
Abstract
The continuous generation of multi-omics and phenotype data is propelling advancements in precision oncology. UCSCXenaShiny was developed as an interactive tool for exploring thousands of cancer datasets available on UCSC Xena. However, its capacity for comprehensive and personalized pan-cancer data analysis is being challenged by the growing demands. Here, we introduce UCSCXenaShiny v2, a milestone update through a variety of improvements. Firstly, by integrating multidimensional data and implementing adaptable sample settings, we create a suite of robust TPC (TCGA, PCAWG, CCLE) analysis pipelines. These pipelines empower users to conduct in-depth analyses of correlation, comparison, and survival in three modes: Individual, Pan-cancer and Batch screen. Additionally, the tool includes download interfaces that enable users to access diverse data and outcomes, several features also facilitate the joint analysis of drug sensitivity and multi-omics of cancer cell lines. UCSCXenaShiny v2 is an open-source R package and a web application, freely accessible at https://github.com/openbiox/UCSCXenaShiny.
Similar content being viewed by others

Introduction
UCSC Xena (https://xena.ucsc.edu/)1 serves as a repository housing multi-omics and phenotype datasets derived from prominent cancer research projects, such as The Cancer Genome Atlas (TCGA)2, Pan-Cancer Analysis of Whole Genomes (PCAWG)3, and Cancer Cell Line Encyclopedia (CCLE)4 (together referred to as TPC), alongside contributions from various independent research entities. This platform facilitates to identify emerging cancer patterns and validates biological findings with a vast collection of 1500+ datasets (https://xenabrowser.net/datapages/) spanning over 50 distinct cancer types.
Over recent years, we have developed the R/CRAN (The Comprehensive R Archive Network) packages UCSCXenaTools5 and UCSCXenaShiny6, featuring one web application with a graphical user interface (UI). These tools have been specifically designed to provide data access, support interactive analysis of UCSC Xena datasets, and aid in creating publication-quality plots, catering to researchers and clinicians without programming skill training. The UCSCXenaShiny tool has garnered substantial recognition, as evidenced by over one hundred citations (Google Scholar) and over 60,000 downloads in the past three years, underscoring its importance in the realm of cancer research. Despite those advantages, it has been noted to possess limitations in terms of unified analysis pipelines and personalized settings, especially for the TPC datasets. Concurrently, there has been a proliferation of online tools dedicated to analyzing public tumor omics data, such as GEPIA7 and GSCA8. Nevertheless, many of these tools lack comprehensive integration of tumor data and customizable functionalities that can adapt to the evolving demands of precision cancer research. In response to these issues, here we introduce UCSCXenaShiny v2, a significant update that provides users with in-depth insights through multidimensional cancer omics analysis, customized sample operations, enhanced analysis functionalities and UI, as well as additional features focusing on cancer pharmacogenomics. Overall, UCSCXenaShiny v2 represents a new milestone in our ongoing efforts to leverage and integrate cancer omics datasets to advance precision oncology. UCSCXenaShiny v2 is open-source and accessible as an R package on GitHub, CRAN, Conda, and a Docker container, alongside being offered as a Shiny web application.
Results
Update overview of UCSCXenaShiny v2
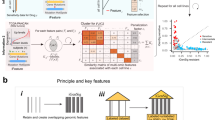
As shown in Fig. 1, the key highlight of UCSCXenaShiny v2 is the proposal and implementation of TPC (TCGA, PCAWG, CCLE) analysis pipelines, featuring exhaustive tumor data compilation from various molecular profiling datasets and phenotypic characteristics, enabling extensive joint analysis. The data subsetting modules enhance precision research by filtering and grouping samples based on user demands, catering to specific subpopulations of interest. The versatile analytical scenarios offer three general analysis methods (correlation, comparison, and survival) across three distinct modes (Individual, Pan-cancer and Batch screen), facilitating investigation into potential biomarkers and therapeutic targets. Additionally, other notable enhancements include organized molecular analysis reports for preliminary pan-cancer exploration, custom download interface for partial dataset retrieval, and pharmacogenomics functionalities for drug analysis based on published studies, collectively improving user experience and accelerating scientific discovery. In detail, Supplementary Fig. 1 illustrates the compositional structure of the project’s framework and Supplementary Fig. 2 summarizes the functional improvements of this updated version.

①Firstly, a set of TPC analysis pipelines are introduced. TPC indicates TCGA, PCAWG and CCLE databases. ②Secondly, the pan-cancer report can be automatically generated for exploratory molecule analysis. ③Thirdly, two download interfaces are designed for custom query of integrative TPC data or general UCSC Xena datasets. ④Fourthly, it provides integrative pharmacogenomics analysis which merges drug sensitivity studies and cancer cell line omics datasets.
Thorough integration of molecular and phenotypic TPC tumor data
In the updated version, we have extensively aggregated TPC datasets about cancer multi-omics (Molecular profile) from UCSC Xena (Supplementary Table 1) and non-omics features (Tumor index, Immune infiltration, Pathway activity, Phenotype data) from other resources (Supplementary Table 2), thereby enhancing the scope of cancer data analysis. Each of these data types comprises multiple subtypes with numerous entities. In the context of TCGA: (1) there are seven data subtypes of Molecular profile (Gene Expression, Transcript Expression, DNA Methylation, Protein Expression, miRNA Expression, Gene Mutation, Copy Number Variation); (2) Alternative datasets with diverse normalization methods or other variables can be easily switched for specific research objectives. For example, in DNA methylation analysis, users can leverage 27 K or 450 K microarray dataset and choose to include or exclude one or multiple CpG loci within a single gene, and to select among various aggregation methods such as mean, median, and maximum; (3) Apart from UCSC Xena, we have also compiled common demographic/clinical (e.g., Gender, Age, Stage) and phenotypic features (e.g., Tumor Purity, Tumor Stemness, Tumor Mutation Burden) from published literature. In addition, 119 immune infiltration estimates based on 7 algorithms were downloaded from the TIMER2 website9, and the expression scores for 500 pathways of 3 categories were calculated in-house (see section “Methods”). (4) The uploading of user-defined metadata and custom molecular signatures is allowed, enhancing analysis flexibility. We concurrently endeavored to gather and integrate multi-omics data for PCAWG and CCLE samples, adhering to the previously described TCGA data hierarchy, thereby establishing uniform TPC pipelines. However, it is noteworthy that some data categories present in TCGA are not available in PCAWG and CCLE (Supplementary Tables 1 and 2).
Data subsetting with customized filtering and grouping
Two data subsetting modules are designed to easily query data of interest with user-defined markers based on the integrated TPC data. The filtering module is utilized to select specific sample subpopulations for analyzing their molecular characteristics. As depicted in Fig. 2A, any supported identifier (such as gene expression or immune infiltration level) can serve as a condition to filter out relevant samples of one cancer type. Six operators (“+”, “-”, “>”, “<”, “%>”, “%<”) have been devised to enable flexible filtering based on the type of selected conditions and expected thresholds. For example, users can conveniently select female samples or those with higher TP53 gene expression (Supplementary Fig. 3A). Combinational filtering based on more than one condition is also feasible for more precise sample selection. In addition, we also designed a supplementary widget for quick filtering based on tissue codes, such as normal or primary tumor samples. The grouping module is used to create two customized groups for comparative purposes (e.g., survival analysis) after optional sample filtering (Fig. 2B). Similar to the filtering operation, customizable parameters are available to stratify samples. For example, users can set two sample subgroups to be compared based on gene expression of TP53, mutation status of TP53 or disease stages (Supplementary Fig. 3B). The integration of these two modules, along with their various combinations, provides users with a highly flexible and expansive toolkit for deeply exploring and leveraging the TPC datasets, offering a multitude of analytical possibilities.

A Filtering module, users firstly need to decide on the initial samples set within one cancer cohort. Then, any identifier from the integrated data can be selected as the filtering condition using six optional data operators (see section “Methods”) to perform sample filtering. The remaining samples can be used for further filtering or downstream analysis. B Grouping module, user can choose to use the initial samples of one cancer cohort or samples remaining after filtering. Then, any one identifier from the integrated data can then be selected as the grouping condition. Finally, one of three types of grouping ranges, based on the selected condition can be set to generate two groups for downstream analysis.
Implementation and demonstration of versatile analysis scenarios
Drawing from the independent analysis modules in the initial version, we have methodically devised several unified pipelines for examining correlations, conducting comparisons, and assessing survival outcomes within TPC datasets subsequent to sample filtering and/or grouping. Within each analysis pipeline, three specific modes have been designed to serve different objectives: individual cancer type analysis, pan-cancer analysis, and batch screen analysis. By amalgamating thorough data integration, personalized sample settings, and versatile analysis methods and modes, the execution of TPC pipelines offers a flexible and comprehensive framework to address the diverse basic and advanced needs of broad community of cancer researchers, compared to other similar TCGA analysis tools (Supplementary Table 3). Presented below are several instances exemplifying the key operations and visualization of TCGA-related pipelines. As Fig. 3A shows, the Spearman correlation between gene expression of TP53 and monocyte infiltration estimated by CIBERSORT algorithm10 among primary tumor samples of CHOL (cholangiocarcinoma) was calculated, showing a certain degree of negative correlation (Spearman, R = −0.299, P = 0.0769). Among the same samples (CHOL primary tumor), it is possible to pinpoint six genes within the apoptosis hallmark pathway were found to exhibit associations with monocytes (Spearman, P < 0.05, Supplementary Table 4). Subsequently, the Pearson coefficients between HDAC1 and APOE were investigated among tumor samples of TCGA pan-cancers based on the log-count gene expression dataset, with the highest correlation observed in OV (ovarian serous cystadenocarcinoma) patients (Fig. 3B).

In each figure panel, the left subpanel indicates the key operational steps, and the right subpanel indicates the corresponding visualization result. A The example correlation analysis in individual mode among CHOL samples (n = 36). B The example correlation analysis in pan-cancer mode (n = 9784). C The example comparison analysis in individual mode among BRCA samples (total n = 1098, Primary n = 1091, Metastatic n = 7). D The example comparison analysis in pan-cancer mode (total n = 2016, PTEN-Wild n = 1843, PTEN-Mut n = 173). The cancer types with a sample size of 3 or more in each group will be retained. All the source data can be downloaded from the Shiny application, which has been uploaded as Supplementary Data.
In the context of comparative analysis, Fig. 3C demonstrates that primary tumor specimens of BRCA (breast invasive carcinoma) exhibit elevated expression of POSTN compared to metastatic samples. Additionally, among 39 xCell11 immune cell subtypes examined, enhanced infiltration of memory B cells was observed in metastatic BRCA samples (Supplementary Table 5). A pan-cancer comparison was performed for primary tumor samples from individuals over 60 years old without CNV alteration in the PTEN gene, evaluating the difference in tumor stemness between groups characterized by wild-type and mutant PTEN (Fig. 3D). In the individual mode of survival analysis, BRCA tumor specimens were stratified based on higher and lower methylation levels at the loci cg15206330. The log-rank test revealed that increased methylation at this site was linked to poorer overall survival (OS) and an optimal cutoff to stratify patient can be determined (Supplementary Fig. 4A). Moreover, a survival analysis was conducted for BRCA across 50 cancer hallmark pathways12, highlighting that different stages of estrogen response could play significant but inconsistent roles in survival outcomes of breast cancers (Supplementary Table 6). Finally, a distinct gene signature comprising TP53 and PTEN was designed, dividing patients into two groups based on the lowest 10% and highest 10% signature scores (Supplementary Fig. 4B). The univariate Cox regression analysis showed a significant association (P < 0.05) between the signature and increased risk in ACC (adrenocortical carcinoma) and PRAD (prostate adenocarcinoma) patients, as evidenced by hazard ratios exceeding 1.
Case study of the TPC pipeline
Here, we present a concise case study using the TPC pipelines to analyze the impact of common signaling pathways on gastric cancer overall survival in the context of elevated immune infiltration. First, we divided TCGA-STAD (stomach adenocarcinoma) samples into two groups according to the median ssGSEA score of each of 186 KEGG pathways and then performed Cox survival analysis using the pathway-specific STAD groupings, respectively. Next, we independently filtered samples using six distinct immune estimations from the TIMER algorithm9, retaining only those subpopulations with higher levels of immune infiltration, defined by a median cutoff. For each sample subpopulation, the Cox analysis of each KEGG pathway was performed in the same way. As shown in Fig. 4A, we found several pathways showing significant prognostic impact under specific immune conditions in the tumor microenvironment. For example, the MAPK (mitogen-activated protein kinase) pathway was associated with poorer survival in the gastric cancer subpopulation with higher CD4+ T cells (Cox regression: P = 0.0070, HR = 1.86, Fig. 4A; log-rank test: P = 0.0061, Fig. 4B) compared with lower infiltration (log-rank test: P = 0.68, Fig. 4C). Interestingly, CD4+ T cell abundance itself cannot stratify STAD patients (log-rank test: P = 0.78, Fig. 4D). Next, pan-cancer analysis revealed similar influence in the MESO (mesothelioma), KIRP (kidney renal papillary cell carcinoma), and LGG (brain lower grade glioma) subpopulations characterized by CD4+ T cells, with STAD showing the highest survival risk (Fig. 4E). Through the molecular association analysis, we found both IFNG and FOXP3 expression showed positive correlation with the MAPK pathway during low-level infiltration of CD4+ T cells (Fig. 4F, G). However, IFNG became negatively correlated to the pathway among tumor samples with high-level CD4+ T cells (Fig. 4H), while FOXP3 exhibited no association (Fig. 4I), indicating different roles in the process.

All the analyses and most visualizations (except for the heatmap via ComplexHeatmap package) were conducted through the TPC pipelines. A The heatmap showing the hazard ratios (HRs) of Cox regression analysis for 186 KEGG pathways among different conditions of TCGA-STAD tumor samples. Apart from the condition of all TCGA-STAD tumor samples, other subpopulations feature higher infiltration of 6 types of immune cells estimated by TIMER algorithm. For one analysis with P value above 0.01, it is considered as insignificant (white color). The pathways with at least one significant condition are showed. (B, C) The log-rank survival analysis for MAPK pathway among STAD tumor samples with higher (B) or lower (C) infiltration of CD4+ T cells. D The log-rank survival analysis for CD4 + T cells among STAD tumor samples. E The log-rank survival analysis for MAPK pathway among pan-cancer tumor samples (n = 4434) with higher infiltration of CD4+ T cells. **P < 0.01; *P < 0.05; ns, P > 0.05. Red and blue color indicate high and low risk of MAPK pathway. F–I The correlation between gene expression of two genes (IFNG and FOXP3) and ssGSEA scores of MAPK pathway among STAD tumor samples with low (n = 205) and high (n = 207) infiltration of CD4+ T cells. All the source data can be downloaded from the Shiny application, which has been uploaded as Supplementary Data.
User-friendly enhancements of data export interface
The updated version is further equipped with various download options to facilitate users in retrieving diverse data to their local computers. Multiple download buttons were available at the last step of the TPC analysis pipelines. Users can generate and save publication-quality plots with adjustable visualization parameters in PNG or PDF formats in both individual and pan-cancer analysis modes. For example, the TCGA cross-omics analysis supports the simultaneously exploration of pan-cancer features of genes or pathways based on multiple omics data and allows users to download the high-quality figures (Supplementary Fig. 5) suitable for publication via funkyheatmap package13. Furthermore, data tables containing analysis results with detailed statistical parameters and original identifier data are also available for download as CSV files to improve the reproducibility and transparency of analysis. Additionally, a molecular pan-cancer analysis report can be quickly generated on the homepage of our web application, enabling users to explore general characteristics of one molecule of interest (Supplementary Fig. 6). The organized report, featuring interactive tables and plots, can be downloaded as a well-structured and self-contained HTML web page file, with the corresponding data also provided. Lastly, two modules were developed to facilitate direct downloading of data subsets for users conducting targeted analysis locally, thereby minimizing unnecessary time and storage costs associated with downloading the entire dataset.
Addition of pharmacogenomics analysis
Apart from utilizing the UCSC Xena resource, we have compiled extensive datasets (Supplementary Table 7) from six prominent pharmacogenomics projects (GDSC1 and GDSC214, CTRP1 and CTRP215,16, PRISM17, gCSI18) focusing on examining the sensitivity of cancer cell lines to diverse chemical agents. This compilation comprises a total of 2123 chemical compounds and 1276 cell lines (Supplementary Fig. 7A, B). Subsequently, diverse functional modules have been developed to investigate this extensive pharmacogenomics resource, integrating multi-omics analyses of cancer cell lines including gene expression, DNA methylation, protein expression, gene fusion, gene mutation and copy number variations. The first module enables users to compare the sensitivity of a particular drug or molecular characteristics of different types of cancer cell lines. For example, SNX-2112 (HSP inhibitor) exhibits stable and replicable sensitivity in leukemia and lymphoma cell lines among 3 datasets (Supplementary Fig. 8A). The second module provides a comprehensive overview of a drug’s performance across various cancer cell lines. For instance, the “MAD&MEDIAN” plot highlights Sangivamycin, an inhibitor of protein kinase C, as exhibiting the most potent and consistent efficacy in the PRISM dataset (Supplementary Fig. 8B). Dimensionality reduction using the t-distributed stochastic neighbor embedding (t-SNE) reveals similar chemical response patterns for two small molecules (Serdemetan and Cisplatin) with anti-cancer activities despite their distinct mechanisms (Supplementary Fig. 8C, D). The third module allows users to explore the relationship between drug sensitivities and omics characteristics of cell lines. Spearman correlation analysis demonstrates a significant correlation between ABCC3 gene expression and YM-155 (Survivin inhibitor) sensitivity (Supplementary Fig. 9A). Moreover, higher sensitivity can be observed in cell lines with TP53 mutations, underscoring the influence of genetic alterations on drug response patterns (Supplementary Fig. 9B). Lastly, the fourth module is tailored to investigate scaling feature associations (e.g., drug-feature pairs or feature-feature pairs) across multiple datasets. Users can identify candidate molecules of a specific omics type robustly associated with a drug response (e.g., Lapatinib) through statistical assessment of drug-molecule associations in each dataset (Supplementary Fig. 9C, D).
Discussion
UCSC Xena has provided great feasibility for cancer research through the compilation of large-scale cancer-related projects. Multi-omics molecular profiling (e.g., genomics, transcriptomics, proteomics, epigenomics) together with various phenotypic features contribute to understanding the complex landscape of cancer biology and discovering novel biomarkers. Considering the multidimensional heterogeneity of tumor tissues, molecular precision cancer research furtherly enables more accurate diagnosis, targeted therapies and thus improved outcomes. For instance, gender as an important phenotypic variable could be an underlying factor in the immunotherapy response of some cancers19,20,21. Another example is that the gene expression of TERT was found to be associated with favorable survival in HNSC (head and neck squamous cell carcinoma) with high B lymphocyte infiltration, indicating a potential mechanism by which TERT interacts with the tumor microenvironment22. Nevertheless, the sophisticated merging and processing of data not only consume significant time and resources but also deter researchers or clinicians lacking certain computer analytical skills. Existing online cancer analysis tools also have limitations in fully leveraging the rich resources of the UCSC Xena repository and providing personalized analysis for in-depth insights. For instance, GEPIA7 adopted three immune infiltration algorithms despite the presence of other emerging deconvolution methods such as xCell. GSCA8 only calculated activity scores of 10 classic pathways and cannot provide more detailed description about molecule-related biological process recorded in public pathway databases (e.g., KEGG23). More importantly, many of these tools do not support flexible sample customization operations for precision research on tumor subpopulations.
To bridge this unmet gap of cancer research, the highlight of UCSCXenaShiny v2 lies in proposing a series of powerful TPC (TCGA, PCAWG, CCLE) pipelines with the following features. Firstly, the vast tumor data integration based on multiple molecular profiling, multi-faceted score annotation and user-defined metadata, makes extensive joint analysis possible. Furtherly, the two data subsetting modules for sample filtering and grouping enable cancer research to achieve enhanced precision and efficiency in analyzing the diverse characteristics of specific subpopulations. Finally, each of the three general analysis methods can be performed via three distinctive modes, facilitating the versatile investigation of potential biomarkers and targets according to differing purposes of users. Following several example analytical operations upon TCGA pipelines, we demonstrated a practical exploration of the prognostic impact of KEGG signaling pathways in gastric tumors under different immune infiltration conditions and revealed the CD4+ T cells-independent influence of MAPK pathway, which exerts a crucial role in cancer by regulating cell proliferation, differentiation, and migration24,25. However, its specific roles under various high immune conditions are less studied. Using our application, we found the MAPK pathway was especially associated with worse survival of gastric tumor subpopulations with higher infiltration of CD4+ T cells. Previous research once reported MAPK signaling pathway could regulate the production of IL-1726, which has been recently validated as a prospective target in gastric tumors through single-cell sequencing technology27. Moreover, the changes of correlation between two markers (IFNG for T-helper 1 (Th1) cells28, FOXP3 for regulatory T (Treg) cells29) of two subtypes of CD4+ T cells and MAPK pathway under different statuses of infiltration were also observed, which further contribute to the potential mechanism of the pathway. Notably, all the above precision analyses were performed in the TCGA pipelines and most visualizations were directly downloaded from the application interface (except for the heatmap from ComplexHeatmap package30), which could be difficult to implement with other tools. Therefore, the robust pipelines can offer a comprehensive approach to tumor data analysis, empowering researchers to gain deeper insights into cancer biology and accelerate the discovery of clinically relevant findings.
We also introduced other remarkable enhancements such as the quick generation of intuitive molecular analysis report, custom downloads for precise subset of matrix datasets, and the supplementary analyses of several pharmacogenomics modules, collectively augmenting the capabilities of the updated version. In addition, some minor improvements like dimension-reduction analysis and daily gene display further contribute to providing a more user-friendly and efficient analysis experience. Meanwhile, substantial efforts are devoted to improving the sustainable data analysis31 of our application. For example, the more intuitive interface with necessary prompts enhances the readability and reduces the learning curve. The availability of detailed results and raw data of analysis benefits the reproducibility and transparency. Furthermore, we have compiled a comprehensive set of resources to guide users, including instructional videos, a concise quick-start guide, and a thorough tutorial book (https://lishensuo.github.io/UCSCXenaShiny_Book/), all detailing the utilization of the Shiny application and R package (see section “Methods”). Lastly, we have introduced a range of refinements to enhance the user experience. For example, recognizing that loading all functional pages of the Shiny application may be time-consuming, we have implemented a startup function with different run modes, enabling a streamlined local launch of a lightweight Shiny application for swift access.
In conclusion, UCSCXenaShiny v2 represents a significant advancement in pan-cancer omics analysis, overcoming many limitations of initial version and providing researchers with a powerful platform for precision cancer research. In the future, UCSCXenaShiny remains committed to facilitating transformative insights that drive advancements in cancer biology and therapeutics with the ever-changing landscape of cancer omics studies.
Methods
Omics datasets curation from UCSC Xena
We have curated multi-omics pan-cancer datasets from UCSC Xena data hubs (https://toil.xenahubs.net, https://gdc.xenahubs.net, https://tcga.xenahubs.net, https://pancanatlas.xenahubs.net) for each of TPC databases. For TCGA, 14 selected datasets involve 7 types of molecular profiling (Gene Expression, Transcript Expression, DNA Methylation, Protein Expression, miRNA Expression, Gene Mutation, Copy Number Variation). There are respectively 3 datasets characterized by different normalization methods (TPM (transcripts per million), FPKM (fragments per kilobase of transcript per million mapped reads), Count) for gene and transcript molecules. In addition, the copy number variation and methylation profiling also have alternative datasets available due to different identification algorithms or sequencing platforms. Regarding the other two projects, we totally selected 8 datasets involving 5 types of molecules (Gene Expression, Promoter Activity, Gene Fusion, miRNA Expression, APOBEC Mutagenesis) for PCAWG and 5 datasets involving 4 types of molecules (Gene Expression, Protein Expression, Gene Mutation, Copy Number Variation) for CCLE.
Non-omics feature collection and calculation
Four categories of non-omics data mainly for TCGA database were collected from UCSC Xena platform or other resource for extensive analysis. Firstly, basic clinical phenotypes of patients (e.g., Age, Gender, Tissue Code, Stage) were incorporated. Then, diverse features for five types of tumor indexes were also compiled, including Tumor Purity, Tumor Stemness, Tumor Mutation Burden, Microsatellite Instability, Genome Instability. Next, we estimated the immune infiltration conditions and pathway expression scores among TCGA samples. In detail, the compositions of immune cells based on 7 types of deconvolution algorithms (CIBERSORT, CIBERSORT-ABS, EPIC, MCPCOUNTER, QUANTISEQ, TIMER, XCELL) were obtained from the TIMER2.0 website9, which were calculated via immunedeconv package32. The expression scores of hundreds of gene sets from three signature resources (HALLMARK, KEGG, IOBR)12,23,33 were calculated through the ssGSEA method of GSVA package34 based on the “TcgaTargetGtex_rsem_gene_tpm” dataset of UCSC Xena. Afterward, we endeavored to collect the same identifiers of non-omics data for PCAWG and CCLE databases. Specifically, the “tophat_star_fpkm_uq.v2_aliquot_gl.sp.log” dataset was utilized to evaluate immune infiltration and pathway activity of PCAWG samples. Data sourced outside of the UCSC Xena platform have been archived on Zenodo (https://zenodo.org/doi/10.5281/zenodo.4625639)35 for accessibility and preservation.
Custom molecular signature design
User-designed molecular signature can be comprised of \({{n}}\) molecules from any one of curated molecular types of TPC databases. For each constituent molecule \({{m}}\), its corresponding coefficient \({{w}}\) can be set and the default value is 1. Then, the signature score is calculated through the aggregation of the products between molecular values and their coefficients.
Two preprocessing modules of the TPC pipelines
The filtering module at the upstream of pipelines enables precise selection of tumor subpopulations with specific characterizations. Any identifier from the integrated TPC data can be used as the condition. Multiple data operators were designed for versatile filtering. In detail, “+” or “-” are used to retain or discard samples for categorical (character) conditions. There are two types of operators to set absolute (“>”, “<”) or percentile (“%>”, “%<”) thresholds for continuous (numeric) conditions. Ordered combinations of multiple conditions are also supported for intricate filtering operations. Another preprocessing module is for grouping samples according to user-defined conditions. Two non-overlapping subgroup ranges can be flexibly set depending on the type of one selected condition.
Three analysis methods of the TPC pipelines
Fundamental tumor data analyses, including correlation, comparison and survival, are generally incorporated with various analysis and visualization parameters, implemented by corresponding R packages. The ggscatterstats and ggbetweenstats functions of the ggstatsplot package36 are applied for correlation and comparison analysis as well as visualization, respectively. Regarding the analysis methods, two correlation coefficients (Pearson, Spearman) and two comparison options (Student’s t-test, Wilcoxon test) are both supported. Two survival analyses (log-rank test and univariate Cox regression) between two groups of samples, are implemented by survdiff and coxph functions of the survival package37. Noteworthily, if the grouping condition is continuous, alternative analyses are supported instead of pre-setting groups: for log-rank test, the optimal cutoff can be automatically decided, while for Cox regression, the continuous variable can be directly included in model. The survival analysis is mainly available in TCGA and PCAWG databases with OS (overall survival), DSS (disease-specific survival), DFI (disease-free interval), PFI (progression-free interval) endpoints for TCGA samples and OS endpoint for PCAWG samples.
Three analysis modes of the TPC pipelines
Depending on different purposes, we have designed three modes for each analysis method, termed as individual mode, pan-cancer mode and batch screen mode. In the basic individual mode, one identifier-specific analysis can be performed in the context of one cancer. In the pan-cancer mode, the same individual analysis can be consecutively performed across multiple cancers. There are various visualization plots for reasonable display of analytical results in above two modes. The batch screen mode is used for identifying statistically significant candidate identifiers for one cancer. In detail, three ways are supported to choose batch identifiers. Except for one-by-one selection, user can upload a text file with eligible identifiers or directly select all identifiers of one data type. We enable users to choose all identifiers in any one pathway gene set curated from the Molecular Signatures Database (MSigDB)38. Finally, three types of results can be downloaded, including the raw data, detailed statistical results and visualization plot.
Well-organized pan-cancer HTML report
The quick generation of pan-cancer analysis report enables the exploration of multi-faceted features of one molecule from seven omics types based on the integrated TCGA data. Given the prepared R markdown script, one well-organized report in HTML format can be rendered via the knitr39 package and it comprises five sections of pan-cancer analysis, involving the relationships of one molecule with clinical phenotypes, survival events, tumor indexes, immune infiltration and pathway activity. The interactive figures and tables embedded in the report are implemented via the DT40 and plotly41 packages, respectively.
Custom download modules
Two download modules are furtherly added to support the custom acquisition of original datasets. The first module is used to directly fetch matrix data of interesting samples and identifiers from the TPC omics tumor data. Other non-omics data, like survival information, can be fully obtained through corresponding buttons. The second module can be generally applied to download the subset of most matrix datasets in UCSC Xena repository, where multiple molecules can be selected through the original identifiers or additional probe map annotation.
Pharmacogenomics data collection
We have totally collected comprehensive drug screening databases from six publicly accessible pharmacogenomics studies (Supplementary Table 7), including two datasets (GDSC1, GDSC2) from the Genomics of Drug Sensitivity in Cancer (GDSC) project14, two datasets (CTRP1, CTRP2) from the Cancer Therapeutics Response Portal15,16, one dataset (PRISM) from the Cancer Dependency Map Consortium’s DepMap portal17, and one dataset (gCSI) from the Genentech Cell Screening Initiative18. Six types of omics profiling (Gene Expression, Protein Expression, Copy Number Variations, DNA Methylation, Gene Fusion, and Gene Mutation) are collected from the Cancer Cell Line Encyclopedia (CCLE) and ORCESTRA portal42. Given that there are overlapping cells in different drug and omics datasets, we have utilized the common data to assess correlations, thereby maximizing the utilization of existing information. For instance, the designation “gdsc_ctrp1” indicates that the omics data is sourced from the GDSC project, while the drug sensitivity data is derived from the CTRP1 project. In the evaluation of projects based on DepMap, GDSC, and CTRP, we employed the reported area under the dose-response curve (AUC) values as a measure of therapeutic efficacy. Conversely, for the gCSI project, the area above the dose-response curve (AAC) served as the indicator for drug sensitivity. Generally, lower values of AUC signifies enhanced sensitivity to drug treatment. To ensure consistency across all datasets, wherein a lower metric reflects higher drug sensitivity, we transformed the AAC values from the gCSI dataset using the formula max(AAC) - AAC.
Implementation of pharmacogenomic modules
The total features for pharmacogenomic analysis include drug sensitivity and multiple molecular information, where the types of drug sensitivity, mRNA expression, DNA methylation, protein expression, and copy number variable are continuous and the types of gene fusion, gene mutation, and gene site mutation are categorical. The first module investigates the relationship between molecular characteristics and drug sensitivity across diverse cell line types. Boxplots with Kruskal-Wallis tests or bar plots with Chi-squared tests are utilized for continuous or categorical features, respectively. To conduct t-SNE analysis, drugs with over 80% missing records and cells with over 50% missing records are excluded from each dataset. Then R package impute43 is then applied to impute remaining missing data using the nearest neighbor averaging function. In the analysis of scaling feature associations, the determination of effect size and statistical significance varies depending on the types of features being compared:
-
1.
For continuous features compared against continuous datasets (e.g., levels of drug A vs. all CNV features), the Spearman correlation coefficient (R) ranging from −1 to 1 is employed.
-
2.
When assessing categorical features against categorical datasets (e.g., TP53 mutation events vs. all recorded gene fusions), the effect size is measured using the log2 odds ratio, with P values computed using the Chi-squared test.
-
3.
In cases where continuous features are compared against categorical datasets or categorical features are compared against continuous datasets, the log2 fold change (events/wildtype) is used as the effect size metric, with P values derived from the Wilcoxon test.
Users have the flexibility to set a threshold to filter the absolute value of the effect size, with default values established at 0.2, 2, and 4 for the aforementioned scenarios. Following table review, users can download the tabular result containing all significant pairs, with statistical significance determined at P < 0.05 and effect size above the user-defined threshold.
Statistics and reproducibility
Three main statistical analyses are supported in the Shiny web. Correlation analysis can be performed using the Spearman or Pearson method. Comparison analysis between two groups can be performed using Wilcoxon test or Student’s t-test. In general, the robust non-parametric tests (Spearman correlation analysis and Wilcoxon comparison analysis) are recommended. The log-rank test and univariate Cox regression can be implemented for survival analysis. The 95% confidence intervals are added into Kaplan–Meier survival curves. All reported P-values are two-tailed, and P value <= 0.05 is considered statistically significant for all analyses (n >= 3). All statistical analyses were conducted using R version 4.2.2.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All supporting data and materials are available in the GitHub repository (https://github.com/openbiox/UCSCXenaShiny), UCSC Xena dataset hubs (https://xenabrowser.net/datapages/), and Zenodo repository for UCSCXenaShiny generated and curated data (https://zenodo.org/doi/10.5281/zenodo.4625639)35. The Shiny application has been deployed at http://shiny.zhoulab.ac.cn/UCSCXenaShiny/, and https://shiny.hiplot.cn/ucsc-xena-shiny. Comprehensive documentation, including tutorial videos, can be found at https://lishensuo.github.io/UCSCXenaShiny_Book/. The source data behind the figures can be found in Supplementary Data.
Code availability
The source code for UCSCXenaShiny v2 is available under the GPLv3 license and can be freely accessed at both GitHub (https://github.com/openbiox/UCSCXenaShiny) and Zenodo platform44. This Shiny application is compatible with Linux, macOS, and Windows operating systems and is written in the R programming language.
References
Goldman, M. J. et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 38, 675–678 (2020).
Weinstein, J. N. et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 45, 1113–1120 (2013).
The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020).
Barretina, J. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012).
Wang, S. & Liu, X. The UCSCXenaTools R package: a toolkit for accessing genomics data from UCSC Xena platform, from cancer multi-omics to single-cell RNA-seq. J. Open Source Softw. 4, 1627 (2019).
Wang, S. et al. UCSCXenaShiny: an R/CRAN package for interactive analysis of UCSC Xena data. Bioinformatics 38, 527–529 (2022).
Li, C., Tang, Z., Zhang, W., Ye, Z. & Liu, F. GEPIA2021: integrating multiple deconvolution-based analysis into GEPIA. Nucleic Acids Res 49, W242–W246 (2021).
Liu, C.-J. et al. GSCA: an integrated platform for gene set cancer analysis at genomic, pharmacogenomic and immunogenomic levels. Brief Bioinform. 24, https://doi.org/10.1093/bib/bbac558 (2023).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48, W509–W514 (2020).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Aran, D., Hu, Z. & Butte, A. J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220 (2017).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Saelens, W., Cannoodt, R., Todorov, H. & Saeys, Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 37, 547–554 (2019).
Iorio, F. et al. A landscape of pharmacogenomic interactions in cancer. Cell 166, 740–754 (2016).
Basu, A. et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154, 1151–1161 (2013).
Rees, M. G. et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 12, 109–116 (2016).
Corsello, S. M. et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 1, 235–248 (2020).
Haverty, P. M. et al. Reproducible pharmacogenomic profiling of cancer cell line panels. Nature 533, 333–337 (2016).
Ye, Y. et al. Sex-associated molecular differences for cancer immunotherapy. Nat. Commun. 11, 1779 (2020).
Li, H. et al. Connecting the mechanisms of tumor sex differences with cancer therapy. Mol. Cell Biochem. 479, 213–231 (2024).
Wang, S., Zhang, J., He, Z., Wu, K. & Liu, X.-S. The predictive power of tumor mutational burden in lung cancer immunotherapy response is influenced by patients’ sex. Int. J. Cancer 145, 2840–2849 (2019).
Xian, S., Dosset, M., Castro, A., Carter, H. & Zanetti, M. Transcriptional analysis links B cells and TERT expression to favorable prognosis in head and neck cancer. PNAS Nexus 2, pgad046 (2023).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592 (2023).
Morgos, D.-T. et al. Targeting PI3K/AKT/mTOR and MAPK signaling pathways in gastric cancer. Int. J. Mol. Sci. 25, https://doi.org/10.3390/ijms25031848 (2024).
Jiang, T. et al. A novel protein encoded by circMAPK1 inhibits progression of gastric cancer by suppressing activation of MAPK signaling. Mol. Cancer 20, 66 (2021).
Noubade, R. et al. Activation of p38 MAPK in CD4 T cells controls IL-17 production and autoimmune encephalomyelitis. Blood 118, 3290–3300 (2011).
Sun, K. et al. scRNA-seq of gastric tumor shows complex intercellular interaction with an alternative T cell exhaustion trajectory. Nat. Commun. 13, 4943 (2022).
Kang, B. et al. Parallel single-cell and bulk transcriptome analyses reveal key features of the gastric tumor microenvironment. Genome Biol. 23, 265 (2022).
Li, Y. et al. Single-cell landscape reveals active cell subtypes and their interaction in the tumor microenvironment of gastric cancer. Theranostics 12, 3818–3833 (2022).
Gu, Z. Complex heatmap visualization. iMeta 1, e43 (2022).
Mölder, F. et al. Sustainable data analysis with Snakemake [version 2; peer review: 2 approved]. F1000Research 10, https://doi.org/10.12688/f1000research.29032.2 (2021).
Sturm, G. et al. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics 35, i436–i445 (2019).
Zeng, D. et al. IOBR: multi-omics immuno-oncology biological research to decode tumor microenvironment and signatures. Front. Immunol. 12, 687975 (2021).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Wang, S. & Li, S. UCSCXenaShiny Extra Data Repository [Data set]. Zenodo https://doi.org/10.5281/zenodo.4625639 (2024).
Patil, I. Visualizations with statistical details: The ‘ggstatsplot’ approach. J. Open Source Softw. 6, 3167 (2021).
Therneau, T. M. A Package for Survival Analysis in R (Version 3.4) [Computer software]. CRAN. https://doi.org/10.32614/CRAN.package.survival (2024).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Xie, Y. knitr: A general-purpose package for dynamic report generation in R (Version 1.45) [Computer software]. CRAN. https://doi.org/10.32614/CRAN.package.knitr (2023).
Xie, Y., Cheng, J. & Tan, X. DT: A Wrapper of the JavaScript Library ‘DataTables’ (Version 0.26) [Computer software]. CRAN. https://doi.org/10.32614/CRAN.package.DT (2022).
Sievert, C. Interactive Web-Based Data Visualization with R, Plotly, and Shiny (Chapman and Hall/CRC, 2020).
Feizi, N. et al. PharmacoDB 2.0: improving scalability and transparency of in vitro pharmacogenomics analysis. Nucleic Acids Res. 50, D1348–D1357 (2022).
Hastie, T., Tibshirani, R., Narasimhan, B. & Chu, G. impute: impute: Imputation for microarray data (Version 1.72.3) [Computer software]. Bioconductor. https://doi.org/10.18129/B9.bioc.impute (2023).
Wang, S. & Li, S. UCSCXenaShiny: an R package for interactively exploring UCSC Xena (Version 2.1.0) [Computer software]. Zenodo. https://doi.org/10.5281/zenodo.13372912 (2024).
Acknowledgements
We gratefully acknowledge the support provided by the Openbiox Bioinformatics Innovation Collaboration Group (https://github.com/openbiox/) and the Open Source Promotion Plan 2023 (https://summer-ospp.ac.cn/, Project ID: 2351d0245). We also extend our thanks to the Hiplot platform (https://hiplot.org/) for freely deploying a backup Shiny application instance of UCSCXenaShiny (https://shiny.hiplot.org/ucsc-xena-shiny/) for general use. Special thanks to Dr. Enrique Medina Acosta (https://github.com/quiquemedina) for his valuable testing and suggestions during the development of this project. We are grateful for resources from the High-Performance Computing Center of Central South University. We are grateful for resources from the Bioinformatics Platform, Furong Laboratory and Bioinformatics Center, Xiangya Hospital, Central South University. This work was funded by the National Natural Science Foundation of China (Grant Nos. 82060475, 81472594, 81770781, 82270825, 82303953), Chunhui program of the Chinese Ministry of Education (Grant No. HZKY20220231), the Natural Science Foundation of Guizhou Province (Grant No. ZK2022-YB632), Guangdong Basic and Applied Basic Research Foundation (Grant No. 2021A1515011743), Youth Talent Project of Guizhou Provincial Department of Education (Grant No. QJJ2022-224), China Postdoctoral Science Foundation (Grant No. 2021M703733), China Lung Cancer Immunotherapy Research Project, Special funds for innovation in Hunan Province (Grant No. 2020SK2062), Excellent Young Talent Cultivation Project of Zunyi City (Zunshi Kehe HZ (2023) 142), Future Science and Technology Elite Talent Cultivation Project of Zunyi Medical University (ZYSE-2023-02) and Collaborative Innovation Center of Chinese Ministry of Education (Grant No. 2020-39).
Author information
Authors and Affiliations
Contributions
S.L. and S.W. conceptualized the study design. Y.X., J.L., P.L., H.W., F.Z. and Q.Z. contributed to the development of the research idea. S.W., J.Z., and S.C. provided supervision throughout the study. S.L., S.W., and Y.P. were responsible for the software development process. S.L. and S.W. led the main updates of the software. Y.P. oversaw the development of the pharmacogenomics modules. S.L., M.C., and Y.Z. were responsible for the creation of tutorial materials. S.L. and Y.P. drafted the initial manuscript. S.W., J.Z., S.C., Y.C., M.C., Y.Z., Y.X., P.L., H.W., F.Z. and Q.Z. contributed to the revision of the manuscript. All authors reviewed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Chunjiang Yu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Aylin Bircan and Mengtan Xing. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, S., Peng, Y., Chen, M. et al. Facilitating integrative and personalized oncology omics analysis with UCSCXenaShiny. Commun Biol 7, 1200 (2024). https://doi.org/10.1038/s42003-024-06891-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06891-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
