
Abstract
Cyanobacteria are important primary producers, contributing to 25% of the global carbon fixation through photosynthesis. They serve as model organisms to study the photosynthesis, and are important cell factories for synthetic biology. To enable efficient genetic dissection and metabolic engineering in cyanobacteria, effective and accurate genetic manipulation tools are required. However, genetic manipulation in cyanobacteria by the conventional homologous recombination-based method and the recently developed CRISPR-Cas gene editing system require complicated cloning steps, especially during multi-site editing and single base mutation. This restricts the extensive research on cyanobacteria and reduces its application potential. In this study, a highly efficient and convenient cytosine base editing system was developed which allows rapid and precise C → T point mutation and gene inactivation in the genomes of Synechocystis and Anabaena. This base editing system also enables efficient multiplex editing and can be easily cured after editing by sucrose counter-selection. This work will expand the knowledge base regarding the engineering of cyanobacteria. The findings of this study will encourage the biotechnological applications of cyanobacteria.
Similar content being viewed by others

Introduction
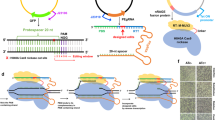
Cyanobacteria are the only oxygenic photosynthetic prokaryotes that can convert CO2 into organic compounds using light as the sole energy source1. Some cyanobacterial strains serve as important model organisms to study the physiological and ecological phenomena, such as photosynthesis, biological nitrogen fixation, and prokaryotic cell differentiation2,3,4. In addition, cyanobacteria have attracted particular attention as promising cell factories for sustainable generation of a large variety of valuable bioproducts, such as biofuels5,6, commercial terpenoids7,8, bioplastics9, bioactive compounds10,11, sugars12,13, and pigments14. To enable efficient genetic dissection and rational metabolic engineering in cyanobacterial strains, reliable genetic manipulation tools are required to allow precise, marker-less, rapid, and multiplex editing of genome. However, the conventional allelic-exchange-based genome editing method available for cyanobacteria is time-consuming, and leaves antibiotic markers at the editing sites15, which can be eliminated by subsequent selection processes16. (Fig. 1a). Thus, lack of reliable genetic manipulation tools has become a major hurdle in understanding the basic processes and metabolic networks of cyanobacteria. This restricts the potential of cyanobacteria in environmental and biotechnological research and applications.

a Traditional homologous recombination method; b CRISPR-Cas9 method; c CRISPR-Cpf1 method; d Cytosine base editor.
The recent developments in the CRISPR-Cas system provide a simple and reliable platform for precise and efficient modification of DNA sequences in a wide range of organisms, including mammals17, plants18,19, and bacteria20,21,22. CRISPR-Cas9 is the most commonly used CRISPR system for genome editing23,24. In this system, the Cas9 nuclease forms a complex with crRNA and tracrRNA, or with an artificial single guide RNA (sgRNA). Then, the complex is guided to the specific complementary target DNA site which must be next to a protospacer adjacent motif (PAM, e.g., 5’-NGG-3’ for Streptococcus pyogenes Cas9). Thus, a blunt double-strand break (DSB) is formed in the genome23,25 (Fig. 1b). The common method to repair the lethal DSBs in prokaryotes is homology-directed repair (HDR). In this method, a repair template is provided exogenously, and sequence-specific deletions and insertions are achieved during the repair process. Cpf1 (also named Cas12a) is another CRISPR-Cas system, which recognizes a 5’ T-rich PAM (e.g., 5’-TTN-3’ for Francisella novicida Cpf1), and generates staggered double-strand DNA break without the assistance of tracrRNA26 (Fig. 1c).
Both Cas9 and Cpf1 have been employed for the genome editing in cyanobacteria27. Cas9-mediated genome editing system was first applied in cyanobacteria, such as Synechococcus sp. PCC 794228, Synechococcus sp. PCC 297329, and Synechocystis sp. PCC 680330. Since high dosage of Cas9 is cytotoxic to cyanobacteria and causes low transformation efficiencies31, inducible promoter was used to strictly control the Cas9 expression, which enabled reliable transformation of the CRISPR-Cas9 vector in cyanobacteria30,32. In contrast, Cpf1 is much less toxic to cyanobacteria, and has been successfully used for different cyanobacterial species, including Synechococcus, Synechocystis, and Anabaena33,34,35. Although Cas9 and Cpf1 enable efficient and scarless genome editing in cyanobacteria, they still rely on the HDR mechanism which can reduce the transformation efficiency and requires multiple cloning steps to assemble the homologous recombination template, especially during multi-site editing.
Recently, the development of “base editors” allows direct base mutagenesis at specific genomic sites, providing a distinctive strategy for genome editing36. Until now, several kinds of base editors have been developed37,38,39,40,41. Among them, the cytosine base editor is the most popular one, which has been applied in a number of organisms37,42,43,44. A cytosine base editor is engineered by fusing a cytidine deaminase with a dead Cas9 (Cas9D10AH840A) or a Cas9 nickase (Cas9D10A). Guided by the Cas9/sgRNA complex, the cytidine deaminase is directed to the specific target site, and a C → T conversion (or G → A in the complementary strand) is achieved (Fig. 1d). By mediating the conversion of CAA, CAG, CGA, or TGG to TAA, TAG, or TGA, cytosine base editor can generate a premature stop codon at the target site, thus inactivating the target gene. This genome editing tool does not create DSBs, and thus it enables efficient genetic manipulation without sacrificing the transformation efficiency. In addition, it does not require the donor repair template for the homologous recombination repair. Therefore, simply by customizing approximately 20 nucleotides (nt) of the sgRNA, the cytosine base editor can edit specific cytosine bases of interest in the genome, which makes it an easily programmable tool for precise genome editing and gene inactivation. Compared with conventional homologous recombination method and Cas9/Cpf1-based genome editing tools, the base editor system has some advantages, such as a simple cloning process, rapid editing speed, and multiplex editing capability (Table 1).
In this study, a highly efficient and convenient cytosine base editing system (pCyCBE) was developed in cyanobacteria. This tool allowed rapid and precise point mutation and gene inactivation in the genome of cyanobacteria. It also enabled efficient multiplexed editing and could be easily cured by sucrose counter-selection. The development of the pCyCBE system will pave the way for future physiological studies and metabolic engineering in cyanobacteria. The findings of this study will provide critical insights into base-editing system development in other microorganisms.
Results
Construction of the pCyCBE plasmid for base editing in cyanobacteria
To harness the cytosine base editor for genome editing in cyanobacteria, a base editor plasmid was constructed by using pBECKP-Km45 and pCpf1b-Sp34 as the PCR template (Fig. 2). The expression cassette of the base editor was amplified from the pBECKP-Km plasmid, a cytidine deaminase-mediated base-editing plasmid in Klebsiella Pneumoniae. The pCpf1b-Sp plasmid is a Cpf1-based plasmid for gene editing in cyanobacteria, and its plasmid backbone was amplified for the replication in cyanobacteria. The two DNA fragments were assembled together via Gibson assembly, resulting in the plasmid pCyCBE. The constructed pCyCBE plasmid contains several components: the broad host range replicon RSF1010 that enables the plasmid to replicate in cyanobacteria; the kanamycin-resistant marker for selection; the cytidine deaminase APOBEC1 linked to the N-terminus of the nickase Cas9D10A via an XTEN linker; the sgRNA, driven by the promoter J23119, a strong constitutive promoter in prokaryotes; and the counter-selection gene sacB for the plasmid curing after editing. In addition, two BsaI sites were designed in the plasmid for convenient and seamless cloning of spacers by Golden Gate assembly (Fig. 2).

The pCyCBE plasmid is constructed by substituting ColE1 origin in pBECKP-Km with the RSF1010 origin of pCpf1b-Sp.
Efficient C → T conversion in Synechocystis sp. PCC 6803 mediated by the pCyCBE plasmid
Base editing capacity of the pCyCBE system was analyzed in the model cyanobacterium Synechocystis sp. PCC 6803. We selected two non-essential genes, hlyD (sll1181) and desB (sll1441), to attempt base editing. Previous studies have shown that HlyD is an important component of the HlyBD-TolC efflux system and is involved in the adaptation of low-iron (Fe) conditions in Synechocystis sp. PCC 680346, while DesB is a fatty acid desaturase and is essentially required for low-temperature adaptation47. Two spacers were designed to target the hlyD and desB genes, and then were assembled into the pCyCBE plasmid, respectively. After transforming the editing plasmids into Synechocystis sp. PCC 6803 by conjugation, individual clones grown on the BG11 agar plate were picked and the editing efficiency was calculated by analysis of the Sanger sequencing chromatogram using the EditR software48. As shown in Fig. 3a, the C at position 8 of the hlyD spacer and the C at position 6 of the desB spacer were successfully mutated to T with editing efficiencies of 93.7% and 92.4%, respectively. This indicated that the pCyCBE system could efficiently mediate the base editing in Synechocystis sp. PCC 6803. To accurately measure the editing efficiency of the pCyCBE base editor, we further amplified the target sites of the mutant strains, and sent the PCR products for deep sequencing. The results showed that the editing efficiencies of the target sites were approximately 99% (Fig. 3b), indicating that almost all of the chromosome copies were mutated.

a HlyD Q58 and DesB Q77 in the genome of Synechocystis sp. PCC 6803 are successfully mutated to stop codon; PAM sites are highlighted in blue color, while mutation sites are shown in red color. Each base editing experiment is repeated three times, and the representative sequencing chromatogram for each mutation is shown. b Deep sequencing results of the hlyD and desB mutant strains. The top 6 most frequent PCR products are displayed. c Growth curves of the wild-type (WT) strain, wild-type strain carrying the pCyCBE plasmid (WT + P), and the hlyD mutant (Mut hlyD) strain grown under standard Fe and Fe-deficient conditions. Photographs of the cultures on the 6th day are presented at the bottom of the growth curve. The growth curve experiments are performed in triplicate, and the data are represented as mean ± SD. d Phototactic movements of the WT strain and Mut hlyD strain grown for 7 days under lateral illumination.
The C → T conversion in hlyD gene generates a premature stop codon (Q58 to stop codon), resulting in the inactivation of the hlyD gene. Since HlyD is involved in the formation of type IV pili and indirectly influences Fe acquisition, the hlyD mutant strain is more sensitive to Fe deficiency and loses the ability of phototactic movement46. To verify the phenotype of the hlyD mutant constructed by the base editor, the growth curve assay was conducted in Fe depletion conditions. As shown in Fig. 3c, the hlyD mutant showed similar growth rate as the unedited strains under standard Fe conditions. However, the hlyD mutant strain grew much slower than the unedited strains under Fe-deficient conditions. In addition, the hlyD mutant lost the ability of phototactic movement (Fig. 3d). These results are consistent with the finding reported by the previous studies based on the traditional homologous recombination method of gene disruption46, suggesting that the hlyD gene was indeed inactivated by the pCyCBE base editing system.
To elucidate the kinetics of base editing, we measured the editing efficiencies of hlyD and desB genes after transformation at different time points (the 7th day, 12th day, and 20th day). As shown in Supplementary Fig. 1, the editing efficiency continued to increase over time. At the 20th day, the editing efficiencies of hlyD and desB genes reached over 90%, indicating that most of the chromosome copies were edited.
To systematically investigated the versatility of the pCyCBE base editor system in Synechocystis sp. PCC 6803, 18 different spacers were designed, targeting different genomic sites (Supplementary Table 1). All of the cyanobacterial colonies grown on the BG11 plate were collected for PCR and the integral editing efficiencies were examined using Sanger sequencing. The results showed that 17 spacers were successful edited (Fig. 4a). Furthermore, we calculated the editing efficiency of all the Cs at the 20 bp spacer by analysis of the Sanger sequencing chromatogram using the EditR software48. As shown in Fig. 4b, editing events occurred across the 20 positions on the spacer. However, these events were particularly concentrated at the 4-9 positions, which indicated that these positions in the spacer were most likely to be edited (referred as “editable window”). All the Cs in the “editable window” were further selected, and the influence of adjacent bases on the editing efficiency was analyzed. The editing efficiencies were found to be in the following order: TC > CC ≈ AC > GC (Fig. 4c), which was consistent with the previous studies37,44. These results suggested that TC is a better mutation site at the “editable window” while designing the suitable spacers for genome editing.

a Sequencing chromatograms of the selected 18 target loci in the pCyCBE system. The edited C(s) in the spacer are colored in red and indicated by red box. b Summary of the base-editing frequency at each cytidine site at the tested sites, with “editable window” being located between positions 4 and 9 in the spacer region, and labeled with red box. c Analysis of the adjacent base preference of the pCyCBE system in Synechocystis sp. PCC 6803.
Efficient multiplex editing in cyanobacteria by the base editor
Considering the high editing efficiencies of the pCyCBE system in Synechocystis sp. PCC 6803, capability of multiplex base editing was further examined using tandem gRNA cassettes. Firstly, two sgRNAs, targeting hlyD and desA (slr1350) genes, were assembled into the pCyCBE plasmid simultaneously (Fig. 5a). The results of Sanger sequencing showed that these two genes were simultaneously mutated with high editing efficiency (Fig. 5a). Another two-site editing plasmid targeting hlyD and tolC (slr1270) genes also showed successful editing at both target sites (Supplementary Fig. 2a). Furthermore, three sgRNAs were assembled into the pCyCBE plasmid, resulting in pCyCBE-3xsgRNA-1, targeting desA, desB, and desD (sll0262) genes, and pCyCBE-3xsgRNA-2, targeting desA, desB, and desC (sll0541) genes. The Sanger sequencing results confirmed successful mutations at the targeted sites (Fig. 5b, Supplementary Fig. 2b). These results indicate that the pCyCBE system is capable of efficient base editing at multiple sites simultaneously.

a The pCyCBE plasmid enables simultaneous base editing at two sites. A Map of pCyCBE-2xsgRNA plasmid is presented in the left, and the representative sequencing chromatograms of two mutation sites are presented in the right. b The pCyCBE plasmid enables simultaneous base editing at three sites. Map of pCyCBE-3xsgRNA plasmid is presented in the left, and the representative sequencing chromatograms of three mutation sites are presented in the right. The mutated Cs are shown in red color and labeled with red squares.
The editing plasmid could be easily cured by sucrose counter-selection
After editing, the plasmid should be cured to construct a marker-less mutant strain for phenotypic analysis without any genetic interference from unrelated genetic background, and for the subsequent editing of other genomic sites. The pCyCBE plasmid contains sacB gene, which acts as a lethal gene in the presence of sucrose. Thus, this plasmid can be easily cured by using agar plates containing sucrose. To cure the pCyCBE plasmid after editing, the hlyD mutant strain was cultured in fresh BG11 medium for 4 generations. Then the culture was diluted 104 and 105 folds with fresh BG11 medium. Subsequently, 50 μL of diluted culture was poured onto the BG11 agar plates in the presence of 5% w/v sucrose. At the same time, diluted culture was also plated on the BG11 agar medium without sucrose. The number of colonies grew on the sucrose-free plate was much more than that on the plate containing sucrose (Fig. 6a). This indicated the elimination of many plasmid-containing colonies by sucrose. Furthermore, 6 colonies were randomly picked from the plates with or without sucrose, and PCR was performed to verify the loss of plasmid using pCyCBE plasmid-specific primers. As shown in Fig. 6b, all colonies picked from the sucrose-free plate contained the editing plasmid, while the colonies from the sucrose plate lost the editing plasmid. Subsequently, the colonies picked from the sucrose plate were streaked on the BG11 agar plate with or without kanamycin, respectively. All the colonies grew well on the antibiotics-free plate, whereas the sucrose selected strains could not grow in presence of kanamycin (Fig. 6c). This confirmed that the editing plasmid was successfully cured in presence of sucrose. Overall, the pCyCBE plasmid could be easily cured by the counter-selection of sucrose after genome editing.

a Photographs of the edited strains grown on the BG11 agar plate in the absence and presence of sucrose. b PCR results of the six colonies randomly chosen from the BG11 plates with or without sucrose. Here, P is the pCyCBE plasmid; WT is the wild-type strain; WT + P is the wild-type strain containing pCyCBE plasmid. c All six colonies chosen from the sucrose plate are sensitive to the antibiotic (kanamycin).
Assessment of the gene editing stability and off-target effect of the pCyCBE system
To assess the gene editing stability of the pCyCBE base editor, we cultured three mutant strains (hlyD, desB, and desA) after plasmid curing in standard BG11 medium for 20 generations, respectively. The Sanger sequencing results showed that all three genes were completely mutated and no unedited event were observed after 20 generations of cultivation (Supplementary Fig. 3a–c), indicating that the mutant strains were stable. In addition, the growth curve assay was performed using the hlyD mutant strain cultured for 20 generations. The results showed that the 20th-generation hlyD mutant showed similar growth rate as the wild-type strain under standard Fe conditions, but grew much slower than the wild-type strain under Fe-deficient conditions (Supplementary Fig. 3d), which is in consistent with the phenotype of the 1st-generation hlyD mutant. Taken together, these results demonstrated that the genomic mutations generated by the pCyCBE base editor is stable, and would not be rapidly lost during the cultivation process.
To assess the off-target effect of the pCyCBE system, we searched the similar genomic sites of four spacers (hlyD, desB, desA, and desC) across the entire genome of Synechocystis sp. PCC 6803 using the CasOFFinder software49. We picked the top five similar sites for each gene (Supplementary Table 2), and amplified these genomic loci of the mutant strains for Sanger sequencing. The sequencing results showed that none of the similar genomic sites were mutated (Supplementary Fig. 4a–d), indicating that the pCyCBE system has good fidelity. Nowadays, the off-target effects of base editors in plant and mammalian cells have been reported50,51,52. Apart from the sgRNA-dependent off-target DNA editing, the cytosine base editor also generates undesired mutations by sgRNA-independent off-target effect, which is caused by random deamination without the participation of sgRNA50,52. Therefore, to comprehensively evaluate the off-target effects of base editor in cyanobacteria, whole genome sequencing might be an effective method.
Application of the pCyCBE system in other cyanobacterial strains
RFS1010 origin is a replicon with broad host range, and it can replicate in different cyanobacterial species. This indicates that the pCyCBE plasmid can also mediate base editing in other cyanobacterial species, apart from Synechocystis sp. PCC 6803. To verify this hypothesis, the pCyCBE system was applied in another model cyanobacterium Anabaena sp. PCC 7120. It is a filamentous strain, which is able to form heterocysts to fix atmospheric nitrogen under the conditions of limited nitrogen in the growth media. Six target sites in the non-essential genes were designed, and the constructed plasmids were transferred into Anabaena sp. PCC 7120 by conjugation. The results of Sanger sequencing showed successful mutations at all selected sites with high editing efficiencies (Fig. 7a, Supplementary Fig. 5). HetF is a membrane protein which is essential to heterocyst formation in Anabaena sp. PCC 7120 and the hetF deletion strain displayed an aberrant cell morphology characterized by enlarged and elongated cells53. The C → T conversion in the hetF (alr3546) gene mutated the Q58 to stop codon, generating hetF mutant strain. Consistent with the previous study53, the hetF mutant strain displayed an aberrant cell morphology, with enlarged and elongated cells in the BG11 medium (Fig. 7b). To seriously analyze the differences of the wild-type and hetF mutant strains in the cell dimensions, we randomly selected 100 cells from each strain, and measured their cell lengths under microscope. The results showed that the lengths of the hetF mutant strain were significantly longer than that of unedited strains (P < 0.001) (Fig. 7c). These results confirmed that the hetF gene in Anabaena sp. PCC 7120 was successfully disrupted by the pCyCBE base editor.

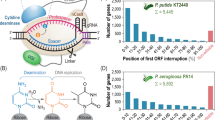
a HetF Q575 in the genome of Anabaena sp. PCC 7120 is successfully mutated to stop codon. b Photographs of Anabaena sp. PCC 7120 wild-type (WT) strain, wild-type strain carrying the pCyCBE plasmid (WT + P), and the hetF mutant strain (Mut hetF). Bars = 10 μm. c The cell lengths of different Anabaena sp. PCC 7120 strains. 100 cells from each strain are measured under microscope. ***p < 0.001 is determined using Student’s t test. d Relative position of the earliest induction of stop codons could be targeted in the ORFs of cyanobacteria (cumulative percentage) by the pCyCBE system. e The portion of the genes targeted by pCyCBE to introduce at least one premature stop codon within the top 25%, 50%, and 75% of the ORFs in Synechocystis sp. PCC 6803 and Anabaena sp. PCC 7120.
To estimate the number of genes that could be inactivated by the pCyCBE system in cyanobacteria, targetable codons in the genome of Synechocystis sp. PCC 6803 and Anabaena sp. PCC 7120 were determined using the CRISPR-CBEI software54. As shown in Fig. 7d, the premature stop codon could be introduced into over 90% genes of both two strains by the pCyCBE base editor. Considering that the relative position of the premature stop codon in an open reading frame (ORF) can significantly affect the effectiveness of gene inactivation, the locations of the introduced premature stop codon in the gene were further analyzed. The results showed that at least one premature stop codon could be introduced in around 93.75%, 87.87%, and 71.58% genes of Synechocystis sp. PCC 6803, within the top 75%, 50%, and 25% of the ORF body, respectively. Whereas in Anabaena sp. PCC 7120, a premature stop codon could be introduced in 87.76%, 80.07%, and 61.12% genes, within the top 75%, 50%, and 25% of the ORF body, respectively (Fig. 7e). These results revealed that most of genes in cyanobacteria could be inactivated by the pCyCBE system.
Discussion
Cyanobacteria are important microorganisms used in basic research and industrial biotechnology. As the first cyanobacterium strain with a sequenced genome55, Synechocystis sp. PCC 6803 has been widely used as a host or model organism in synthetic biology studies. On the other hand, filamentous cyanobacterium Anabaena sp. PCC 7120 can form heterocysts for nitrogen fixation, and serves as the prominent model cyanobacterium to study nitrogen fixation and cell division. In this study, the pCyCBE plasmid was engineered, which enabled efficient C → T base editing in Synechocystis sp. PCC 6803 and Anabaena sp. PCC 7120. The base editing plasmid contains replicon RSF1010, which has a broad host range and is known to replicate well in diverse prokaryotes, such as Synechococcus33, Pseudomonas56, Salmonella57, Streptomyces58, Rhizobium59, and Mycobacterium58. Therefore, it is assumed that the versatility of this editing system can be extended to other cyanobacterial species and even other prokaryotes. In the traditional allelic-exchange-based genome editing method, target genes are knocked out by inserting an antibiotic selection cassette, which may have polar effects on downstream genes in the operon. Using base editing, only a single nucleotide could be mutated, and marker-less gene inactivation was achieved without causing any polar effect. In addition, single base mutation by traditional method requires complex cloning steps and the selection of antibiotics marker60, while the base editor could achieve scarless and precise base editing only by assembling a spacer at the target site. This greatly simplifies the construction of the editing plasmid. Besides the promising efficiency and precision of the base editing system, clean edits were observed in most colonies at the first round of selection. This suggested that the fully mutated strains could be quickly obtained without extra segregation steps. Moreover, the base editor harbored a defective Cas9 protein as the CRISPR effector to recognize the target site and did not generate DSBs in the cyanobacterial genome. Therefore, it exhibited low toxicity compared to CRISPR-Cas9 systems.
Recently, Wang et al. developed a base-editing system which employs dCas9 and the activation-induced cytidine deaminase (AID)61. This gene-editing tool could achieve efficient “C” to “T” base conversion at the positions 2 to 5 of the target site in the model cyanobacterium Synechococcus elongatus PCC 7942. In addition, a dCas12a-mediated base editing was also developed in S. elongatus PCC 7942 with a broader editable window (from positions 4 to 16 in the target spacer)62. In this study, we engineered the pCyCBE base editor plasmid by fusion of the cytidine deaminase APOBEC1 with the nickase Cas9D10A. This base editor enables efficient C → T conversion in Synechocystis sp. PCC 6803 and Anabaena sp. PCC 7120, and could achieve three-site editing simultaneously. These studies altogether demonstrate that base-editing is another powerful genetic tool for cyanobacteria.
Given that many cyanobacteria are polyploids and have a long generation time, serial genome editing of multiple genes in cyanobacteria is time-consuming. Multiplexed, simultaneous editing could drastically accelerate the genome-editing process. The pCyCBE system enables efficient base editing in multiplex sites simultaneously by simply assembling the tandem sgRNA cassettes. Thus, the pCyCBE system shows great potential to accelerate the metabolic engineering and synthetic biology research in cyanobacteria. Combined with massive parallel oligomer synthesis and high-throughput sequencing, the development of the pCyCBE system allows accurate genome-wide or defined gene library screening, which has been previously applied in human cells, Corynebacterium glutamicum, and Bacillus subtilis63,64.
In summary, an efficient and convenient base editing system of pCyCBE plasmid is developed in cyanobacteria, which enables rapid C → T mutations at the specific genomic sites and introduces premature stop codon for gene inactivation. The pCyCBE system also enables simultaneous mutations at multiple genomic sites and can be cured easily by sucrose counter-selection. The pCyCBE system will not only expand the genome editing toolbox of cyanobacteria, but may also be helpful in the metabolic engineering and fundamental researches in cyanobacteria. The developed plasmid and the editing strategies presented in this study could be readily applied in other microorganisms and to other CRISPR systems.
Methods
Strains and culture conditions
All the strains used in this study are listed in Supplementary Table 3. Escherichia coli DH5α was used for molecular cloning, while E. coli DH10B carrying pRL443 and pRL623 was used for conjugation. All E. coli strains were cultivated in LB medium and on LB agar plates at 37 °C. The sub-strain of Synechocystis sp. PCC 6803 used in this study is a mobile and glucose-tolerant strain which is initially obtained from Professor Jindong Zhao’s lab at Peking University. Synechocystis sp. PCC 6803 and Anabaena sp. PCC 7120 strains were grown in BG11 liquid at 30°C under the light density of 30 μmol photons m-2 s-1. The final Fe concentrations were 21.4 μM and 4 nM for the standard Fe and Fe-deficient conditions, respectively. BG11 plates were prepared by adding 0.3% Na2S2O3, 1.4% agar, and 8 mM TES (pH 8.2) to the BG11 liquid medium. Antibiotics were used as follows: 50 μg/mL kanamycin, 50 μg/mL chloramphenicol, and 100 μg/mL ampicillin for the E. coli strains; 30 μg/mL kanamycin for the Synechocystis sp. PCC 6803 strains; and 50 μg/mL neomycin for the Anabaena sp. PCC 7120 strains.
Construction of editing plasmids
All the primers used in this study are listed in Supplementary Data 1. To construct the pCyCBE plasmid, components of cytosine base editor were amplified using the pBECKP-Km plasmid, and the RSF1010 origin was amplified using the pCpf1b-Sp plasmid (Supplementary Fig. 6a). Then the two amplified DNA fragments were assembled via Gibson assembly (Supplementary Fig. 6b). The successful construction of the pCyCBE plasmid was verified by PCR and Sanger sequencing.
To assemble the target spacer, a 20 bp-spacer sequence before NGG in the target gene of cyanobacteria was selected. The spacers were prepared by annealing a pair of complementary oligonucleotides, and ligating them into the BsaI sites of the pCyCBE plasmid by Golden Gate assembly. Successful construction of plasmids was confirmed by PCR and Sanger sequencing (Supplementary Fig. 6c).
For the construction of two-site base editing plasmid, the DNA fragment of sgRNA1 was amplified from the constructed pCyCBE-sgRNA1 plasmid with the primers CBE2spF/CBE2spR (Supplementary Data 1). The pCyCBE-sgRNA2 plasmid was digested with the enzymes XbaI/NotI, and the digested product was purified using SanPrep Column PCR Product Purification Kit (Sangon, Shanghai, China). Then the DNA fragment of sgRNA1 and the digested pCyCBE-sgRNA2 plasmid were ligated together via Golden Gate assembly. The constructed pCyCBE-2xsgRNA plasmid was verified by PCR and Sanger sequencing.
The construction procedure of three-site base editing plasmid was similar with that of two-site base editing plasmid. Briefly, the DNA fragments of sgRNA1 and sgRNA2 were amplified from the constructed pCyCBE-sgRNA1 and pCyCBE-sgRNA2 plasmids with the primers CBE3spF1/CBE3spR1 and CBE3spF2/CBE3spR2, respectively (Supplementary Data 1). Then the two DNA fragments were assembled into the XbaI/NotI sites of the pCyCBE-sgRNA3 plasmid via Golden Gate assembly.
Base editing in cyanobacterial strains
The constructed plasmids were transformed into Synechocystis sp. PCC 6803 by conjugation, as previously described33. The editing plasmid was first transformed into the DH10B strain carrying pRL443 and pRL623. Then the DH10B strain was cultured in LB broth containing 50 μg/mL kanamycin, 50 μg/mL chloramphenicol, and 100 μg/mL ampicillin, and was grown to OD600 ≈ 0.6. 5 mL culture was harvested by centrifugation at 5000 rpm for 5 min, and was resuspended with 200 μL fresh LB medium after washing with fresh LB medium three times. 50 mL culture of wild-type Synechocystis sp. PCC 6803 (OD730 = 1.0~1.5) was harvested by centrifugation, and was resuspended with 2 mL BG11 medium after washing with BG11 medium three times. Then 100 μL of bacterial cells were mixed with 200 μL of Synechocystis sp. PCC 6803 strain, and the mixture was diluted 104 folds with fresh LB medium. 200 μL diluted mixture was poured onto the HAF Milipore filters (82 mm) overlayed on the BG11 agar plate. After 24 h incubation at 30°C under the light density of 20 μmol photons m–2 s–1, the conjugation filters were transferred onto BG11 agar plate supplemented with 30 μg/mL kanamycin. Approximately 1000 colonies could be observed on the plate about one week later. The conjugation process of Anabaena sp. PCC 7120 strains was the same as that of Synechocystis sp. PCC 6803, except that the Anabaena cells were selected on the BG11 agar plate containing 50 μg/mL neomycin. Approximately 100 colonies of Anabaena sp. PCC 7120 could be observed on the plate one week later.
To assess the editing efficiency of the pCyCBE system, the colonies grown on the BG11 plate were collected as PCR template. PCR products covering the editing sites were sent to Shanghai Sangon for Sanger sequencing, and the editing efficiency was determined using EditR software48. The EditR software analyzes the fluorescence area of all four bases in the Sanger sequencing chromatogram to delineate the composition and frequency of base mutations.
Deep sequencing
The genomic DNA of mutant strains was extracted using a FastPure Bacteria DNA Isolation Mini Kit (Vazyme). About 400 bp regions surrounding the target loci were amplified using primers with barcodes. The PCR products were merged and gel-purified. The amplicon-seq library was prepared by using a TruSeq DNA Sample Prep Kit (Illumina). The library was subjected to Illumina MiSeq sequencing by Shanghai Majorbio Bio-pharm Technology Co. Ltd. The sequencing data were demultiplexed according to the barcodes, and the mutation efficiencies were analyzed using Cas-Analyzer65. The primers used in this study are listed in Supplementary Data 1.
Growth curve assay
For the Fe-deplete assay, glassware, tips, and bottles were all soaked in 6 M HCl for at least 12 h and subsequently rinsed with Milli-Q water six times before use. The Synechocystis sp. PCC 6803 wild-type (WT) strain and Mut hlyD strain were grown to logarithmic growth phase in BG11 media, and harvested by centrifugation, followed by washing with Fe-deplete BG11 medium (twice) to remove extracellular Fe. The WT and Mut hlyD strains were diluted using BG11 medium and Fe-deplete BG11 medium, respectively, with the final OD730 of 0.02. The growth curves of the cyanobacterial strains were monitored by measuring turbidity (OD730) of three independent biological replicates every 2 days until the 12th day. The photographs of the strains were taken on the 6th day.
Phototaxis assay
The phototaxis assay was performed by following the method described previously46. Briefly, Synechocystis sp. PCC 6803 WT and Mut hlyD strains were both grown on solid BG11 plates containing 1.5% (w/v) agar, and then streaked onto solid BG11 plates containing 0.8% (w/v) agar with a sterile toothpick. The plates were then wrapped in black cardboard and incubated at 30°C with only one side exposed to unilateral light (5 μmol photons m-2 s-1). After 7 days of incubation, the movement of Synechocystis strains on the plates was observed and photographs were taken.
Plasmid curing
To cure the pCyCBE plasmid after successful editing, the strain to be cured was grown in antibiotic-free BG11 liquid medium till 4th generation. The cell culture (OD730 = 1.0 ~ 1.5) was then serially diluted and spread onto the sucrose-free BG11 plate and the BG11 plate containing 5% w/v sucrose, simultaneously. To check the loss of the editing plasmid, colonies formed on the sucrose plate were randomly picked, and streaked on the antibiotics-containing plates. Loss of the editing plasmid was further confirmed by PCR.
Statistics and reproducibility
The statistical analyses conducted on the data in each figure were described in their respective figure captions. All analyses were conducted with the software package GraphPad Prism 8.2.1 (GraphPad, San Diego, CA). Experiments were performed with three independent repeats.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are available in the main text or the supplementary materials. The raw data of deep sequencing were deposited in the NCBI Sequence Read Archive (SRA) with the accession number SAMN41576882. The source data underlying Figs. 3, 4, 7, and Supplementary Fig. 3 can be found in Supplementary Data 2.
References
Knoot, C. J., Ungerer, J., Wangikar, P. P. & Pakrasi, H. B. Cyanobacteria: Promising biocatalysts for sustainable chemical production. J. Biol. Chem. 293, 5044–5052 (2018).
Cassier-Chauvat, C. & Chauvat, F. Cyanobacteria: Wonderful Microorganisms for Basic and Applied Research. (Cyanobacteria: Wonderful Microorganisms for Basic and Applied Research, 2018).
Suga, M. et al. Native structure of photosystem II at 1.95 Å resolution viewed by femtosecond X-ray pulses. Nature 517, 99–103 (2015).
Herrero, A., Stavans, J. & Flores, E. The multicellular nature of filamentous heterocyst-forming cyanobacteria. FEMS Microbiol. Rev. 40, 831–854 (2016).
Wang, W., Liu, X. & Lu, X. Engineering cyanobacteria to improve photosynthetic production of alka(e)nes. Biotechnol. Biofuels 6, 69 (2013).
Yunus, I. S. et al. Improved bioproduction of 1-Octanol Using Engineered Synechocystis sp. PCC 6803. ACS Synth. Biol. 10, 1417–1428 (2021).
Lin, P. C., Zhang, F. & Pakrasi, H. B. Enhanced limonene production in a fast-growing cyanobacterium through combinatorial metabolic engineering. Metab. Eng. Commun. 12, e00164 (2021).
Rodrigues, J. S. & Lindberg, P. Metabolic engineering of Synechocystis sp. PCC 6803 for improved bisabolene production. Metab. Eng. Commun. 12, e00159 (2021).
Ciebiada, M., Kubiak, K. & Daroch, M. Modifying the Cyanobacterial Metabolism as a Key to Efficient Biopolymer Production in Photosynthetic Microorganisms. Int. J. Mol. Sci. 21, 7204 (2020).
Singh, R. K., Tiwari, S. P., Rai, A. K. & Mohapatra, T. M. Cyanobacteria: an emerging source for drug discovery. J. Antibiot. 64, 401–412 (2011).
Kachel, B. & Mack, M. Engineering of Synechococcus sp. strain PCC 7002 for the photoautotrophic production of light-sensitive riboflavin (vitamin B2). Metab. Eng. 62, 275–286 (2020).
Song, K., Tan, X., Liang, Y. & Lu, X. The potential of Synechococcus elongatus UTEX 2973 for sugar feedstock production. Appl. Microbiol. Biotechnol. 100, 7865–7875 (2016).
Qiao, Y., Wang, W. & Lu, X. Engineering cyanobacteria as cell factories for direct trehalose production from CO(2). Metab. Eng. 62, 161–171 (2020).
Diao, J. et al. Tailoring cyanobacteria as a new platform for highly efficient synthesis of astaxanthin. Metab. Eng. 61, 275–287 (2020).
Koksharova, O. A. & Wolk, C. P. Genetic tools for cyanobacteria. Appl. Microbiol. Biotechnol. 58, 123–137 (2002).
Vasudevan, R. et al. CyanoGate: A modular cloning suite for engineering Cyanobacteria based on the plant MoClo Syntax. Plant Physiol. 180, 39–55 (2019).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Zhou, H., Liu, B., Weeks, D. P., Spalding, M. H. & Yang, B. Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Res. 42, 10903–10914 (2014).
Zhang, Z. et al. A multiplex CRISPR/Cas9 platform for fast and efficient editing of multiple genes in Arabidopsis. Plant Cell Rep. 35, 1519–1533 (2016).
Jiang, W., Bikard, D., Cox, D., Zhang, F. & Marraffini, L. A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31, 233–239 (2013).
Chen, W., Zhang, Y., Yeo, W. S., Bae, T. & Ji, Q. Rapid and efficient genome editing in Staphylococcus aureus by using an engineered CRISPR/Cas9 system. J. Am. Chem. Soc. 139, 3790–3795 (2017).
Jiang, Y. et al. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 81, 2506–2514 (2015).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Wang, H., La, R. M. & Qi, L. S. CRISPR/Cas9 in genome editing and beyond. Annu. Rev. Biochem. 85, 227–264 (2016).
Anders, C., Niewoehner, O., Duerst, A. & Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513, 569–573 (2014).
Zetsche, B. et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771 (2015).
Pattharaprachayakul, N., Lee, M., Incharoensakdi, A. & Woo, H. M. Current understanding of the cyanobacterial CRISPR-Cas systems and development of the synthetic CRISPR-Cas systems for cyanobacteria. Enzym. Microb. Technol. 140, 109619 (2020).
Li, H. et al. CRISPR-Cas9 for the genome engineering of cyanobacteria and succinate production. Metab. Eng. 38, 293–302 (2016).
Racharaks, R., Arnold, W. & Peccia, J. Development of CRISPR-Cas9 knock-in tools for free fatty acid production using the fast-growing cyanobacterial strain Synechococcus elongatus UTEX 2973. J. Microbiol. Methods 189, 106315 (2021).
Xiao, Y. et al. Developing a Cas9-based tool to engineer native plasmids in Synechocystis sp. PCC 6803. Biotechnol. Bioeng. 115, 2305–2314 (2018).
Wendt, K. E., Ungerer, J., Cobb, R. E., Zhao, H. & Pakrasi, H. B. CRISPR/Cas9 mediated targeted mutagenesis of the fast growing cyanobacterium Synechococcus elongatus UTEX 2973. Microb. Cell Fact. 15, 115 (2016).
Cengic, I., Cañadas, I. C., Minton, N. P. & Hudson, E. P. Inducible CRISPR/Cas9 allows for multiplexed and rapidly segregated single-target genome editing in Synechocystis Sp. PCC 6803. ACS Synth. Biol. 11, 3100–3113 (2022).
Ungerer, J. & Pakrasi, H. B. Cpf1 is a versatile tool for CRISPR genome editing across diverse species of cyanobacteria. Sci. Rep. 6, 39681 (2016).
Niu, T. C. et al. Expanding the potential of CRISPR-Cpf1-based genome editing technology in the Cyanobacterium Anabaena PCC 7120. ACS Synth. Biol. 8, 170–180 (2019).
Baldanta, S., Guevara, G. & Navarro-Llorens, J. M. SEVA-Cpf1, a CRISPR-Cas12a vector for genome editing in cyanobacteria. Microb. Cell Fact. 21, 103 (2022).
Anzalone, A. V., Koblan, L. W. & Liu, D. R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38, 824–844 (2020).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016).
Gaudelli, N. M. et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017).
Kurt, I. C. et al. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 39, 41–46 (2021).
Tong et al. Programmable A-to-Y base editing by fusing an adenine base editor with an N-methylpurine DNA glycosylase. Nat. Biotechnol. 41, 1080–1084 (2023).
Chen, L. et al. Adenine transversion editors enable precise, efficient A•T-to-C•G base editing in mammalian cells and embryos. Nat. Biotechnol. 42, 638–650 (2024).
Zong, Y. et al. Efficient C-to-T base editing in plants using a fusion of nCas9 and human APOBEC3A. Nat. Biotechnol. 36, 950–953 (2018).
Gu, T. et al. Highly efficient base editing in Staphylococcus aureus using an engineered CRISPR RNA-guided cytidine deaminase. Chem. Sci. 9, 3248–3253 (2018).
Chen, W. et al. CRISPR/Cas9-based genome editing in Pseudomonas aeruginosa and Cytidine Deaminase-Mediated Base Editing in Pseudomonas species. iScience 6, 222–231 (2018).
Wang, Y. et al. CRISPR-Cas9 and CRISPR-assisted Cytidine Deaminase enable precise and efficient genome editing in Klebsiella pneumoniae. Appl. Environ. Microbiol. 84, e01834–18 (2018).
Liu, L. M., Li, D. L., Deng, B., Wang, X. W. & Jiang, H. B. Special roles for efflux systems in iron homeostasis of non-siderophore-producing cyanobacteria. Environ. Microbiol. 24, 551–565 (2022).
Sakamoto, T., Shen, G., Higashi, S., Murata, N. & Bryant, D. A. Alteration of low-temperature susceptibility of the cyanobacterium Synechococcus sp. PCC 7002 by genetic manipulation of membrane lipid unsaturation. Arch. Microbiol. 169, 20–28 (1998).
Kluesner, M. G. et al. EditR: A method to quantify base editing from Sanger sequencing. CRISPR J. 1, 239–250 (2018).
Bae, S., Park, J. & Kim, J. S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014).
Jin, S. et al. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science 364, 292–295 (2019).
Kim, D. et al. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat. Biotechnol. 35, 475–480 (2017).
Zuo, E. et al. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science 364, 289–292 (2019).
Risser, D. D. & Callahan, S. M. HetF and PatA control levels of HetR in Anabaena sp. strain PCC 7120. J. Bacteriol. 190, 7645–7654 (2008).
Yu, H., Wu, Z., Chen, X., Ji, Q. & Tao, S. CRISPR-CBEI: a designing and analyzing tool kit for cytosine base editor-mediated gene inactivation. mSystems 5, e00350–20 (2020).
Kaneko, T. et al. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions (supplement). DNA Res. 3, 185–209 (1996).
Nagahari, K. & Sakaguchi, K. RSF1010 plasmid as a potentially useful vector in Pseudomonas species. J. Bacteriol. 133, 1527–1529 (1978).
Yau, S., Liu, X., Djordjevic, S. P. & Hall, R. M. RSF1010-like plasmids in Australian Salmonella enterica serovar Typhimurium and origin of their sul2-strA-strB antibiotic resistance gene cluster. Microb. Drug Resist. 16, 249–252 (2010).
Gormley, E. P. & Davies, J. Transfer of plasmid RSF1010 by conjugation from Escherichia coli to Streptomyces lividans and Mycobacterium smegmatis. J. Bacteriol. 173, 6705–6708 (1991).
Labes, M., Pühler, A. & Simon, R. A new family of RSF1010-derived expression and lac-fusion broad-host-range vectors for gram-negative bacteria. Gene 89, 37–46 (1990).
Xu, N. et al. Identification of an iron permease, cFTR1, in cyanobacteria involved in the iron reduction/re-oxidation uptake pathway. Environ. Microbiol. 18, 5005–5017 (2016).
Wang, S. Y., Li, X., Wang, S. G. & Xia, P. F. Base editing for reprogramming cyanobacterium Synechococcus elongatus. Metab. Eng. 75, 91–99 (2023).
Lee, M., Heo, Y. B. & Woo, H. M. Cytosine base editing in cyanobacteria by repressing archaic Type IV uracil-DNA glycosylase. Plant J. 113, 610–625 (2023).
Hanna, R. E. et al. Massively parallel assessment of human variants with base editor screens. Cell 184, 1064–1080.e1020 (2021).
Wang, Y. et al. In-situ generation of large numbers of genetic combinations for metabolic reprogramming via CRISPR-guided base editing. Nat. Commun. 12, 678 (2021).
Park, J., Lim, K., Kim, J. S. & Bae, S. Cas-analyzer: an online tool for assessing genome editing results using NGS data. Bioinformatics 33, 286–288 (2017).
Acknowledgements
We are very thankful to Prof. Chengcai Zhang (Institute of Hydrobiology, Chinese Academy of Sciences) for presenting the Anabaena sp. PCC 7120 strain and E. coli DH10B carrying pRL443 and pRL623 as the gifts. This study was funded by the National Natural Science Foundation of China (Grant Nos. 32170108, 42188102), the Ningbo Youth Leading Talent Project (Grant no. 2024QL060), the Ningbo Public Welfare Science and Technology Program (Grant Nos. 2023S040, 2023S068), the Independent Research Projects of Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai, Grant No. SML2021SP204), the Science and Technology Innovation 2025 Major Project of Ningbo City (Grant no. 2022Z189) and Ningbo University Startup Funding.
Author information
Authors and Affiliations
Contributions
H.B.J. and W.C. conceived and designed the experiments. X.D.L., L.M.L., Y.C.X., Q.W.S., Z.L., H.L.H, X.W.W, and W.C. performed the experiments. X.D.L. and W.C. analyzed the data. X.D.L., H.B.J., and W.C. wrote the manuscript. H.B.J. and W.C. supervised the research project. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Conrad Mullineaux, Ju-Yuan Zhang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Tobias Goris. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, XD., Liu, LM., Xi, YC. et al. Development of a base editor for convenient and multiplex genome editing in cyanobacteria. Commun Biol 7, 994 (2024). https://doi.org/10.1038/s42003-024-06696-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06696-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
