscopy (AFM) technique that measures the contact potential difference (CPD) between the AFM tip and sample. Using KPFM, we can obtain information about the work function and electrical potential distribution of sample surfaces16,17. In this work, we continuously obtained CPD images in the cathode composite region (indicated in Fig. 1b) during CV operation of the ASS-LIB. The cathode has a composite structure that is a mixture of LiCoPO\({}_{4}\) (LCP) as an active material, Li\({}_{1+x}\)Al\({}_{x}\)Ti\({}_{2-x}\)(PO\({}_{4}\))\({}_{3}\) (LATP) as a solid electrolyte, and Pd as a conductive additive, as depicted in Fig. 1b (see Methods for the entire battery structure). Figure 1c shows the CV curve during the KPFM measurements. Oxidation and reduction current peaks represent the charge and discharge reactions of the cathode active materials, respectively18,19,20,21. The shape of the peaks during the forward and backward sweeps was asymmetric. During the forward sweep, a single peak was observed at 2.55 V, whereas during the backward sweep, in addition to a peak at 2.36 V, a shoulder feature appeared at \(\sim\)2.03 V.

a Schematic illustration of cross-sectional KPFM setup equipped with a source measure unit for CV operation of the ASS-LIB. The anode-side current collector was electrically connected to the ground. The voltage for CV operation was applied to the cathode-side current collector with respect to the ground. b Schematic structure of the ASS-LIB. The region measured by KPFM is enclosed by a dashed square. c Cyclic voltammogram of the ASS-LIB during the KPFM measurement. The scan rate was 0.1 mV s\({}^{-1}\), and the voltage range was 0.2–2.7 V.
Observing surface potential distribution during CV operation
Figure 2 shows selected CPD images of the cathode composite region (10 \(\mu\)m \(\times\) 4 \(\mu\)m) obtained during a voltage sweep from 0.2 to 2.7 V (a–l) and the reverse sweep (m–x) (see also Supplementary Movie 1, which contains all the CPD images acquired during CV operation). The positions of each material in the composite electrode were identified by scanning electron microscopy with energy-dispersive X-ray spectrometry (SEM-EDS) after the KPFM measurements9 and are marked by dashed lines in Fig. 2a. No apparent change in the image contrast was observed at the beginning of the voltage sweep from 0.2 to \(\sim\)2.2 V, during which the current measured by CV was nearly zero. When the current increased slightly at approximately 2.3 V (which is a signature of the charge reactions), the LCP and Pd particles on the left side became brighter, as shown in Fig. 2d–f. Subsequently, the brightly imaged regions rapidly broadened to the right from 2.4 to 2.6 V with a sharp increase in the current (Fig. 2f–l). The left is the current collector side (the current collector was located approximately 1 μm to the left of the edge of the image). Therefore, these results revealed that the charge reactions started at the LCP particles close to the current collector and gradually proceeded to the solid electrolyte side. This finding means that although the Li\({}^{+}\) ion conductivity of the cathode composite was sufficient, the electronic conductivity was too poor to ensure homogeneous electrochemical reactions over the composite electrode. That is, continuous electronic conductive paths from the current collector were not well formed across the composite electrode, and thus only LCP particles on the left side, which were electrically connected to the current collector, were able to react electrochemically at the beginning of the charge process. The extension of the electrochemical reactions to the right side indicated that the conductive path network gradually broadened to the right.

The image size is 10 \(\mu\)m \(\times\) 4 \(\mu\)m. a–l Images obtained during the forward voltage sweep. m–x Images obtained during the backward sweep. LCP and Pd particles are enclosed by yellow and light blue dashed lines in (a), respectively. The remaining parts correspond to the LATP region. Cell voltages at the end of CPD imaging are displayed in the upper right corner of each image.
During the backward sweep, the CPD images gradually became darker homogeneously over the entire composite region with decreasing sweep voltage from \(\sim\)2.6 to \(\sim\)1.5 V, as shown in Fig. 2m–r. During this period, a reduction current flowed from \(\sim\)2.5 to \(\sim\)1.8 V (a signature of discharge reactions), but the image contrast showed no discernible correlation with the current flow (see also Supplementary Fig. 1), in contrast to the forward sweep (the charge process). These findings suggested that the discharge reactions proceeded homogeneously over the entire composite electrode. Subsequently, the brightness of the LCP and Pd particles did not decrease, except for those on the left side of the image (close to the current collector), as shown in Fig. 2s–x. This phenomenon implies that charges (electrons/holes) in those particles could not escape, and thus the electrical potential could no longer decrease, which can be attributed to the collapse of the conductive network, as will be discussed in detail below.
Change in local surface potential during CV operation
To examine the variation of the local surface potential during CV operation in detail, we quantitatively analyzed the data set of the CPD images. Figure 3g–i displays the change in the CPD at the positions of the Pd, LCP, and LATP particles with elapsed time (the corresponding sweep voltages are also indicated on the upper x axis). The current and sweep voltage of the CV data are also plotted in Fig. 3d–f and Fig. 3g–i, respectively. At the Pd particle positions labeled 1–4 in Fig. 3a, the CPD remained constant during a voltage sweep from 0.2 to \(\sim\)2.3 V, as shown in Fig. 3g. Next, it increased steeply at a sweep voltage of approximately 2.3 V within \(\sim\)10 min at each position. The increase occurred first in particles a short distance from the current collector (see also Supplementary Fig. 2c). Subsequently, the CPD curves followed the shape of the sweep voltage (the CPD changed linearly in proportion to the sweep voltage) in the voltage range from \(\sim\)2.3 V in the forward sweep to \(\sim\)1.5 V in the backward sweep. Then, at a sweep voltage of approximately 1.5 V (sometime after the discharge current became nearly zero at \(\sim\)1.8 V), the curves again started to deviate from linear behavior. The rate of decrease gradually slowed, and the CPD approached constant values. A clear sequence in the order of deviation depending on the distance from the current collector was not observed, except for the CPD curve of the particle labeled 1.

The positions where the CPD was taken are indicated in (a), (b), and (c) for Pd, LCP, and LATP particles, respectively. In (d–f), the current from the CV data is plotted as a function of elapsed time (the corresponding sweep voltage is also plotted on the upper x axis). (g), (h), and (i) show the change in CPD at the positions of Pd, LCP, and LATP, respectively, plotted as a function of elapsed time (and also the corresponding sweep voltage, dashed line). Period (and voltage range) in which current flows during the CV measurement is shaded in dark yellow.
Pd is a metal added to ensure sufficient electronic conductivity of the composite electrode. In principle, the potential (and also the CPD) of the Pd particles has to change linearly in proportion to the sweep voltage. However, the constant CPD observed from 0.2 to \(\sim\)2.3 V was inconsistent with this expectation. Because the CPD during the period was independent of the applied voltage, we considered that these Pd particles were in an electrically floating state owing to incomplete formation of the electronic conductive path network in the composite. The sequential increase in the CPD from positions 1 to 4 confirmed that the electronic conduction paths between the current collector and these particles formed gradually from the current collector side, resulting in the formation of an electronic conductive path network across the composite electrode. The state of the network was thought to be maintained in the voltage range between \(\sim\)2.3 V in the forward sweep and \(\sim\)1.5 V in the backward sweep because the CPD curves followed the sweep voltage. This explains why the discharge reactions proceeded homogeneously over the composite electrode. The manner in which the CPD curves again deviated from linear behavior suggested that the Pd particles again entered an electrically floating state after approximately 1.5 V owing to the collapse of the electronic conductive network. In addition, the collapse seemed to occur almost simultaneously over the composite electrodes except for the regions close to the current collector.
A comparison of the CPD changes in Fig. 3g with the current from CV data in Fig. 3d suggests that the formation and collapse of the electronic conductive network were strongly correlated with the current flow, that is, the charge/discharge reactions of the cathode active material. More specifically, the conductive network formed during the period in which Li ions were extracted from the active material. Thus, we deduced that the state of the conductive network varied depending on the charge state of the active material. The initial incomplete formation of the conductive network arose from the low electronic conductivity of LCP particles20, and the state of the conductive network was improved by the increase in electronic conductivity due to the charge reactions (in which Li\({}^{+}\) ions are extracted, and the oxidation state of Co ions is changed from Co\({}^{2+}\) to Co\({}^{3+}\)). The collapse of the conductive network was induced by the re-lowering of the electronic conductivity of the LCP particles owing to the discharge reactions (Li\({}^{+}\) ion insertion and reduction of Co ions).
At the positions of the LCP particles labeled 1–4 in Fig. 3b, the overall tendency of the CPD was similar to that at the Pd particle positions. The CPD remained constant during the voltage sweep from 0.2 to \(\sim\)2.3 V. Subsequently, it increased steeply with increasing current within \(\sim\)10 min at each position. The increase proceeded in order from the particles labeled 1 to 4 (see also Supplementary Fig. 2d). For LCP, the increase in the CPD was \(\sim\)0.5 V smaller than that expected from the applied voltage. We attribute this discrepancy mainly to the change in the work function followed by the stoichiometric change of LCP owing to the charge reactions9. In the voltage range between \(\sim\)2.3 V in the forward sweep and \(\sim\)1.5 V in the backward sweep, the CPD changed linearly with the sweep voltage. Then, it deviated from the linear behavior after the current became nearly zero. At the end of the voltage sweep, the CPD increased slightly, which can be attributed to the effect of overvoltage. In principle, the electrical potential of the active material also has to change linearly according to the applied voltage. The deviation from linear behavior therefore indicated that the potential applied to the outer electrode was not transmitted to the LCP particles owing to incomplete formation of the electronic conductive network in the composite electrode.
At the LATP positions labeled 1–4 in Fig. 3c, the behavior of the CPD curves was almost the same in every region of the composite electrode. The CPD was nearly constant during the voltage sweep from 0.2 to \(\sim\)2.3 V. In the voltage range from \(\sim\)2.3 V in the forward sweep to \(\sim\)2.0 V in the backward sweep, the CPD changed linearly in proportion to the sweep voltage. Then it returned to the level equivalent to the initial value and did not change subsequently.
When a cell voltage is applied between the cathode and anode of an ASS-LIB, the voltage drop is expected to occur mainly at the two interfaces, i.e., the interface between the cathode and the solid electrolyte, and that between the solid electrolyte and the anode. In this experiment, because the anode-side current collector was electrically connected to the ground, the CPD in the solid electrolyte regions is assumed to change with the magnitude of the voltage drop at the solid electrolyte/anode interface. Therefore, during the forward sweep, the CPD is expected to monotonically increase with the sweep voltage. In contrast to this expectation, the CPD remained constant from 0.2 to \(\sim\)2.3 V. Because this behavior is similar to that seen at the Pd and LCP positions, it might also originate from the electrical floating of the LATP particles due to the incomplete formation of the electronic conductive path network. However, the behavior of the CPD curves at the LATP positions differed from that at the Pd and LCP positions in several ways. No difference was found to depend on the position in the composite electrode and no steep increase in CPD as seen in supplementary Fig. 2c and d was observed. These differences are inconsistent with the gradual formation of the electronic conductive path network from the current collector side discussed above. Thus, in addition to the incomplete formation of the electronic conductive path network, another scenario may be needed to explain the behavior of the CPD curves at the LATP positions.
Discussion
LCP is known to exhibit two oxidation peaks and two reduction peaks in the CV curve arising from two distinct two-phase reactions18,20,21,22,23. However, the CV curve of the ASS-LIB showed only a single peak during the forward sweep, whereas a peak and shoulder features were observed during the backward sweep, resulting in asymmetric peak shapes. Asymmetric behavior was also observed in the variation of the local potential distribution between the charge and discharge processes, which can be explained by the difference in the state of the electronic conductive network in the composite electrode. We therefore expect that the asymmetric peak shape in the CV curve is also related to the state of the electronic conductive network.
Next, we will discuss temporal resolution of our technique. Usually, it takes several seconds to several minutes to obtain one CPD image with KPFM. Therefore, if the electrical potential changes faster than the scan speed, the contrast of a CPD image can be different at the bottom and top sections of the image, leading to an apparent non-uniformity of the potential distribution. In fact, it can be seen that the brightness of the Pd particle on the left side of Fig. 2e changed during the scan (it went brighter as the scan proceeded from bottom to top). Importantly, such an apparent non-uniformity itself has no substantial influences on the quantitative analysis of the local CPD change at a fixed point, as depicted in Fig. 3g–i. Furthermore, we can improve the temporal resolution of the CPD measurements by decreasing the scan size and the number of pixels (the number of lines) of a CPD image. For example, if the number of lines is halved, measurement can be performed in half the time. Ultimately, it is possible to measure the changes in CPD with a temporal resolution of \(\sim\)1 ms in our experimental setup by performing KPFM with an AFM tip fixed at a certain point.
Another point that should be discussed concerns the difference between the surface and the bulk potential. Although what we would like to know is the change in the bulk potential during the electrochemical operation of the device, KPFM can only measure the surface potential. Unfortunately, in most cases, the bulk and surface potentials are different due to e.g. the occurrence of the band bending close to the surface region, formation of a surface layer made of a material different from the bulk, and so on. However, it is important to note that the surface potential does not change independently of the bulk potential. Since the surface and the bulk are continuously connected, it is highly unlikely that only the surface potential remains constant while the bulk potential is changing. In general, when the bulk potential changes, the surface potential also does by the same amount while maintaining the relationship between the surface and bulk potential (for example, when the band bending exists, the amount of the bending does not change). This means that as long as the change in CPD (surface potential) is measured rather than the absolute CPD at a certain time, the amount of the change is expected to be equal to the change in the bulk potential. In other words, although the information about the bulk potential are difficult to obtain by analyzing only a single CPD image, it is possible to examine the change in the bulk potential by measuring the change in CPD (surface potential). In fact, the CPD at the Pd particles changed in proportion to the applied external potential while the electronic conductive path network was formed, as shown in Fig. 3g, which demonstrates that the change in the bulk potential was obtained correctly.
Methods
Fabrication of ASS-LIB
The ASS-LIB consists of composite electrodes, Li\({}_{1+x}\)Al\({}_{x}\)Ge\({}_{2-x}\)(PO\({}_{4}\))\({}_{3}\) (LAGP) as a solid electrolyte, and Pd as a current collector, as schematically illustrated in Fig. 1b. The cathode and anode electrodes both have the same composite structure, i.e., a mixture of LCP, LATP, and Pd. In the cathode, LCP works as an active material, LATP acts as a solid electrolyte, and Pd acts as a conductive additive. In the anode, LATP works as an active material24. This battery operates at \(\sim\)2.3 V [from LCP (4.8 V vs. Li/Li+)25,26 to LATP (2.5 V vs. Li/Li+)]. LCP and LATP (a NASICON-type solid electrolyte) are known to react with each other during co-sintering at high temperature27,28. To suppress this reaction, we used Co-doped LATP and LAGP. The ASS-LIB structure was fabricated by stacking green ceramic sheets on which each material was printed using polyvinyl butyral as a binder and subsequent co-sintering at up to \(\sim\)750 \({}^{\circ }\)C.
Cross section preparation
To prepare a sample for the cross-sectional KPFM measurements, an ASS-LIB cell was first cut into a 1 mm \(\times\) 0.5 mm piece in an Ar-filled glove box (O\({}_{2}\): \(<\)0.8 ppm, H\({}_{2}\)O: \(<\)0.8 ppm). Then, we transferred it into a vacuum chamber (\(\sim\)10\({}^{-4}\) Pa) without exposure to air using a transfer vessel to polish the cross-sectional surface by Ar ion milling (JEOL, IB-09020CP, 8 kV). This process exposed cross sections of the particles of each material in the composite electrode and flattened the entire cross-sectional surface. We confirmed that this process did not alter the LIB performance9. After milling, the sample was again transferred to the Ar-filled glove box and mounted on a KPFM holder on which the anode- and cathode-side current collectors were clamped between two stainless steel plates, as shown in Fig. 1a. Finally, the sample was transferred to a N\({}_{2}\) flow glove box for KPFM measurements using aluminum-laminated bags.
KPFM measurements
KPFM measurements were performed at room temperature using a commercial atomic force microscope (Park Systems, XE-100), which was placed in a N\({}_{2}\) flow glove box (O\({}_{2}\): \(<\)40 ppm, H\({}_{2}\)O: \(<\)40 ppm). We used Cr/Pt-coated Si cantilevers (BudgetSensors, Multi75E-G) with a resonance frequency of \(\sim\)65 kHz and spring constant of \(\sim\)3 N m\({}^{-1}\). The CPD was measured in sideband frequency modulation mode29,30. The amplitude and frequency of the modulation voltage were 1 V and 5 kHz, respectively. The modulation voltage and a dc voltage to minimize the electrostatic force between the tip and the sample were applied to the tip. The sample was set so that the anode-side current collector was electrically connected to the ground. The slow scan direction was from the bottom to the top of the images. A source measure unit (ADCMT, 6241A) was used for CV operation of the ASS-LIB. The cell voltage was applied to the cathode-side current collector with respect to the ground, as depicted in Fig. 1a. A CV measurement was performed with home-made LabVIEW-based software. The scan rate was 0.1 mV s\({}^{-1}\), and the voltage range was 0.2–2.7 V. Because the acquisition of a CPD image took \(\sim\)4 min, the difference in cell voltage between the first and last scan lines of a CPD image was \(\sim\)24 mV.
Data availability
The authors declare that all data supporting the findings of this study are available within the article and its Supplementary Information Files. Additional data related to this study are available from the corresponding author upon reasonable request.
References
Deng, D. Li-ion batteries: basics, progress, and challenges. Energy Sci. Eng. 3, 385–418 (2015).
Blomgren, G. E. The development and future of lithium ion batteries. J. Electrochem. Soc. 164, A5019–A5025 (2017).
Li, M., Lu, J., Chen, Z. & Amine, K. 30 years of lithium-ion batteries. Adv. Mater. 30, 1800561 (2018).
Bard, A. J. & Faulkner, L. R. Electrochemical Methods: Fundamentals and Applications. (Wiley, New York, 2001).
Yamamoto, K. et al. Dynamic visualization of the electric potential in an all-solid-state rechargeable lithium battery. Angew. Chem. Int. Edn. 49, 4414–4417 (2010).
Nagpure, S. C., Bhushan, B. & Babu, S. Surface potential measurement of aged Li-ion batteries using Kelvin probe microscopy. J. Power Sources 196, 1508–1512 (2011).
Zhu, J., Zeng, K. & Lu, L. In-situ nanoscale mapping of surface potential in all-solid-state thin film Li-ion battery using Kelvin probe force microscopy. J. Appl. Phys. 111, 063723 (2012).
Luchkin, S. Y., Amanieu, H.-Y., Rosato, D. & Kholkin, A. L. Li distribution in graphite anodes: a Kelvin probe force microscopy approach. J. Power Sources 268, 887–894 (2014).
Masuda, H., Ishida, N., Ogata, Y., Ito, D. & Fujita, D. Internal potential mapping of charged solid-state-lithium ion batteries using in situ Kelvin probe force microscopy. Nanoscale 9, 893–898 (2017).
Wu, J. et al. Multi-characterization of LiCoO2 cathode films using advanced AFM-based techniques with high resolution. Sci. Rep. 7, 11164 (2017).
Zhu, X., Revilla, R. I. & Hubin, A. Direct correlation between local surface potential measured by Kelvin probe force microscope and electrochemical potential of LiNi0.80Co0.15Al0.05O2 cathode at different state of charge. J. Phys. Chem. C 122, 28556–28563 (2018).
Nomura, Y. et al. Direct observation of a Li-ionic space-charge layer formed at an electrode/solid-electrolyte interface. Angew. Chem. Int. Edn. 58, 5292–5296 (2019).
Sun, C., Liu, J., Gong, Y., Wilkinson, D. P. & Zhang, J. Recent advances in all-solid-state rechargeable lithium batteries. Nano Energy 33, 363–386 (2017).
Zheng, F., Kotobuki, M., Song, S., Lai, M. O. & Lu, L. Review on solid electrolytes for all-solid-state lithium-ion batteries. J. Power Sources 389, 198–213 (2018).
Takada, K. Progress in solid electrolytes toward realizing solid-state lithium batteries. J. Power Sources 394, 74–85 (2018).
Nonnenmacher, M., O’Boyle, M. P. & Wickramasinghe, H. K. Kelvin probe force microscopy. Appl. Phys. Lett. 58, 2921–2923 (1991).
Sadewasser, S. & Glatzel, T. (eds) Kelvin Probe Force Microscopy (Springer, 2003).
Strobridge, F. C. et al. Identifying the structure of the intermediate, Li2/3CoPO4, formed during electrochemical cycling of LiCoPO4. Chem. Mater. 26, 6193–6205 (2014).
Ikuhara, Y. H. et al. Atomic level changes during capacity fade in highly oriented thin films of cathode material LiCoPO4. J. Mater. Chem. A 5, 9329–9338 (2017).
Zhang, M., Garcia-Araez, N. & Hector, A. L. Understanding and development of olivine LiCoPO4 cathode materials for lithium-ion batteries. J. Mater. Chem. A 6, 14483–14517 (2018).
Wang, Y. et al. Cr-substituted LiCoPO4 core with a conductive carbon layer towards high-voltage lithium-ion batteries. J. Solid State Chem. 258, 32–41 (2018).
Dimesso, L. et al. Developments in nanostructured LiCoPO4 (m = Fe, Co, Ni, Mn) composites based on three dimensional carbon architecture. Chem. Soc. Rev. 41, 5068–5080 (2012).
Kaus, M. et al. Electrochemical delithiation/relithiation of LiCoPO4: a two-step reaction mechanism investigated by in situ x-ray diffraction, in situ x-ray absorption spectroscopy, and ex situ 7Li/31P NMR spectroscopy. J. Phys. Chem. C 118, 17279–17290 (2014).
Iriyama, Y., Yada, C., Abe, T., Ogumi, Z. & Kikuchi, K. A new kind of all-solid-state thin-film-type lithium-ion battery developed by applying a d.c. high voltage. Electrochem. Commun. 8, 1287–1291 (2006).
Padhi, A. K., Nanjundaswamy, K. S. & Goodenough, J. B. Phospho]olivines as positive electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 144, 1188–1194 (1997).
Amine, K., Yasuda, H. & Yamachi, M. Olivine licopo4 as 4.8 v electrode material for lithium batteries. Electrochem. Solid St. 3, 178–179 (2000).
Nagata, K. & Nanno, T. All solid battery with phosphate compounds made through sintering process. J. Power Sources 174, 832–837 (2007).
Xie, J. et al. Li-ion diffusion kinetics in LiCoPO4 thin films deposited on nasicon-type glass ceramic electrolytes by magnetron sputtering. J. Power Sources 192, 689–692 (2009).
Zerweck, U., Loppacher, C., Otto, T., Grafström, S. & Eng, L. M. Accuracy and resolution limits of Kelvin probe force microscopy. Phys. Rev. B 71, 125424 (2005).
Bergmann, V. W. et al. Real-space observation of unbalanced charge distribution inside a perovskite-sensitized solar cell. Nat. Commun. 5, 5001 (2014).
Acknowledgements
The Ar ion milling and SEM-EDS measurements were performed at the Battery Research Platform of the National Institute for Materials Science. This work was partially supported by CREST, JST (JPMJCR1322), the MEXT Program for the “Development of Environmental Technology using Nanotechnology” of the Ministry of Education, Culture, Sports, Science and Technology of Japan, JSPS KAKENHI (JP17K18347), and JSPS KAKENHI (JP17K06366).
Author information
Authors and Affiliations
Contributions
H.M. and N.I. conceived and designed the experiment. N.I. constructed the KPFM setup. D.I. fabricated the ASS-LIB. H.M. and K.M. prepared the sample for KPFM measurements and performed the KPFM experiments. H.M., K.M., and N.I. analyzed the data. H.M. and N.I. wrote the paper. D.F. provided funding. All the authors read the paper and commented on the contents.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Masuda, H., Matsushita, K., Ito, D. et al. Dynamically visualizing battery reactions by operando Kelvin probe force microscopy. Commun Chem 2, 140 (2019). https://doi.org/10.1038/s42004-019-0245-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-019-0245-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
