
Abstract
A critical step for SARS-CoV-2 assembly and maturation involves the autoactivation of the main protease (MProWT) from precursor polyproteins. Upon expression, a model precursor of MProWT mediates its own release at its termini rapidly to yield a mature dimer. A construct with an E290A mutation within MPro exhibits time dependent autoprocessing of the accumulated precursor at the N-terminal nsp4/nsp5 site followed by the C-terminal nsp5/nsp6 cleavage. In contrast, a precursor containing E290A and R298A mutations (MProM) displays cleavage only at the nsp4/nsp5 site to yield an intermediate monomeric product, which is cleaved at the nsp5/nsp6 site only by MProWT. MProM and the catalytic domain (MPro1-199) fused to the truncated nsp4 region also show time-dependent conversion in vitro to produce MProM and MPro1-199, respectively. The reactions follow first-order kinetics indicating that the nsp4/nsp5 cleavage occurs via an intramolecular mechanism. These results support a mechanism involving an N-terminal intramolecular cleavage leading to an increase in the dimer population and followed by an intermolecular cleavage at the C-terminus. Thus, targeting the predominantly monomeric MPro precursor for inhibition may lead to the identification of potent drugs for treatment.
Similar content being viewed by others

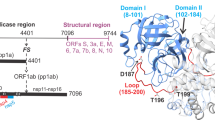

Introduction
Specific cleavages of the polyproteins catalyzed by the virally encoded protease to release the mature structural and functional proteins are indispensable for viral assembly and maturation1,2,3. A single copy of the main protease of SARS-CoV2 (nsp5 or MPro) is translated from overlapping reading frames in two large polyproteins (pp) 1a and 1ab that encompass all non-structural proteins (nsps)4,5,6. A critical step in the regulation of MPro is the formation of a fully active mature MPro via autoprocessing at its termini2,7. MPro of SARS-CoV and SARS-CoV-2 are 96% identical in sequence and thus are similar in structure and function8. Both enzymes are ~68 kDa homodimers with each protomer having one active site C145-H41 catalytic dyad7,9. The protomer is composed of 306 amino acids and organized in three domains, I-III. Domains I (residues 8–101) and II (102–184) exhibit a chymotrypsin-like fold and connected to the helical domain III (residues 201–306) through a long loop region (residues 185–200)9,10. Structural studies indicate that monomeric forms are similar in their tertiary fold to the wild-type dimer except for differences in the active site oxyanion loop, the N-finger region and its interface, and domain III orientation7,8,11. Upon dimerization, the oxyanion loop (residues 137–144) assumes an active wound conformation (E*), whereas it is predominantly in an inactive unwound conformation (E) in the monomeric form7,11,12. Dimer interface mutants that are monomeric or predominantly monomeric bind to known inhibitors of wild-type MPro accompanied by the reorganization of the oxyanion loop to the active E* conformation typical of the dimer11,13. Also, inhibitor or substrate favor binding to the dimeric form of the enzyme13,14.
The spatio-temporal regulation of MPro release from its polyprotein precursor in vivo is predicted to be complex because of the association of its flanking nsp4 and nsp6 proteins with membranes6,15,16,17. Studies aimed at understanding the early mechanisms of maturation of the viral proteases from polyproteins are also complicated by the fact that polyproteins contain multiple cleavage sites and undergo autoprocessing during expression and purification6. In addition, our attempts to refold MPro from chemical denaturants were unsuccessful for in vitro studies. In this study, we have engineered several model precursors containing mutations that restrict dimer formation and thus facilitate the accumulation and purification of intact precursors from E. coli. These precursors undergo time-dependent autoprocessing reaction mediating cleavages at the termini of MPro in E. coli and in vitro and thus, provide simple models to study the mechanisms of MPro autocatalytic maturation.
Results
Order of cleavage at the termini of MPro from model precursors in E. coli
Our simplified strategy entails expressing MPro (nsp5) containing flanking region sequences of nsp4 and nsp6 as a precursor mimetic. Recent studies indicate that the addition of non-native residues to the N-terminus of MPro results in a drastic decrease in catalytic activity as compared to such residues appended to the C-terminus of MPro18. The decrease in catalytic activity is consistent with our earlier observation that the addition of six native nsp4 residues (TSAVLQ) to the N-terminus of MPro increases the dimer dissociation constant12. These results imply that the reorganization of the free N-terminal residues of MPro upon N-terminal autoprocessing (nsp4/nsp5 site) through specific key interactions with domain III leads to enhanced dimer stability and appearance of mature-like catalytic activity, similar to that previously described for SARS-CoV MPro7,19. Thus, our design strategy for MPro precursor contained 25 C-terminal residues of nsp4 appended to the N-terminus of MPro [(−25)MPro] to enable discerning the migration of the precursor from the mature MPro upon autoprocessing by SDS-PAGE without compromising solubility. Likewise, extending the length of the flanking nsp4 even up to 102 residues (−102)MPro resulted in the production of soluble protein and with a Kdimer that is predicted to be like that of (−25)MPro11,13. Constructs and their designations used in this study are shown in the corresponding main figures and listed in Figure S1. Since appending even 25 residues of nsp6 to MPro causes severe insolubility of the expressed protein, the soluble immunoglobulin binding domain B1 of Protein G [GB120,], preceded by 3 N-terminal residues of nsp6 (SAV) to retain nsp5/nsp6 cleavage site specificity, were used (Fig. 1). This strategy allows monitoring native cleavages at both termini of MPro as a fully soluble precursor in E. coli and in vitro. Expression of PrecursorWT results in rapid autoprocessing at both sites within 20 min and no full-length precursor or intermediate product is observed (Fig. 1b). The products corresponding to mature MProWT (33.8 kDa, Fig. 1a) and the terminal fragments were identified (Fig. 1b), and the purified terminal fragments were confirmed by mass spectrometry.

The MPro model precursor comprising the nsp4/nsp5 and nsp5/nsp6 cleavage sites is shown on top. a–f 12 ml of cells were harvested at the time points indicated below the lanes, subjected to NAC and equal amounts (3–4 µg) of the flow-through (FT) and bound (B) fractions were analyzed by SDS-PAGE. Molecular weight standards (M) are indicated in kDa. a Migration of purified mature MProWT (33.8 kDa). b Autoprocessing of precursorWT results in 33.8 kDa mature MProWT in the FT, and 3.9 kDa and 7.5 kDa products in the B fractions. Cleavage products were verified by mass spectrometry. c Autoprocessing of precursorM (45.2 kDa) results in the appearance of an intermediate product (41.2 kDa, MProM-IP) upon cleavage at the N-terminus of MProM, seen as a doublet together with the full-length protein at 45 min of induction, and only that of MProM-IP at 2 hr of induction in the bound B fractions. d No C-terminal cleavage occurs even after 4 h of induction as indicated by the absence of the mature MProM (33.7 kDa) in the FT. e, f Autoprocessing of PrecursorR298A and PrecursorE290A. g Fractionation of the bound fractions of PrecursorE290A at 15, 30 (f) and 60 min (e) on Superose 12 (1 × 30 cm column) in buffer A confirming the stepwise cleavage at the N-terminus first, followed by the cleavage at the C-terminus as seen on gels. The precursor and products of cleavage were verified by mass spectrometry. h Cleavage of the nsp5/nsp6 site of MProM-IP by MProWT.
Since the catalytic activity inversely correlates with the dimer dissociation constant [Kdimer11,21,], it is predictable that mutations of residues contributing to dimer formation would enable the production of a functional precursor for monitoring the autoprocessing reaction. Thus, interface residues E290 and R298 were targeted for mutations in PrecursorWT. In earlier studies, we had shown that these mutations together increase the Kdimer of mature MPro by 5000-fold13 and accordingly this mutant was termed MProM (where M denotes monomer). PrecursorR298A undergoes rapid autoprocessing at both termini, like that of PrecursorWT, i.e., complete conversion within 1 h of expression to release the mature MProR298A, 3.9 kDa and 7.5 kDa products (Figs. 1e, 2b). In contrast, PrecursorE290A processing is observed to be much slower than PrecursorWT and PrecursorR298A allowing the transient observation of the time-dependent stepwise autoprocessing at both termini (see Fig. 1f). The expressed protein being fully soluble enabled the purification of the full-length protein and its product by nickel-affinity chromatography (NAC) expeditiously under native conditions. At 15 min, only PrecursorE290A (45.1 kDa) is detectable in the bound (B) fraction, followed by the appearance of the intermediate species of 41.2 kDa and the 3.9 kDa corresponding to the products of the N-terminal cleavage at 30 min. The 41.2 kDa product is then converted to the mature MProE290A and the 7.5 kDa product upon C-terminal cleavage at 1 h of induction (Fig. 1e). This is further confirmed by analyses of the bound fractions at 15, 30 and 60 min by size-exclusion chromatography (Fig. 1g) and mass spectrometry.

The N-terminal cleavage site is indicated with an upward black arrow. Precursor, products released upon cleavage at the N-terminus of MPro and molecular weight standards (M) are indicated in kDa. The gels show the time course of the autoprocessing reaction of MProM (a) and single mutants R298A and E290A (b) with 102 amino acids of the nsp4 sequence appended to the N-terminus of MPro. Cells (12 ml) were harvested at the time points indicated, subjected to NAC, and analyzed by SDS-PAGE. FT and B denote flow-through and bound fractions, respectively, after NAC. The 11.8 kDa product is not observed in the bound fraction because it lacks the 6His-tag. c–f Catalytic activity of mature MProR298A and MProE290A. c, d Linear relationship between the rate vs the square of the protein concentration assayed with 200 µM substrate. e, f Lineweaver-Burk plots for the hydrolysis of substrate by 0.2 μM MProR298A and 3.5 μM MProE290A in the presence of equimolar concentration of GC373. These concentrations represent the Kd estimated by ITC like that described for MProM 13. The kcat/Km was calculated as described13, and compared with previously characterized constructs shown in Table 211,13.
The double mutant E290A and R298A precursor (PrecursorM) also exhibits its accumulation in E. coli and time-dependent autoprocessing reaction, but at a slower rate than that of PrecursorE290A. Analysis of the fractions indicates cleavage at the N-terminus of MProM leading to the accumulation of the intermediate product (MProM-IP, 41.2 kDa, Fig. 1c) after 2 h of expression with no detectable product indicative of a C-terminal cleavage even after 4 h (Fig. 1d). The identity of MProM-IP was verified by mass spectrometry (41,180 Da, calculated = 41,180 Da). Nevertheless, incubation of MProM-IP with MProWT at a ratio of 50:1 produced the previously characterized mature MProM 13 indicating that the C-terminal cleavage site is accessible for reaction (Fig. 1h).
N-terminal autoprocessing of MPro precursor analogues in E.coli
Like PrecursorWT, expression of MProWT with only the native nsp4/nsp5 cleavage site also results in the lack of accumulation of the precursor, with autoprocessing complete within 15 min of expression to release mature MProWT [Fig. S2a11,]. An active site C145A mutation abolishes this cleavage (Fig. S2a11,).
We had previously reported the expression of a construct consisting of 25 amino acids of the nsp4 sequence appended to the N-terminus of the double mutant MProM as a miniprecursor, (−25)MProM, which undergoes time-dependent autoprocessing upon expression in E. coli [Fig. S2b11,]. The 37.4 kDa precursor is converted to the 34.6 kDa and 2.8 kDa products at a much slower rate relative to the MProWT precursor. Only the 34.6 kDa product (MProM) containing the 6H-tag allows its detection together with the precursor after NAC. To better resolve the separation between the starting material and products of the autoprocessing reaction by SDS-PAGE, a similar set of constructs comprising 102 amino acids of nsp4 appended to the N-terminus of MProWT, MProE290A, MProR298A, and MProM were expressed in E. coli (Fig. 2). Like PrecursorWT and its mutants, expression of (−102)MProWT and (−102)MProR298A (Fig. 2b) resulted in observing only the processed products released upon N-terminal autoprocessing. Time course of the autoprocessing of (−102)MProWT parallels that of the wild-type precursor shown in Figure S2A. Precursor (−102)MProE290A and (−102)MProM exhibit transient accumulation of the full-length precursor followed by N-terminal cleavage to yield products (Fig. 2a, b) like that of PrecursorE290A and PrecursorM (and (−25)MProM, Fig. S2c), respectively, undergoing N-terminal cleavage.
N-terminal autoprocessing of MPro precursor analogues in vitro
Interestingly, like (−25)MProM, precursor (−25)MPro1–199 lacking the entire helical domain (residues 200–306), as a monomer, also undergoes time-dependent N-terminal autoprocessing upon expression in E. coli to produce MPro1–199 [Fig. S2c11,]. Transient accumulation of full-length precursors (−102)MProM, (−25)MProM and (−25)MPro1–199 allowed their purification in a small scale to yield sufficient protein for in vitro analyses. Details of the reaction set-up are shown in Tables S1, S2. Samples collected at various time points were subjected to SDS-PAGE. Figure 3 shows the time course of the reactions. For (−102)MProM and (−25)MPro199, the starting material and the products are well separated on gels allowing quantifying the band intensities. The percent conversion at 15 and 24 h of incubation are calculated from each initial protein concentration and shown in Table 1. In addition, precursor (−102)MProM-catalyzed hydrolysis of the FRET substrate was monitored. The intrinsic catalytic activity was estimated to be 0.0204 μM/h when assayed with 50 μM precursor and 200 μM substrate. After incubation of (−102)MProM for 24 h, the observed value is 0.0384 μM/h, pointing to a 75% increase in catalytic activity attributable to the N-terminal cleavage of (−102)MProM.

a, b Precursor constructs showing the products released upon N-terminal autoprocessing. Time course of (c) (−102)MProM and (d) (−25)MPro1–199 autoprocessing. Reactions were initiated at the indicated concentration in buffer B at 28 °C (see Tables S1, S2 for details). Aliquots of the reaction were drawn at the indicated times and terminated by the addition of gel sample buffer. Samples (2.6–3.6 µg/lane) were subjected to SDS-PAGE and visualized by staining. Precursor, reaction products, and molecular weight standards (M) are indicated in kDa.
Like MProM 13, the autoprocessing of (−25)MProM precursor is accelerated in a concentration-dependent manner by GC373 up to an equimolar concentration (Fig. 4a). Figure 4b, c show qualitatively that, similar amount of MProM is produced from (−25)MProM in 15 h in the presence of equimolar amount of GC373 as compared to that in 47 h without GC373.

a Autoprocessing monitored by varying the inhibitor GC373 concentration. Time course of autoprocessing in the absence (b) and presence (c) of GC373. Reactions were initiated at the indicated concentration in buffer B at 28 °C. Aliquots of the reaction were drawn at the indicated times, terminated by the addition of gel sample buffer, subjected (3.5 µg/lane) to SDS-PAGE and visualized by staining. Precursor, reaction product, and molecular weight standards (M) are indicated in kDa.
Characterization of products of the autoprocessing reactions
The intermediate product MProM-IP resulting from the N-terminal autoprocessing of PrecursorM, was purified from cells expressed for 2.5 h as described in Methods. It is monomeric in solution as shown by SV-AUC up to a concentration of 85 μM (Fig. 5a). It catalyzes the hydrolysis of the FRET substrate, and the rate of hydrolysis of the peptide substrate displays first-order kinetics in protein concentration, i.e., a linear dependency on the protein concentration at a constant substrate concentration (Fig. 5b). This further confirmed that the observed catalytic activity is that of a monomer, and no detectable amount of dimer is present in solution. Unlike mature MProM, the kinetics of MProM-IP catalyzed hydrolysis is similar to that of monomeric MPro1–199 [11 and Table 2)]. However, like MProM 13, MProM-IP binds to GC373 with concomitant dimer formation and enhanced catalytic activity up to an equimolar concentration of the MProM-IP and GC373 (Fig. 5c). Above the equimolar amount, the catalytic activity is inhibited by increasing the concentration of GC373. The kinetic parameter kcat/Km for MProM-IP in the absence and presence of GC373 as well as the binding constant to GC373 determined by SV-AUC and ITC are listed in Table 2 (Fig. 5d, S3, S4) along with those of MProWT, MProM and MPro1–199. Although the 7.5 kDa SAV-GB1-6H fragment corresponding to the product of the C-terminal nsp5/nsp6 cleavage of MProM-IP by MProWT is easily observed (Fig. 1h), surprisingly no cleavage is detected upon incubation of MProM-IP in the absence or presence of equimolar amount of GC373 up to 48 h.

a Normalized sedimentation velocity absorbance c(s) distributions in the absence (red) and presence (black) of GC373. b Linear relationship between the rate of catalyzed hydrolysis vs concentration. c A plot of the rate as a function of increasing GC373 mixed with a constant amount of MProM-IP. d Lineweaver-Burk plot for the hydrolysis of substrate by 10 μM MProM-IP in the presence of equimolar concentration of GC373. The kcat/Km was calculated as described13 and listed in Table 2. e Room temperature structure of GC373 bound MProM (PDB 8FIG). The positions of mutations E290A and R298A used to create MProM with a very high Kdimer13 and GC373 are shown.
The 6H-tag at the C-terminus facilitates the initial purification of the MPro products derived from the autoprocessing of (−102)MProE290A and (−102)MProR298A and was removed to produce mature MProE290A MProR298A as previously described for MProWT 9,13. MProR298A and MProE290A exhibit a dimer dissociation constant (Kdimer) of 7.1 ± 0.4 µM and 353 ± 21 µM, respectively, compared to 6.6 mM for the double mutant MProM (Table 2 and Fig. S5). Purified mature MProE290A and MProR298A catalyze the hydrolysis of the peptide substrate, and the rate of hydrolysis is linearly dependent on the square of the protein concentration, i.e., second-order kinetics, indicating that the observed catalytic activity is that of a dimer like that of MProWT and MProM (Fig. 2c, d). The kinetic parameters kcat/Km of mature MProE290A MProR298A- catalyzed hydrolysis of substrate as well as the inhibitor GC373 binding constants are listed in Table 2 (Figs. 2e, f, S4a, b, S5c). The monomer-dimer distributions of MProR298A and MProE290A in the absence of inhibitor or substrate by SV-AUC are shown in Figure S5a, b.
Room-temperature structure of GC373 bound MProM
Since MProM is predominantly monomeric in solution, the effect of the mutation of residues E290A and R298A on the overall structure of the protein particularly in the active site region and the monomer-dimer interface regions that include the N- and C-termini is examined by X-ray crystallography. Attempts to grow crystals of monomeric MProM and MProM-IP were unsuccessful, although a crystal structure of the monomeric SARS-CoV-1 MProR298A single mutant was attained previously22. However, we succeeded in obtaining a room-temperature crystal structure of MProM in complex with the reversible covalent inhibitor GC373 at 1.75 Å resolution (Table 3). MProM crystallizes in the monoclinic unit cell (space group I2) with one protomer present in the asymmetric unit. MProM-GC373 complex is homodimeric, with the quaternary structure virtually identical to that of the MProWT [Fig. 5e10,23]. We modeled residues 1-303 in MProM-GC373 structure, whereas the rest of the C-terminal residues are disordered and are not visible in the electron density map. GC373 is covalently bonded to the sulfur of the catalytic Cys145 generating the hemithioacetal conjugate. Interestingly, the nucleophilic attack of the sulfur of Cys145 on the carbonyl carbon of GC373 is not stereospecific and occurs from both sides of the carbonyl carbon such that the hemithioacetal hydroxyl group is observed in two alternate orientations (Fig. 6a). In one orientation, the hydroxyl is directed into the oxyanion hole created by residues Leu141-Cys145, whereas in the other it faces the catalytic His41. In addition, the phenyl tail of GC373 occupies two alternate conformations. In the previous room temperature X-ray structure of MProWT-GC373 complex11, the ligand was found in one orientation, bound in a stereospecific fashion with the hemithioacetal hydroxyl directed into the oxyanion hole. GC373 makes five direct hydrogen bonds with MProM residues (Fig. 6b). The shortest and possibly the strongest hydrogen bond of 2.5 Å is formed between the ligand’s P1 γ-lactam and imidazole of His163 and is known to be a critical interaction for inhibitors with the MPro active site24,25,26. The remaining four hydrogen bond distances are significantly longer, 2.8–3.4 Å. It should be noted that the lack of stereospecificity in the reaction of the sulfur of C145 with the carbonyl carbon of GC373 is not caused by the mutations because (1) the isomeric hemithioacetals have been previously reported in the cryo-structure of MProWT-GC373 complex27 and (2) the room temperature structure of MPro1–199-GC373 complex shows only the S-configuration for the hemithioacetal carbon11.

a 2FO-FC electron density of GC373 covalently bonded to C145 is shown at 1.5 σ level. GC373 binds in two alternate conformations shown with green carbon atoms and in gray with the refined occupancies of 51/49%, respectively. b Hydrogen bonds formed by GC373 with the MProM active site residues. Active site and oxyanion hole residues are colored by atom type with cyan and magenta carbon atoms, respectively. c 2FO-FC electron density for the interprotomer interface near E290A mutation site shown at 1.5 σ level. Protomer A carbon atoms are colored cyan, whereas protomer B carbon atoms are colored slate. d Superposition of MProM (cyan and slate carbon atoms) and MProWT (gray carbon atoms) near E290A mutation site demonstrating the loss of the E290…R4’ salt bridge. e 2FO-FC electron density for the interprotomer interface near R298A mutation site is shown at 1.5 σ level. f Superposition of MProM and MProWT near R298A mutation site showing a conformational reorientation of the C-terminal residues (black curved arrow). Atoms are colored in the same way as in panel d. All distances are shown in Angstroms.
The electron density map for the E290A mutation site located at the dimer interface is shown in Fig. 6c where interactions with the N-terminal finger (residues 1–6) occur. Four water molecules (W1-W4) are clearly visible in the vicinity of Ala290, making a tight hydrogen-bonded network with distances between the waters of 2.4–2.6 Å. The water structure intervenes between the stretch of residues 288–291 of protomer A and Arg4 and Lys5 of protomer B. W1 hydrogen bonds with Arg131 (protomer A), and W3 makes a hydrogen bond with Lys5 (protomer B). Thus, the waters mediate the connection between the two protomers. Arg4 of protomer B forms hydrogen bonds with the main chain carbonyl of Glu127 of protomer A. Evidently, there are no direct hydrogen bonds between the protomers in this region. When MProM-GC373 is superimposed with the room-temperature joint X-ray/neutron structure of MProWT [PDB ID 7JUN23,] or with the room-temperature X-ray structure of MProWT-GC373 complex [PDB ID 7UKK11,], several important structural differences relative to MProM can be noted (Figs. 6d and S6). In both MProWT structures, Glu290 of protomer A makes strong 2.6–2.8 Å hydrogen bond interactions with Arg4 of protomer B through a salt bridge structure. In addition, Glu288 of protomer A forms 2.9–3.0 Å hydrogen bonds with Lys5 of protomer B in the inhibitor-free and GC373-bound MProWT structures. In MProM-GC373, these hydrogen bonds are severed due to shifts of Glu288 of protomer A and Lys5 of protomer B away from each other, resulting in their separation of 5.5 Å. Moreover, to stabilize its position in the absence of the Glu290 carboxylic group, the Arg4 of the protomer B side chain flips towards the main chain of Glu127. W1 is conserved between inhibitor-free MProWT and MProM-GC373 structures and is replaced with the alternate conformation of Glu290 in MProWT-GC373 complex. W1 makes a 2.6 Å hydrogen bond with Glu290 in MProWT. Thus, because of the E290A mutation, three water molecules W2, W3, and W4 invade the dimer interface disrupting several hydrogen bonds between the protomers and contributing to the increase in the Kdimer.
The electron density map for the MProM residues around the R298A mutation site located where C-terminal residues interact with the other protomer and specifically with Ser139-Leu141 turn at the start of the oxyanion loop is shown in Fig. 6e. There is one direct interprotomer hydrogen bond of 2.8 and 2.7 Å from Gln299 of protomer A to Ser139 of protomer B in inhibitor-free MProWT and MProWT-GC373, respectively. This hydrogen bond is retained in MProM-GC373 compared to MProWT when the structures are superimposed. In both MProWT structures, Arg298 is situated more than 5 Å from the nearest residue of the other protomer. However, it makes hydrophobic interactions with the C-terminal residues from the same protomer, which are lost when this Arg is mutated to Ala. In both MProWT, the C-terminal residues starting from Cys300 located at the end of a short α-helix of residues 293–300 make a 90° turn towards the interface between the catalytic domains. In MProM-GC373 instead, apparently caused by the R298A mutation, the C-terminus flips in the opposite direction towards the helical domain so that Val303 side chain, the last residue visible in the electron density, is as close as 4 Å to Ser1 side chain of the same protomer (Figs. 6f and S6). Although C-terminal residues do not make hydrogen bonds at the dimer interface, they interact through hydrophobic interactions with the other protomer in MProWT. These interactions are lost in MProM-GC373 due to the drastic conformational change of the C-terminus in the double mutant.
Discussion
All precursor analogues used in this study autocatalyze the hydrolytic cleavage at the N-terminus (nsp4/nsp5 site) of MPro including the monomeric (−25)MPro1–199 which lacks the entire helical domain (domain III). E. coli expression of constructs comprising MProWT fused to native cleavage sites at the N- and C-termini results in processed mature MProWT without observing any full-length or intermediate products, consistent with previous reports9,11,28. Similarly, full-length precursors are not observed upon expression of PrecursorR298A and (−102)MProR298A. In contrast, full-length precursors accumulate in E. coli upon expression of PrecursorE290A, PrecursorM, (−102)MProE290A, (-102)MProM, (−25)MProM, and (−25)MPro199 that undergo time-dependent autoprocessing reaction at the N-terminus (Figs. 1, 2 and S2). We isolated the full-length precursors and the products of the autoprocessing reactions from E. coli. The results in Table 2 show the effect of mutations on the kinetic parameter kcat/Km, binding constants to the known covalent inhibitor GC373, and the dimer dissociation constants. Mutations E290A and R298A affect both kcat/Km and Kdimer to a different extent. MProR298A exhibits a kcat/Km of 0.31 μM−1 min−1, i.e., about half that of MProWT, and a Kdimer of 7.13 μM, i.e., about 5.5 times larger than that of MProWT. E290A mutation has a much larger effect on kcat/Km and Kdimer compared to the R298A mutation. The observed kcat/Km for MProE290A is within the range of the double mutant MProM. The observed Kdimer of 353 μM is about 19 times smaller than that of MProM and about 270 times larger than that of MProWT (Table 2). These results are consistent with the observed accumulation profile of the precursor in E. coli and highlight the role of dimer formation in the autoprocessing reaction. Precursor comprising MProR298A does not accumulate in E. coli and only the fully processed mature enzyme upon cleavages at both termini is observed. While the intrinsic catalytic activity (kcat/Km) of MProE290A is nearly identical to that of MProM, the precursor comprising MProE290A undergoes stepwise autoprocessing at both termini at a much faster rate because of its smaller Kdimer. In contrast, precursors comprising MProM and MPro1–199 accumulate and undergo autoprocessing in the monomeric form at a much slower rate in cells which allows their purification for in vitro reactions. It is intriguing, however, that the monomeric precursor comprising MProM with only the nsp5/nsp6 cleavage site, even in its dimer form, failed to promote cleavage at the C-terminus of MProM despite the accessibility for cleavage of this site by provided MProWT.
Similarly, E290A and R298A mutations affect the binding of the transition state analog GC373 to varying extent. While the binding constant of GC373 to MProR298A is within the experimental error to that of MProWT, MProE290A is 23 times larger relative to MProWT and comparable to that of MProM. Furthermore, the mutant enzymes bind the aldehyde inhibitor GC373 with a binding constant inversely proportional to kcat/Km which is consistent with previously reported studies [see Table 2 and ref. 11,13].
Room temperature crystal structure of MproM shows that E290A and R298A mutations destabilize the dimer by excluding all direct hydrogen bonding between the protomers in the vicinity of residue 290 and retaining one hydrogen bond near residue 298. In the presence of the inhibitor, the dimer is stabilized through hydrophobic interactions, bridged by hydrogen bonding through water molecules and a H-bond between Gln299 of protomer A with Ser139 of protomer B. Destabilization of the dimer form is reflected in the catalytic activity, the Kdimer, binding of the covalent inhibitor GC373 (Table 2) as well as the accumulation of the full-length precursor upon expression in E. coli. Both mature MPro mutants, MProE290A and MProR298A, follow second-order kinetics in protein concentration for catalyzing the hydrolysis of the peptide substrate (Fig. 2c, d) indicating that the observed catalytic activity is that of a dimer, like that of MProWT and MProM and in contrast to that of the monomeric catalytic activity of MPro1–199 11. Recently, we reported the room temperature X-ray structures of MPro1–199 and MPro1–196-GC373 complexes11. The structure of the monomeric MPro1–199 is nearly identical to that of the dimeric wild-type except for the oxyanion-loop being in an unwound conformation (inactive E-state), whereas MPro1–196-GC373 covalent complex displays a wound oxyanion loop (active E*-state) typical of the dimeric form of inhibitor-free mature MPro. The observed catalytic activities of MProM and MPro1–199 indicated that the E-E* equilibrium is dynamic in which the E state predominates in the monomer while the E* is the catalytically active species and favored in the dimer11,12,13.
Although we were unable to isolate PrecursorE290A in sufficient purity and quantity for in vitro analysis due to the relatively fast autoprocessing reaction, small-scale isolation of the full-length precursor and its products enabled verification by mass spectrometry. PrecursorE290A exhibits time-dependent autoprocessing via initial cleavage at the N-terminus to produce MProE290A-IP followed by the cleavage at the C-terminus to produce mature MProE290A (Fig. 1e-g). This order of cleavage is consistent with the observed cleavage order of a construct comprising 10 amino acid of the native flanking sequence at the N- and C-termini of MPro of SARS-CoV having an active site C145A mutation catalyzed intermolecularly by mature MProWT 29.
As indicated above, (−102)MProM and (−25)MPro199 undergo time-dependent autoprocessing at the N-terminus to produce MProM and MPro199 in vitro (Fig. 3). The results in Table 1 show that the percent conversion of the precursor to products at a given time is independent of the initial protein concentration indicating that the reactions follow first-order kinetics, i.e., unimolecular. The percent conversion of the two precursors at a given time are similar, if not identical. Thus, the cleavage at the N-terminus of MPro occurs via an intramolecular mechanism from a monomer in these two model precursors. Importantly, the addition of GC373 to the precursor analogue, (−25)MProM, increases the rate of autoprocessing and the increase in rate of autoprocessing is dependent on the GC373 concentration. This attribute is similar to that reported for the activation of MProM by GC373 and can be explained by a similar mechanism13. In that mechanism, MProM is predominantly in the monomer form with an oxyanion loop in the E conformation. Upon binding GC373, MProM forms a dimer concomitant with the reorganization of the oxyanion loop to the active E* conformation. In the presence of the equimolar amount of protein and GC373, three major protein species are present in equilibrium: monomer, dimer with one of the active sites occupied by GC373, and a dimer with both active sites occupied by GC373. The increase in catalytic activity is attributed to the presence of the population of a dimer with one active site occupied by GC373, leaving the other active site with an oxyanion loop in the active E* conformation for catalytic function. Thus, the increase in the reaction rate of (−25)MProM autoprocessing in the presence GC373 must be due to the formation of a dimer. Accordingly, the autoprocessing of the model precursor is initiated by N-terminal intramolecular cleavage from a monomer or a dimer. The enhanced dimer stability in the context of a wild-type precursor, relative to MProM, suggests that the initial cleavage at the N-terminus most likely occurs from the dimer because it is more reactive than the monomer. This conclusion is in accordance with the precursor accumulation profile in E.coli described above.
N-terminal cleavage of (−102)MProM to produce MProM is accompanied by an increase in catalytic activity as well as the binding affinity to GC373 increasing by about 3-fold (Table 2, Fig. S4c). As previously reported, the observed catalytic activity of MProM is that of a dimer indicating that N-terminal cleavage will favor dimer formation. The involvement of the N-finger in dimer formation has been examined in detail by structural and mutational studies. Deletion and mutational studies of the N-finger residues 1–9 show drastic effect on dimer formation and catalytic activity, relative to MProWT, consistent with the critical interfaces involving the N-finger with domains II and III8,11. In recent studies, the effect of flanking nsp4 residues, contrary to deleting the N-terminal residues of MPro, on Kdimer was also examined12. The active site H41A mutation to restrict N-terminal autoprocessing enabled the analyses of such precursor mimetics. Even the addition of six residues of the native nsp4 sequence to mature MProH41A ((−6)MProH41A) increases the Kdimer by >85-fold. Structural studies revealed asymmetry, semi-open conformation and disorder of the terminal residues of the dimeric (−6)MProH41A 12. Thus, the product of the autoprocessing reaction to liberate the free N-terminus of MPro is critical for dimer formation.
The product MProM-IP resulting from the autoprocessing of PrecursorM containing the nsp5/nsp6 cleavage site at the C-terminus of MProM exhibits catalytic activity, which is that of a monomer and is activated and inhibited by GC373 like that of mature MProM. Activation by GC373 is concomitant with dimer formation. However, it does not autocatalyze the nsp5/nsp6 junction indicating that the monomer is incapable of this cleavage either in the absence, or presence of GC373 in its dimer form. But treatment of the monomeric MProM-IP with MProWT produces MProM indicative of the site being accessible for cleavage in its monomeric form. Failure of MProM-IP to process its own C-terminus could be due to its inability to form a dimer. The requirement of a dimer for C-terminal cleavage is consistent with the observations in crystal structures showing the C-terminal residues of one dimer of MProC145A bound to the active site of a second dimer30,31,32. It is unclear as to why MProM-IP fails to undergo C-terminal cleavage in the presence of equimolar amount of GC373 when both monomer and the enzymatically active dimer coexist. This may point to the dynamics of this system which is incapable of forming a productive reaction complex. It is worth noting, however, that lack of C-terminal autoprocessing is limited only to MProM-IP having both mutations E290A and R298A, whereas MProE290A-IP exhibits C-terminal cleavage likely due to its ability to form a significant population of dimer even in the absence of inhibitor GC373.
Transient accumulation of PrecursorE290A and its conversion to MProE290A-IP and mature MProE290A indicates that cleavage at the N-terminus precedes the C-terminal cleavage. A proposed mechanism for MPro autoprocessing from a model precursor is shown in Fig. 7. In this mechanism, the MPro region of the monomeric precursor exhibits a tertiary fold like that of the mature subunit with an oxyanion loop predominantly in the E-state. This inactive state is in equilibrium with a minor population having the active E* conformation, which undergoes slow unimolecular N-terminal autoprocessing. The monomeric precursor is also in equilibrium with its homodimer. This is supported by the observation that the presence of N- and C-terminal flanking sequences increase the Kdimer significantly but do not exclude the precursor from becoming a dimer. In the transient precursor dimer, oxyanion loop dynamics favor an E* conformation leading to a faster rate of cleavage of the N-terminal site than that of the monomeric precursor. Results presented here show that the monomer and dimer precursor forms can autocatalyze N-terminal cleavage and that the dimer undergoes significantly faster reaction, compared to the monomer. The observed first-order kinetics of the monomeric model precursors indicate that the cleavage at the N-terminus proceeds via an intramolecular mechanism. N-terminal cleavage of a monomer or a dimer would lead to an increase in the dimer population as the newly formed free N-finger adopts a native-like interface with the second protomer. A heterodimer comprising a free N-terminus is likely to be more stable than the precursor homodimer and hence, being more reactive leads to an even faster rate of cleavage of the second N-terminus resulting in further stabilization of the dimer interface and decrease in Kdimer. It is likely that the precursor protomer containing the N-terminal flanking sequence is responsible for the cleavage of its own N-terminus because it conforms with the correct orientation for substrate binding visualized in structures33,34. Since the N-terminal flanking sequence has a larger effect on the Kdimer as well as the GC373 binding constant (Table 2 and Fig. S4c) than the C-terminal flanking sequence29, upon N-terminal cleavage, a more stable population of homo- and heterodimers favoring the E* conformation of the oxyanion loop leads to the processing of the C-terminal nsp5/nsp6 site via an intermolecular mechanism to release the mature enzyme.

Catalytic (domains I and II) and helical (domain III) regions are shown as red and blue ovals, respectively. Solid black lines denote the N-terminal residues (N-finger) of MPro. Dashed black and solid blue lines represent the truncated nsp4 and nsp6 regions flanking the N- and C-termini of MPro. Catalytic dyad H41 and C145 residues are indicated as white circles in the top left monomer cartoon.
The above mechanism of activation is consistent with reports on the self-cleavage of picornaviral replicase precursors35,36 and early cleavages of 3CL protease of coronavirus MHV-A5937. Both nsp4 and nsp6 are shown to have multiple transmembrane domains and thus, MPro is suggested to be anchored on both sides to membranes. The resulting major products nsp5-nsp10 and nsp5-nsp16 generated from pp1a and pp1ab, respectively, by cleavage at the N-terminus may be transported through membrane association17 for processing of the nsp5/nsp6 site through a concentration-dependent intermolecular mechanism at a later stage of viral polyprotein maturation.
Also, the proposed mechanism of MPro autoprocessing is strikingly similar to that of the dimeric aspartic acid protease of HIV-1 from its precursor1,3,38. The main difference lies in the cleavage at the N-terminus of HIV protease occurring from the dimer form because each of the monomer possesses only half of the catalytic residues of the active site. Thus, it appears that intramolecular cleavage at the N-terminus followed by intermolecular cleavage at the C-terminus is a general mechanism for autoprocessing of proteases of RNA viruses from their polyprotein precursor.
Methods
Construction and designation of MPro constructs
Expression and purification of MProWT (GenBank ID: MN908947.3), MProM and MPro1–199 were described before11,13,39. New constructs prepared for this work are: PrecursorWT, PrecursorM and its single mutants, precursor (−102)MProWT, precursor (−102)MProM and its single mutants, precursor (−25)MPro1–199, mature MProR298A and mature MProE290A. All constructs were synthesized and cloned into pJ414 vector (ATUM, Newark, CA). Designations and amino acid sequences of the constructs are listed in Figure S1.
Expression and purification
Plasmids were transformed into BL21-DE3 cells (Agilent) and induced for expression at 0.7–0.8 optical density with 1 mM isopropyl β-d-1-thiogalactopyranoside, typically for 3 h at 37 °C. Proteins were purified from the cell lysate by nickel-affinity chromatography (NAC, step 1). The bound fraction was subjected to isocratic fractionation on Superose-12 column (step 2, Cytiva Life Sciences) in a final buffer of 25 mM Tris-HCl, pH 7 or 7.2, 150 mM NaCl and 1 mM TCEP (buffer A). Peak fractions were pooled and concentrated to the desired concentration using Amicon Ultra – 15 or 0.5 ml centrifugal filters (Merck Millipore Ltd.) and stored in aliquots at −30 °C and for long-term storage at −80 °C. Purity was verified both by SDS-PAGE on 4–20% gradient mini-protean TGX precast gel (Bio-Rad) and reverse-phase liquid chromatography with in-line electrospray ionization mass spectrometry11. Protein concentrations were measured before storage and prior to the experiment at least in duplicate based on the extinction coefficient (Fig. S1) at 280 nm.
Purifications of mature MProR298A and MProE290A were carried out with an additional HRV-3C protease cleavage step to remove the C-terminal 6His-tag as described previously for MProWT and MProM 11. To isolate precursors (−102)MProM and (−25)MPro1–199, cell cultures (<100 ml) were induced for <30 min and chilled immediately prior to harvesting. Cell pellets were subjected to the same purification scheme as above not exceeding a total time of 4 h at 4 °C due to autoprocessing at native conditions. The final column step on Superose-12 was carried out in buffer B (25 mM Tris-HCl, pH 7, 50 mM NaCl and 1 mM TCEP). The full-length protein, representing only a small fraction of the total load on the column, with minimal contamination of the products were pooled and stored in aliquots at −70 °C.
Autoprocessing
Time course of the autoprocessing reaction was carried out as follows. Cells (12 ml) were harvested at the indicated time points, chilled on ice, and subjected to NAC on spin columns. Equal volumes of the bound fraction were analyzed by SDS-PAGE. But for side-by-side comparison of the flow through (FT) and bound (B) fractions and monitoring the autoprocessing reactions in vitro (Tables S1, S2), equal amounts of proteins11 were used for gel fractionation. Molecular weight markers (in kDa) are shown for all gel panels except in few panels when a protein of similar mass (Figs. 2 and S1) or protein from the same stock solution are analyzed (Fig. 4). Uncropped gel images of Figs. 1–4 and S2 are shown in Fig. S7.
Enzyme kinetics
Activity assays using the FRET substrate Dabcyl-KTSAVLQ/SGFRKM-E(Edans)-NH2, where (/) denotes the scissile peptide bond, were performed in a total volume of 100 µl in buffer B (25 mM Tris-HCl, pH 7, 50 mM NaCl and 1 mM TCEP) at 28 °C as previously described9,13. Assays were carried out with equimolar amount of GC373 and enzyme and varying the substrate concentration from 5–200 µM. kcat/Km was calculated from plots of 1/v vs 1/S and the GC373 binding constant was determined by ITC. Plots of rate vs the square of the protein concentration at 200 μM were linear, and the slope of the line is 2 kcat[S]/Kdimer(Km + [S]). Since the Km value is about the same as the substrate concentration used, the slope of the line is simplified to kcat/KmKdimer that allowed the calculation of Kdimer. For details, see reference 13. The substrate was custom synthesized (Biomatik, Ontario, Canada), and GC376 was purchased from Selleckchem, Houston, TX.
Sedimentation velocity analytical ultracentrifugation (SV-AUC) and Isothermal titration calorimetry (ITC)
SV-AUC (in buffer B at 25 °C) and ITC in buffer C (25 mM Tris-HCl, pH 7.2, 20 mM NaCl and 1 mM TCEP) at 28 °C were carried out as described in references 11,12,13.
Protein crystallization and room-temperature X-ray crystallography
MProM protein sample was concentrated to 7 mg/ml. GC376 stock was prepared at 10 mM GC376 in 25 mM Tris-HCl, pH 7.2, 20 mM NaCl and 1 mM TCEP for crystallization purposes and stored at −30 °C. GC376 is converted to the reactive aldehyde GC373 when mixed with an aqueous solution. For co-crystallization, MProM was mixed with GC376 at 1:5 molar ratio and allowed to incubate at room temperature for at least 30 min before setting up crystal trays. Crystals of GC373-bound MProM were grown at 14 °C by sitting drop vapor diffusion methodology with 18–21% PEG3350, 0.1 M Bis-Tris pH 6.5, or pH 7.0 (1 mL) as the precipitant solution. Crystallization drops of 20 µL at 1:1 ratio were seed struck using the crystals of the native MPro in complex with a covalent ligand NBH2 as described in references 26,39. The crystals suitable for X-ray diffraction measurements were mounted in MiTeGen (Ithaca, NY) room-temperature capillary setups for data collection.
Room temperature X-ray crystallographic data were collected on a Rigaku HighFlux HomeLab instrument equipped with a MicroMax-007 HF X-ray generator, Osmic VariMax optics, and a DECTRIS Eiger R 4 M hybrid photon counting detector. X-ray diffraction data were integrated using the CrysAlis Pro software suite (Rigaku Inc., The Woodlands, TX), then reduced and scaled using Aimless40 from the CCP4 suite41. Structures were solved by molecular replacement using Phaser42. MProWT-GC373 complex structure (PDB code 7UKK)11, was used as a search model to solve the structure of MProM-GC373 complex. The model was iteratively refined with phenix.refine from the PHENIX suite43 and COOT44. Geometry validation was aided by Molprobity45. GC373 restraints were generated with eLBOW46 using geometry optimized by quantum chemical calculations in Gaussian16 at B3LYP/6-31 g(d,p) level of theory47. Final data collection and refinement statistics can be found in Table 3.
Statistics and reproducibility
Expressed proteins were verified both by DNA sequencing and mass spectrometry. The reproducibility of enzyme kinetics was tested at least 2–3 times with freshly prepared enzyme and stock solutions of the substrate and inhibitor. Once this was determined to provide consistent reaction rates within an error limit of 5%, the final experiment for the data displayed in the manuscript was carried out in duplicate and 4 reads per well for each time point. The mean of the data points was used for fitting. The same stock solutions of enzyme and inhibitor were used for SV-AUC and ITC analyses to determine the dimer dissociation constant (Kdimer) and the binding constant of the inhibitor to the enzyme (Kd), respectively. Kdimer, and Kd were determined with multiple protein constructs (Fig. S1) and concentrations. Each ITC experiment was carried out with a minimum of 20 injections. The apparent dimer dissociation constants were determined by Lamm equation modeling of the absorbance and interference data. X-ray diffraction data and refinement statistics are shown. Gel images are best representative for each of the construct analyzed.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The structure and corresponding structure factors have been deposited into the protein data bank with the PDB accession code 8FI9 for MProM/GC373 complex. Source data files are provided in Supplementary Data 1, 2, and Supplementary information. All other data that support this study are available from the corresponding authors upon reasonable request.
References
Louis, J. M., Weber, I. T., Tozser, J., Clore, G. M. & Gronenborn, A. M. HIV-1 protease: maturation, enzyme specificity, and drug resistance. Adv. Pharm. 49, 111–146 (2000).
Groneberg, D. A., Hilgenfeld, R. & Zabel, P. Molecular mechanisms of severe acute respiratory syndrome (SARS). Respir. Res. 6, 8 (2005).
Louis, J. M., Ishima, R., Torchia, D. A. & Weber, I. T. HIV-1 protease: structure, dynamics, and inhibition. Adv. Pharm. 55, 261–298 (2007).
Mariano, G., Farthing, R. J., Lale-Farjat, S. L. M. & Bergeron, J. R. C. Structural Characterization of SARS-CoV-2: Where We Are, and Where We Need to Be. Front Mol. Biosci. 7, 605236 (2020).
Wang, C. et al. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J. Med. Virol. 92, 667–674 (2020).
Yoshimoto, F. K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 39, 198–216 (2020).
Xia, B. & Kang, X. Activation and maturation of SARS-CoV main protease. Protein Cell 2, 282–290 (2011).
Goyal, B. & Goyal, D. Targeting the Dimerization of the Main Protease of Coronaviruses: A Potential Broad-Spectrum Therapeutic Strategy. ACS Comb. Sci. 22, 297–305 (2020).
Zhang, L. et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 368, 409–412 (2020).
Kneller, D. W. et al. Structural plasticity of SARS-CoV-2 3CL M(pro) active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 11, 3202 (2020).
Nashed, N. T. et al. Autoprocessing and oxyanion loop reorganization upon GC373 and nirmatrelvir binding of monomeric SARS-CoV-2 main protease catalytic domain. Commun. Biol. 5, 976 (2022).
Kovalevsky, A. et al. Unmasking the Conformational Stability and Inhibitor Binding to SARS-CoV-2 Main Protease Active Site Mutants and Miniprecursor. J. Mol. Biol. 434, 167876 (2022).
Nashed, N. T., Aniana, A., Ghirlando, R., Chiliveri, S. C. & Louis, J. M. Modulation of the monomer-dimer equilibrium and catalytic activity of SARS-CoV-2 main protease by a transition-state analog inhibitor. Commun. Biol. 5, 160 (2022).
Cheng, S. C., Chang, G. G. & Chou, C. Y. Mutation of Glu-166 blocks the substrate-induced dimerization of SARS coronavirus main protease. Biophys. J. 98, 1327–1336 (2010).
Oostra, M. et al. Localization and membrane topology of coronavirus nonstructural protein 4: involvement of the early secretory pathway in replication. J. Virol. 81, 12323–12336 (2007).
Oostra, M. et al. Topology and membrane anchoring of the coronavirus replication complex: not all hydrophobic domains of nsp3 and nsp6 are membrane spanning. J. Virol. 82, 12392–12405 (2008).
Ricciardi, S. et al. The role of NSP6 in the biogenesis of the SARS-CoV-2 replication organelle. Nature 606, 761–768 (2022).
Rong, Y., Zhang, C., Gao, W. C. & Zhao, C. Optimization of the expression of the main protease from SARS-CoV-2. Protein Expr. Purif. 203, 106208 (2023).
Chen, S., Jonas, F., Shen, C. & Hilgenfeld, R. Liberation of SARS-CoV main protease from the viral polyprotein: N-terminal autocleavage does not depend on the mature dimerization mode. Protein Cell 1, 59–74 (2010).
Gronenborn, A. M. et al. A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science 253, 657–661 (1991).
Lin, P. Y., Chou, C. Y., Chang, H. C., Hsu, W. C. & Chang, G. G. Correlation between dissociation and catalysis of SARS-CoV main protease. Arch. Biochem Biophys. 472, 34–42 (2008).
Shi, J., Sivaraman, J. & Song, J. Mechanism for controlling the dimer-monomer switch and coupling dimerization to catalysis of the severe acute respiratory syndrome coronavirus 3C-like protease. J. Virol. 82, 4620–4629 (2008).
Kneller, D. W. et al. Unusual zwitterionic catalytic site of SARS-CoV-2 main protease revealed by neutron crystallography. J. Biol. Chem. 295, 17365–17373 (2020).
Owen D. R. et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 274, 1586–1593 (2021).
Qiao, J. et al. SARS-CoV-2 M(pro) inhibitors with antiviral activity in a transgenic mouse model. Science 371, 1374–1378 (2021).
Kneller, D. W. et al. Covalent narlaprevir- and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease. Nat. Commun. 13, 2268 (2022).
Ma, C. et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 30, 678–692 (2020).
Xue, X. et al. Production of authentic SARS-CoV M(pro) with enhanced activity: application as a novel tag-cleavage endopeptidase for protein overproduction. J. Mol. Biol. 366, 965–975 (2007).
Hsu, M. F. et al. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 280, 31257–31266 (2005).
Noske, G. D. et al. A Crystallographic Snapshot of SARS-CoV-2 Main Protease Maturation Process. J. Mol. Biol. 433, 167118 (2021).
Muramatsu, T. et al. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl Acad. Sci. USA 113, 12997–13002 (2016).
Lee, J. et al. Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nat. Commun. 11, 5877 (2020).
Kneller, D. W., Zhang, Q., Coates, L., Louis, J. M. & Kovalevsky, A. Michaelis-like complex of SARS-CoV-2 main protease visualized by room-temperature X-ray crystallography. IUCrJ 8, 973–979 (2021).
MacDonald, E. A. et al. Recognition of Divergent Viral Substrates by the SARS-CoV-2 Main Protease. ACS Infect. Dis. 7, 2591–2595 (2021).
Palmenberg, A. C. & Rueckert, R. R. Evidence for intramolecular self-cleavage of picornaviral replicase precursors. J. Virol. 41, 244–249 (1982).
Blair, W. S. & Semler, B. L. Self-cleaving proteases. Curr. Opin. Cell Biol. 3, 1039–1045 (1991).
Lu, X., Lu, Y. & Denison, M. R. Intracellular and in vitro-translated 27-kDa proteins contain the 3C-like proteinase activity of the coronavirus MHV-A59. Virology 222, 375–382 (1996).
Louis, J. M., Nashed, N. T., Parris, K. D., Kimmel, A. R. & Jerina, D. M. Kinetics and mechanism of autoprocessing of human immunodeficiency virus type 1 protease from an analog of the Gag-Pol polyprotein. Proc. Natl Acad. Sci. USA 91, 7970–7974 (1994).
Kneller, D. W. et al. Structural, Electronic, and Electrostatic Determinants for Inhibitor Binding to Subsites S1 and S2 in SARS-CoV-2 Main Protease. J. Med. Chem. (2021).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr D. Biol. Crystallogr 69, 1204–1214 (2013).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D. Biol. Crystallogr 67, 235–242 (2011).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl Crystallogr 40, 658–674 (2007).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D. Struct. Biol. 75, 861–877 (2019).
Casanal, A., Lohkamp, B. & Emsley, P. Current developments in Coot for macromolecular model building of Electron Cryo-microscopy and Crystallographic Data. Protein Sci. 29, 1069–1078 (2020).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D. Biol. Crystallogr 66, 12–21 (2010).
Moriarty, N. W., Grosse-Kunstleve, R. W. & Adams, P. D. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr D. Biol. Crystallogr 65, 1074–1080 (2009).
Frisch M. J. et al. Gaussian 16, Revision B.01. Gaussian Inc., Wallingford, CT. (2016).
Acknowledgements
This research used resources at the Spallation Neutron Source, and the High Flux Isotope Reactor, which are DOE Office of Science User Facilities operated by the Oak Ridge National Laboratory. The Office of Biological and Environmental Research supported research at ORNL’s Center for Structural Molecular Biology (CSMB), a DOE Office of Science User Facility. ORNL is managed by UT-Battelle LLC for DOE’s Office of Science, the single largest supporter of basic research in the physical sciences in the United States. We thank John Lloyd and the NIDDK mass spectrometry core facility. This work was supported by the Intramural Research Program of National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), NIH.
Funding
Open Access funding provided by the National Institutes of Health (NIH).
Author information
Authors and Affiliations
Contributions
N.T.N., A.K. and J.M.L. designed the study; N.T.N., A.A. and J.M.L. carried out enzyme kinetics; A.A. and J.M.L. constructed, expressed, and purified proteins; D.W.K. and A.K. crystallized the protein and inhibitor complexes; D.W.K., L.C. and A.K. collected X-ray data, reduced the data and refined the structures; R.G. performed SV-AUC and analyzed data; J.M.L. performed ITC and analyzed data; N.T.N., A.K. and J.M.L. wrote the paper with help from all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Yu Chen and Iñaki Tuñón for their contribution to the peer review of this work. Primary Handling Editors: Min Zhuang, Gene Chong, and David Favero.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aniana, A., Nashed, N.T., Ghirlando, R. et al. Insights into the mechanism of SARS-CoV-2 main protease autocatalytic maturation from model precursors. Commun Biol 6, 1159 (2023). https://doi.org/10.1038/s42003-023-05469-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-023-05469-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
