
Abstract
Aromatic chemicals play indispensable roles in our daily lives, having broad applications in household goods, textiles, healthcare, electronics and automotive, but their production currently relies on fossil resources that have heavy environmental burdens. Synthesis of aromatic chemicals from bio-based resources would be a viable approach to improve their sustainability. However, very few methods are available for achieving this goal. Here we present a strategy to synthesize aromatics from 5-hydroxymethylfurfural (HMF), an organic compound derived from sugars under mild conditions. HMF was first converted in two high-yielding steps into 2,5-dioxohexanal (DOH), a novel C6-compound containing three carbonyl groups. Subsequently, acid-catalysed intramolecular aldol condensation of DOH in the presence of secondary amines selectively produced a range of bio-based 4-dialkylamino substituted phenols and 1,4-di-(dialkylamino)benzenes (Wurster’s blue analogues) in 15–88% yields. In the absence of amines, the industrially important hydroquinone was also synthesized from DOH under acidic conditions. Using a similar approach where 4,5-dioxohexanal was the intermediate, we were also able to prepare catechol, a compound with important industrial applications, from HMF. The proposed approach can pave the way for the production of sustainable aromatic chemicals and move their industrial applications closer to achieving a bioeconomy.
Similar content being viewed by others


Main
Benzenoid aromatics are ubiquitous in human society, with many applications in household goods, textiles, healthcare, electronics, automotive and others. The importance of aromatics in industry is further manifested by the size of the benzene, toluene, xylenes (BTX) market, which was 95 million metric tons per year in 2012 and has been growing at an average of 3% per year1,2. The current production of benzenoid aromatics is still completely dependent on fossil oil, which is associated with a number of environmental problems, in particular the formation of the greenhouse gas CO2. Thus, there is a clear need for new methodology that allows the production of aromatics based on renewable resources. Lignocellulose in the form of wood or agro-waste, as well as its constituents including cellulose, hemicellulose, the carbohydrates that form their building blocks and lignin can be considered as sustainable raw materials. In this Article, we report on a new methodology to produce aromatics from sugars.
Whereas many economic routes for the production of aliphatic chemicals from biomass have been developed over the past decades3,4,5,6, very few high-yielding methods are available for the sustainable production of aromatics such as BTX, phenols and anilines from renewable resources7,8,9.
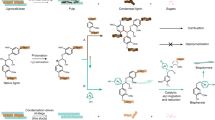
Catalytic pyrolysis of lignocellulosic biomass delivers a mixture of products around 23% of which is aromatics10. Aromatics could in principle also be produced from lignin11,12, but this requires a yet not commercially available clean source of lignin (organosolv); even then, yields of pure products are still rather low13. Aromatization of renewable hydrocarbons such as biogas14 or alcohols15 is another possibility, but here, the main obstacle is the rapid deactivation of the catalyst through coking. Methyl ketones such as acetone can be trimerized to 1,3,5-trisubstituted benzenes via an aldol-based cyclotrimerization reaction16,17,18. Perhaps the most successful approach towards renewable aromatics has been via Diels–Alder (DA) reactions on alkylfurans derived from 5-hydroxymethylfurfural (HMF) or furfural (Fig. 1a)19,20. With all six carbons originating from glucose or fructose, HMF is considered to be an outstanding candidate among the platform chemicals21. A range of aromatic compounds have been synthesized via the DA approach, such as, p-xylene22,23, toluene23, benzene23, phthalic anhydride24 and m-xylylenediamine25. However, DA reactions are highly dependent on the alkyl substituents in the furan ring, hence the yields of p-xylene are quite good, whereas the yields of toluene from 2-methylfuran and benzene from furan are much lower23. Since p-xylene is mainly used as a precursor for terephthalic acid, the initial reduction of the side chains followed by the later oxidation seems rather wasteful; however, the DA reaction either does not work or works very poorly on oxygenated variants of 2,5-dimethylfuran26. Besides, the electron-deficient nature of dienophiles required in the DA reactions limits the scope of accessible aromatic products.

a, DA/dehydration reaction. b, Catalytic pyrolysis. c, Deuss’ method. d, This work. The coloured balls signify different substituents.The dotted boxes signify the essence of this paper.
Bio-based aromatics have also been prepared through catalytic pyrolysis of HMF derivatives, including furan, 2-methylfuran and 2,5-dimethylfuran10,27,28,29,30. However, this method produces a bio-oil containing BTX and other alkylated benzenes and naphthalenes with an overall aromatics yield below 20% (Fig. 1b)10,28,29,30. In 2018, Deuss and co-workers reported the selective formation of 1,2,4-benzenetriol directly from HMF (Fig. 1c)31. At high temperature (400 °C), an aqueous HMF solution produced 1,2,4-benzenetriol at 54% yield under high pressure (>120 bar) in the presence of ZnCl2 as catalyst31. This is a clear improvement from a previously reported methodology32. In 2020, a similar method to produce aromatics from HMF at 300 °C was reported, catalysed by excess acid (>1,000 equivalents of acetic acid (HOAc))33. Besides 51% of 1,2,4-benzenetriol, 4% of hydroquinone was also observed33. Despite the good selectivity, this method suffers from harsh conditions (high temperature and a large excess of HOAc). In addition, the obtained 1,2,4-benzentriol was found to be unstable under air (dimerization) and thus far has not been put to any commercial applications34.
Here we describe the development of a new pathway to bio-based aromatics from HMF via 2,5-dioxohexanal (DOH) as intermediate. In the presence of trifluoroacetic acid (TFA), DOH reacted with secondary amines and produced a series of 4-substituted phenols tolerating various functional groups in high selectivity. When TFA was replaced by HOAc, DOH reacted with two molecules of amine and formed a couple of 1,4-di-(dialkylamino)-substituted benzenes, commonly identified as Wurster’s blue analogues. In the absence of amine, DOH was converted to hydroquinone, an important bulk chemical that has application in photography and as polymerization inhibitor, as anti-oxidant and raw material for dyes, pigments, agrochemicals and pharmaceuticals35,36. Similarly, aldol condensation of 4,5-dioxohexanal (DOA) under acidic condition led to the formation of catechol, a synthetic precursor in the fine chemical industry37. Bio-based catechol can also be prepared by microbial conversion of d-glucose followed by chemical decarboxylation at 190–310 °C at 24–43% yield38. Synthesis of benzenoid aromatics by intramolecular aldol condensation of a C6 fragment is unprecedented.
Results
Following the procedures reported recently by our group39,40, DOH was synthesized from HMF via 1-hydroxy-2,5-hexanedione (HHD) as intermediate through Ir-catalysed hydrogenation followed by Cu(I)-catalysed oxidation in 70% and 93% isolated yields, respectively. It is worth noting that HHD had already been synthesized from HMF at 98% yield41.
The proposed pathway to form aromatics from DOH is shown in Fig. 1d: aldol condensation and concomitant cyclization of DOH leads to intermediate I, which would immediately undergo dehydration to intermediate II. Subsequently, keto-enol tautomerization would result in the formation of hydroquinone (Fig. 1d).
4-Substituted phenols and 1,4-di-(dialkylamino)-benzenes from DOH
Our attempts to cyclize DOH started by screening a range of organic and inorganic bases to effect the intramolecular aldol condensation (Supplementary Table 1). l-Proline (Supplementary Table 2) and thiourea catalysts (Supplementary Table 3) were also tested. In all trials, neither the desired hydroquinone nor cyclic intermediate I (Fig. 1d) was observed. Excitingly, when pyrrolidine 1a was added to the solution of DOH in tetrahydrofuran (THF) at room temperature (Supplementary Table 2, entry 32–33), 1,4-dipyrrolidinylbenzene (2a) was observed in 18% yield along with 2% of 4-pyrrolidinylphenol (3a).
Encouraged by this result, we evaluated various parameters to improve the yield and selectivity towards 2a and 3a (Fig. 2a). Among all the tested solvents, 1,4-dioxane provided the same yields of 2a and 3a as THF (Fig. 2b). When the solvent was replaced by toluene, ether (Et2O), methyl tert-butylether (MTBE), deuterochloroform (CDCl3) or pyridine, the yield of 2a dropped to 3–17% and 3a was produced at only 1–2% yield. Using dimethylformamide (DMF) or dimethylsulfoxide (DMSO) as solvent, the yield of 3a increased to 9 and 5%, respectively, while that of 2a decreased to 8 and 11%, respectively. No desired product was observed with H2O and acetone as reaction medium. We then tested the effects of additives on the reaction of DOH and two equivalents of 1a in THF (Fig. 2c). The total aromatic yield was reduced when either acid or base was added. Interestingly, addition of one equivalent of HOAc to the solution of DOH and two equivalents of 1a in THF at 66 °C improved the yield of 3a and 2a to 9 and 21%, respectively. However, the reaction without any additives at 66 °C did not raise the yield compared with the reaction at room temperature, suggesting that hot HOAc had a positive effect on the reaction. It is interesting to note that addition of one equivalent of TFA altered the selectivity towards 3a.

a, Reaction of DOH and 1a. b, Solvent. Reaction conditions: DOH (0.1 mmol), 1a (2.0 equiv), solvent (0.5 ml), r.t., overnight. c, Additives. Reaction conditions: DOH (0.1 mmol), 1a (2.0 equiv), THF (0.5 ml), additives (1.0 equiv), r.t., overnight. a66 °C; b1,4-dioxane (0.5 ml), 101 °C. d, Amount of 1a with HOAc (1.0 equiv) as catalyst. Reaction conditions: 1st step: 1a, THF (0.2 ml), HOAc (1.0 equiv), r.t., 5 min; 2nd step: DOH (0.1 mmol), THF (0.3 ml), 66 °C, overnight. e, Amount of HOAc. Reaction conditions: 1st step: 1a (6.0 equiv), THF (0.2 ml), HOAc, r.t., 5 min; 2nd step: DOH (0.1 mmol), THF (0.3 ml), 66 °C, overnight. f, Amount of 1a with TFA (1.0 equiv) as catalyst. Reaction conditions: DOH (0.1 mmol), THF (0.5 ml), 1a, TFA (1.0 equiv), 66 °C, overnight. g, Comparison of reaction in THF and 1,4-dioxane. Reaction conditions: DOH (0.1 mmol), solvent (0.5 ml), 1a (6.0 equiv), TFA (1.0 equiv), 66 °C (101 °C using 1,4-dioxane as solvent), overnight.
Inspired by the fact that ammonium salts are known to enhance aldol-type reactions, we modified the reaction procedure and optimized ratios between 1a and HOAc (Fig. 2d,e). A mixture of 1a and HOAc in THF was added to the solution of DOH in THF at 66 °C. After overnight reaction, gas chromatographic (GC) analysis shows higher selectivity towards 2a and a very low yield of 3a (5–10%, Fig. 2d). The yield of 2a increased from 7 to 31% when the amount of 1a was increased from one and a half to four equivalents and slightly improved further to 33% with six equivalents of 1a. Meanwhile, the overall aromatic yield was increased from 17 to 41%. The varying amounts of HOAc from half to four equivalents slightly promoted the yield of 2a from 28 to 37% and 3a from 4 to 18% (Fig. 2e). The total aromatics reached its highest yield (54%) using four equivalents of HOAc. Increasing the amount of HOAc further decreased the total aromatic yields and the selectivity towards 2a. The replacement of THF by 1,4-dioxane and an increase in temperature to 101 °C had no obvious effects on the overall yields (49%) but slightly decreased the selectivity to 2a (35%; Supplementary Table 6, entry 16).
As the result in Fig. 2c suggested that TFA altered the selectivity of this reaction to 3a, we then optimized the reaction conditions by adjusting the amount of 1a and TFA (Fig. 2f,g). Increasing the amount of 1a from one to six equivalents slowly improved the yields of 2a and 3a, where the proportion of 3a to 2a declined drastically (Fig. 2f). Using six equivalents of 1a in THF at 66 °C with one equivalent of TFA maximized the yield of 3a to 41% along with a 25% yield of 2a, providing a 66% total aromatic yield. Switching THF to 1,4-dioxane as reaction medium and increasing the temperature from 66 °C to 101 °C slightly improved the overall aromatic yield (66 to 72%) and dramatically increased the selectivity to 3a (Fig. 2g). When the reaction was performed at 190 °C using diethylene glycol diethyl ether as solvent, an 84% yield of aromatics was produced, consisting of 69% of 3a and 15% of 2a (Supplementary Table 7, entry 20). Decreasing the amount of TFA to 50 mol% and 25 mol% resulted in an aromatics yield of 70% and 69%, respectively (Supplementary Table 7, entries 22 and 25).
On the basis of our results and the known aldol condensation chemistry, we propose a plausible reaction mechanism and computed the reaction energies. All computational details are given in Supplementary Information and the simplified results are shown in Fig. 3. In our computations, we used DOH and 1a as starting materials. Since several acids were found active for the reaction and 1a was used in excess, we used pyrrolidin-1-ium ion (1aH+) as acid. Our results show that the formation of 2a and 3a is highly exergonic by 17.9 and 21.3 kcal mol−1 (Supplementary Information 6), and thus favoured thermodynamically.

The network includes the computed Gibbs free energy (ΔG, in kcal mol−1).
The first step is the nucleophilic addition of 1a to the aldehyde group in DOH under acidic conditions producing intermediate III, which was confirmed by gas chromatography–mass spectrometry (GC–MS) analysis (Supplementary Information 9) and this step is exergonic by 3.0 kcal mol−1 (Supplementary Information 7). The second step is the nucleophilic addition of a second molecule of 1a to either the 5-keto group or the iminium group in intermediate III, resulting in the formation of intermediates IV or IV-iso, respectively. We found that IV-iso is more stable than IV by 12.6 kcal mol−1 (Supplementary Information 8). We also observed isomer IV-iso for the piperidine derivate in the GC–MS analysis (Supplementary Information 9). The detection of intermediates III and IV supports the computed exergonic reaction steps. Dehydration of intermediate IV produces terminal olefin intermediate V and internal olefin intermediate V-iso and the latter is more stable than the former by 9.2 kcal mol−1. We noted that intermediates III, IV and V have gauche-chair conformations due to the intramolecular electrostatic interaction of the termini, and their chain-like conformers are less stable by 6.2, 9.9 and 4.9 kcal mol−1, respectively (Supplementary Information 7 and 8). This interaction also explains the catalytic function of the cyclic amine: the cationic intermediate favours this interaction, which would be much higher in energy in a simple base-catalysed aldol condensation because of the unfavourable cis conformation of the ketoaldehyde. The superior behaviour of TFA can be ascribed to its ability to enhance the formation of intermediates III, IV and V, as well as the cyclization of V due to its strong acidity.
The V-gauche-chair has the appropriate conformation for the C-C coupling and ring closure to produce intermediate VI-chair. Due to intramolecular electrostatic interaction of the termini and the resulting very short C-C distance of 2.423 Å, it is not possible to locate the transition state for the C-C coupling and ring closure. This step, forming VI-chair, is exergonic by 12.7 kcal mol−1 and favourable thermodynamically (Supplementary Information 9). This shows that the intermediate V-gauche-chair, once formed, can be easily transformed to the VI-chair intermediate without any barrier. Under acidic conditions, the VI-chair intermediate releases one molecule of 1a and produces intermediate VII and this step is exergonic by 3.3 kcal mol−1. Subsequent deprotonation of VII by 1a leads to intermediate VIII and this step is endergonic by 3.6 kcal mol−1.
Intermediate VIII can undergo two different transformations (Supplementary Information 10). One is keto-enol tautomerization leading to the desired mono-substituted phenol 3a. This step is exergonic by 14.3 kcal mol−1. The other one is an acid-catalysed reaction with 1a resulting in intermediate IX and this step is exergonic by 12.2 kcal mol−1. The subsequent deprotonation of intermediate IX by 1a forms the desired disubstituted product 2a and this step is exergonic by 5.5 kcal mol−1. Use of strong acid (TFA) enhances the tautomerization and results in high selectivity to 3a.
Figure 3 shows that V-gauche-chair represents the highest energy point on the potential energy surface (Supplementary Fig. 3) and the apparent Gibbs free energy barrier is 8.8 kcal mol−1. It is clear from these calculations that pyrrolidine (1a) plays a double role as both reactant and catalyst.
To explore the synthetic application of this approach to bio-based aromatics, we subjected a series of secondary amines to the optimized conditions (Fig. 4). The reaction between DOH and cyclic amines produced a range of bio-based phenols with 4-dialkylamino substituents tolerating various functional groups, including a hydroxyl group (3c, 3l, 3m), an ester (3j, 3o) and a nitrile (3k). In general, the reaction of DOH with pyrrolidines produced higher yields of aromatics than the reaction with piperidines. In both the reactions with 5-membered and 6-membered cyclic amines, substituents in the 3-position led to higher overall aromatic yields (3c, 3d, 3e, 3i, 3l, 3m) whereas 2-subsitution led to decreased yields (3b) and 4-substitution had little effect (3g, 3j, 3n). We attributed these results to the steric effects of the amines on the nucleophilic addition of DOH. Compounds 3q and 3r, containing a crucial N-phenyl-piperazinyl moiety that has been used in drug molecules (Supplementary Fig. 2)42,43, were also synthesized through this approach. Remarkably, it was also possible to prepare 1,4-dipyrrolidinylbenzenes (2a–d) in high selectivity from DOH and pyrrolidines simply by switching the acid catalyst to 2.0 equivalents of HOAc. These compounds may find application as fluorescence quenchers and liquid crystals as Wurster’s blue analogues44,45.

Reaction conditions: DOH (1.0 mmol), amine (6.0 equiv), TFA (1.0 equiv), 1,4-dioxane (5 ml), 101 °C, overnight, isolated yield. aDOH (1.0 mmol), amine (6.0 equiv), HOAc (2.0 equiv), THF (5 ml), 67 °C, overnight. bNMR yield using 1,4-dinitrobenzene as standard reference. cDOH (0.5 mmol), HOAc (2.0 equiv), 1,4-dioxane (2.5 ml), 101 °C, overnight.
To improve the sustainability of this approach, we examined the recycling of solvent, excess amine and acid used. We performed the reaction of 1.0 mmol DOH and 1a in 1,4-dioxane with 1.0 equivalent of TFA and adjusted the purification procedure by employing Kugelrohr distillation and reverse-phased column chromatography to isolate compound 2a and 3a (Supplementary Fig. 1). Meanwhile, 79% of the remaining pyrrolidine (1a), 99% of TFA and all of the1,4-dioxane were recycled. The recycled mixture was used directly for the next run by adding 1.0 equivalent of DOH and 2.0 equivalents of 1a.
Synthesis of hydroquinone from DOH
Hydroquinone is widely used as a photographic developer, as a polymerization inhibitor, as an anti-oxidant and as a skin-lightening agent35,36. The industrial production of hydroquinone still largely relies on fossil resources, mainly through oxidation of aniline, hydroxylation of phenol and hydroxyperoxidation of p-di-isopropylbenzene. Considering its irreplaceable role in industry, we continued our endeavour towards the production of hydroquinone from renewable DOH under acidic conditions (Table 1). After 26 h of reaction at 100 °C, 5% hydroquinone was detected in the aqueous solution of DOH and 2.0 equivalents of TFA (entry 3). We then investigated solvent effects on the reaction. In general, non-polar solvents gave improved yields of hydroquinone (5–19%, entries 11–14), except cyclohexane (entry 12), perhaps due to its low boiling point. We attributed the relatively low yields of hydroquinone to the poor solubility of DOH and hydroquinone in non-polar solvents and undesired intermolecular reactions. Reducing the concentration of DOH in xylene to 0.01 M improved the yield of hydroquinone to 32% (entry 15). However, further dilution produced lower yields of hydroquinone, even with prolonged reaction times (Supplementary Table 9). High-temperature reactions in an autoclave using H2O as solvent were then investigated (an elevated pressure of N2 was used to make sure all components stay in solution rather than in the gas phase), which showed a positive correlation between the yields of hydroquinone and temperature (entries 20–23). Interestingly, d4-hydroquinone was obtained when D2O replaced H2O as solvent (8%; Supplementary Information S-34–35).
Synthesis of catechol from HMF following the same strategy
To investigate the applicability of this new aldol-cyclization strategy for the syntheses of bio-based aromatics, we prepared DOA, a DOH analogue, through ozonolysis of 2-methyl-2-cyclopenten-1-one (MCO)46. MCO is naturally existing in Panax ginseng and cigarette smoke47 and is also accessible from HMF as reported (Supplementary Information 5)48,49,50. Similar to DOH, DOA was successfully converted to catechol under acidic condition using water as solvent at 100 °C (Supplementary Table 11). Among all the tested acids, 2.0 equivalents of HCl gave the best yield (18%, entry 2). A comparable yield of catechol was also observed when 2.0 equivalents of p-toluene sulfonic acid was used instead of HCl (16%, entry 1). When HCl was substituted by TFA, the yield dropped to 6% (entry 3). Replacement of H2O by 1,4-dioxane or 1,2-diethoxyethane as solvent reduced the yield to 2–3% (entry 14–15). Optimization of the concentration of DOA and the amount of HCl resulted in 34% yield of catechol when the solution of DOA in H2O (0.05 M) was heated at 100 °C for 24 h in the presence of 2.0 equivalents HCl (Supplementary Table 12, entry 7). The practical application of this approach was then evaluated (Fig. 5). After ozonolysis of MCO followed by reductive work-up, the obtained crude DOA was dissolved in H2O and stirred at 100 °C for 24 h in the presence of 2.0 equivalents of HCl (Fig. 5). After flash column chromatography, this two-step reaction afforded catechol in 42% isolated yield.

Discussion
HMF, which is already produced at ton-scale on the basis of either sugars or cellulose, could be converted into the C6 tricarbonyl synthon DOH via an iridium-catalysed hydrogenation to HHD, followed by a highly selective copper-catalysed oxidation in 65% overall yield. This can be further improved to 91% by using the methodology of Fu and co-workers41 for the hydrogenation step. Although DOH seems perfectly set up for a base-catalysed intramolecular aldol condensation reaction, this reaction failed due to the unfavourable cisoid 6-membered transition state, which is too high in free energy. However, DOH reacted with secondary amines at 101 °C, catalysed by 1.0 equivalent of TFA to produce a range of bioderived 4-dialkylamino substituted phenols tolerating various functional groups. Substitution of TFA by HOAc resulted in the production of a number of 1,4-dipyrrolidinylbenzenes from the reaction of DOH and pyrrolidine derivatives. Excess reagents as well as solvents can be recovered and reused.
The reason that the aldol condensation works in the presence of the secondary amine lies in the formation of the cationic iminium intermediate, which is formed from the reaction of the secondary amine with the aldehyde group of DOH, which makes the cisoid cyclic transition state much more favourable. Although the need for the presence of secondary amines is a limitation of the method, it was nevertheless possible to achieve TFA-catalysed ring closure of DOH to hydroquinone at 32% yield using either H2O or xylene as solvent. When H2O was used as solvent, the yield of hydroquinone showed a positive correlation with the temperature (100–265 °C). This result implies that this reaction can possibly be optimized further by applying highly stable heterogeneous acids at even higher temperatures. It was also possible to utilize the intramolecular aldol-condensation strategy to produce catechol from HMF via DOA as intermediate.
Our study clearly shows the potential of sugar-based DOH as sustainable starting point for the production of bio-based aromatics. Further improvements of the methodology seem to be possible via reduction of the number of steps. Indeed, it has already been shown that it is possible to produce a 55% yield of HHD in a single step from fructose in a combined dehydration–hydrogenation reaction using Amberlyst-15 and Pd/C as catalysts51,52.
This new methodology opens the door to the sustainable production of para-disubstituted benzenoid aromatic compounds based on renewable resources.
Methods
All methods used are described in the electronic Supplementary Information.
Materials
HMF was purchased from AVA-Biochem and used directly without further purification. All other commercially available reagents were purchased from Sigma-Aldrich, Strem, TCI and ABCR. Solvents used in reactions were obtained from Acros Organics, or a solvent purification system (MBraun SPS). All solvents used for work-up or column chromatography were purchased from Walter.
Synthesis of DOH from HMF
To a 300 ml Hastelloy autoclave was added 5-HMF (2.5 g, 20.0 mmol), 100 ml phosphate-buffered saline solution (pH 2.49), Ir catalyst (8.0 mg, 0.075 mol%; see Supplementary Information 2 for the structure) and a magnetic stirring bar. The autoclave was flushed with N2 three times, H2 twice and pressurized with 30 bar H2. The reaction was stirred at 140 °C for 1 h (temperature achieved after 30 min). The reaction was allowed to cool down to room temperature and the solvent was removed by evaporation. The crude product was purified through column chromatography (silica gel) using ethyl acetate/cyclohexane (1/1 to 3/1) as eluent to give a yellow solid. Further recrystallization with CH2Cl2 and pentane under −20 °C gave the desired HHD (1.8 g, 70%) as yellow needles. To a 1 l Schlenk flask was added HHD (10.0 mmol, 1.3 g), MS 4 Å (powdered, 2 g) and a magnetic stirring bar. The flask was evacuated for 0.5 h and then flushed with O2 three times. To the flask equipped with an O2 balloon was added dry MeOH (80 ml), Cu(CH3CN)4PF6 (5 mol%, 186 mg) and pyridine (10 mol%, 81 µl). The reaction was stirred at room temperature for 100 min. The reaction mixture was filtered and diluted with 200 ml dichloromethane (DCM). Subsequently, this mixture was washed with 20 ml NaEDTA solution (1% in H2O) followed by 10 ml NaEDTA solution (1% in H2O, prepared by neutralizing 1 g of EDTA in 100 ml H2O with sodium hydroxide). Then the blue-green aqueous solution was counter-extracted using DCM (2 × 50 ml) and the combined organic layers were dried over anhydrous sodium sulfate. Removal of the solvent through evaporation afforded 1.2 g (93%) of a yellowish oil.
Reaction of DOH with pyrrolidine catalysed by TFA in 1,4-dioxane
To a 35 ml pressure tube was added DOH (1.0 mmol, 128 mg), a magnetic stirring bar and 1,4-dioxane (3 ml). To the solution was added the mixture of amine (6.0 equivalents) and TFA (1.0 equivalent, 80 µl) in 1,4-dioxane (2 ml). The reaction was stirred at 101 °C overnight. After cooling down to room temperature and removal of the solvent by evaporation, the reaction mixture was purified by column chromatography (SiO2). Elution with EtOAc/cyclohexane (2/23) gave 35 mg (13%) of 1,4-di(pyrrolidin-1-yl)benzene (2a) as a white solid. Further elution with EtOAc/cyclohexane (3/17) gave 95 mg (58%) of 4-(pyrrolidin-1-yl)phenol (3a) as yellow crystals.
Reaction of DOH with pyrrolidine catalysed by HOAc in THF
To a 4 ml vial was added pyrrolidine (6.0 equivalents, 480 µl), a magnetic stirring bar, THF (2 ml) and HOAc (2.0 equivalents, 120 µl). After stirring at room temperature for 5 min, the mixture was added to the solution of DOH (1.0 mmol, 128 mg) in THF (3 ml) in a 35 ml pressure tube at 66 °C. The reaction was stirred at 66 °C overnight. After cooling down to room temperature, the solvent was evaporated. Column chromatography (SiO2; EtOAc/cyclohexane, 2/23) afforded the desired 1,4-di(pyrrolidin-1-yl)benzene (2a) as a white solid at 42% yield (90 mg). Further elution with EtOAc/cyclohexane (3:17) gave 18 mg (11%) of 4-(pyrrolidin-1-yl)phenol (3a) as yellow crystals.
Synthesis of hydroquinone
To a 35 ml pressure tube was added DOH (2.0 mmol, 256 mg), xylene (10 ml) and a magnetic stirring bar. To the solution was added TFA (2.0 equivalents, 306 µl) and the reaction was stirred at 140 °C for 30 min. After cooling down to room temperature, the solvent was evaporated. Column chromatography (SiO2; EA/cyclohexane, 1/4) afforded hydroquinone as a white solid (26 mg, 12% yield).
Synthesis of catechol
To a 25 ml Schlenk flask was added MCO (1.0 mmol, 98 µl), a magnetic stirring bar, DCM (2.5 ml) and a trace amount of Sudan III. A glass pipette (ozone inlet) was inserted into the Schlenk flask and a tube was connected as a gas outlet. The mixture was cooled down to −78 °C and ozone (generated by an Erwin Sander ozonisator 301.7, flow rate of inlet compressed air was 150 l h−1) was bubbled through the reaction. The reaction was stirred at −78 °C for 5 min until the red reaction solution turned blue. Compressed air was bubbled into the reaction mixture until the solution turned yellow, followed by addition of dimethyl sulfide (200 µl). After stirring at room temperature for 2 h, the reaction mixture was diluted with DCM and washed with brine twice. The organic layer was dried over Na2SO4 and concentrated to around 1 ml. The obtained DOA solution was used directly for the next step. To a 50 ml Schlenk flask was added a magnetic stirring bar, degassed H2O (20 ml) and HCl (37%, 2 equivalents, 200 µl) under Ar. The above solution of DOA was added into the stirred aqueous solution. The reaction was stirred at 100 °C for 24 h. After cooling down to room temperature and removal of all volatiles in vacuo, column chromatography (SiO2; EA/cyclohexane, 1/4) afforded catechol as a white solid (46 mg, 42% yield for two steps).
All other synthetic procedures as well as spectra of products can be found in Supplementary Information.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated or analysed during this study are included in this paper and its Supplementary Information.
References
Franck, H.-G. & Stadelhofer, J. W. Industrial Aromatic Chemistry 1st edn (Springer, 1988).
Bender, M. Global aromatics supply—today and tomorrow. In DGMK Conference: New Technologies and Alternative Feedstocks in Petrochemistry and Refining (2013); https://www.osti.gov/etdeweb/servlets/purl/22176034
Corma, A., Iborra, S. & Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 107, 2411–2502 (2007).
Tuck, C. O., Pérez, E., Horváth, I. T., Sheldon, R. A. & Poliakoff, M. Valorization of biomass: deriving more value from waste. Science 337, 695–699 (2012).
Stadler, B. M., Wulf, C., Werner, T., Tin, S. & de Vries, J. G. Catalytic approaches to monomers for polymers based on renewables. ACS Catal. 9, 8012–8067 (2019).
Mika, L. T., Cséfalvay, E. & Németh, Á. Catalytic conversion of carbohydrates to initial platform chemicals: chemistry and sustainability. Chem. Rev. 118, 505–613 (2018).
Dodds, D. & Humphreys, B. in Catalytic Process Development for Renewable Materials (eds Imhof, P. & van der Waal, J. C.) 183–237 (Wiley, 2013).
Cavani, F., Albonetti, S. & Basile, F. in Chemicals and Fuels from Bio‐Based Building Blocks (eds Cavani, F. et al.) 33–50 (Wiley, 2016).
Nekhaev, A. I. & Maksimov, A. L. Production of aromatic hydrocarbons from biomass. Pet. Chem. 61, 15–34 (2021).
Cheng, Y. T., Jae, J., Shi, J., Fan, W. & Huber, G. W. Production of renewable aromatic compounds by catalytic fast pyrolysis of lignocellulosic biomass with bifunctional Ga/ZSM-5 catalysts. Angew. Chem. Int. Ed. 51, 1387–1390 (2012).
Sun, Z., Fridrich, B., de Santi, A., Elangovan, S. & Barta, K. Bright side of lignin depolymerization: toward new platform chemicals. Chem. Rev. 118, 614–678 (2018).
Liu, X. et al. Recent advances in the catalytic depolymerization of lignin towards phenolic chemicals: a review. ChemSusChem 13, 4296–4317 (2020).
Liao, Y. et al. A sustainable wood biorefinery for low-carbon footprint chemicals production. Science 367, 1385–1390 (2020).
Spivey, J. J. & Hutchings, G. Catalytic aromatization of methane. Chem. Soc. Rev. 43, 792–803 (2014).
Ni, Y. et al. High selective and stable performance of catalytic aromatization of alcohols and ethers over La/Zn/HZSM-5 catalysts. J. Ind. Eng. Chem. 16, 503–505 (2010).
Adams, R., Hufferd, R. W., Clarke, H. T. & Hartman, W. W. Mesitylene. Org. Synth. 2, 41 (1922).
Wu, Z. et al. Selective conversion of acetone to mesitylene over tantalum phosphate catalysts. Chem. Commun. 58, 2862–2865 (2022).
Reif, P., Gupta, N. K. & Rose, M. Liquid phase aromatization of bio-based ketones over a stable solid acid catalyst under batch and continuous flow conditions. Catal. Commun. 163, 106402 (2022).
Kucherov, F. A., Romashov, L. V., Galkin, K. I. & Ananikov, V. P. Chemical transformations of biomass-derived C6-furanic platform chemicals for sustainable energy research, materials science, and synthetic building blocks. ACS Sustain. Chem. Eng. 6, 8064–8092 (2018).
Kucherov, F. A., Romashov, L. V., Averochkin, G. M. & Ananikov, V. P. Biobased C6-furans in organic synthesis and industry: cycloaddition chemistry as a key approach to aromatic building blocks. ACS Sustain. Chem. Eng. 9, 3011–3042 (2021).
van Putten, R.-J. et al. Hydroxymethylfurfural, a versatile platform chemical made from renewable resources. Chem. Rev. 113, 1499–1597 (2013).
Teixeira, I. F. et al. From biomass-derived furans to aromatics with ethanol over zeolite. Angew. Chem. Int. Ed. 55, 13061–13066 (2016).
Chang, C.-C., Green, S. K., Williams, C. L., Dauenhauer, P. J. & Fan, W. Ultra-selective cycloaddition of dimethylfuran for renewable p-xylene with H-BEA. Green Chem. 16, 585–588 (2014).
Thiyagarajan, S. et al. A facile solid-phase route to renewable aromatic chemicals from biobased furanics. Angew. Chem. Int. Ed. 55, 1368–1371 (2016).
Scodeller, I. et al. Synthesis of renewable meta-xylylenediamine from biomass-derived furfural. Angew. Chem. Int. Ed. 57, 10510–10514 (2018).
Pacheco, J. J. & Davis, M. E. Synthesis of terephthalic acid via Diels–Alder reactions with ethylene and oxidized variants of 5-hydroxymethylfurfural. Proc. Natl Acad. Sci. USA 111, 8363–8367 (2014).
Cheng, Y.-T., Wang, Z., Gilbert, C. J., Fan, W. & Huber, G. W. Production of p-xylene from biomass by catalytic fast pyrolysis using ZSM-5 catalysts with reduced pore openings. Angew. Chem. Int. Ed. 51, 11097–11100 (2012).
Uslamin, E. A., Kosinov, N. A., Pidko, E. A. & Hensen, E. J. M. Catalytic conversion of furanic compounds over Ga-modified ZSM-5 zeolites as a route to biomass-derived aromatics. Green Chem. 20, 3818–3827 (2018).
Zheng, A. et al. Maximum synergistic effect in the coupling conversion of bio-derived furans and methanol over ZSM-5 for enhancing aromatic production. Green Chem. 16, 2580–2586 (2014).
Cheng, Y.-T. & Huber, G. W. Production of targeted aromatics by using Diels–Alder classes of reactions with furans and olefins over ZSM-5. Green Chem. 14, 3114–3125 (2012).
Kumalaputri, A. J., Randolph, C., Otten, E., Heeres, H. J. & Deuss, P. J. Lewis acid catalyzed conversion of 5-hydroxymethylfurfural to 1,2,4-benzenetriol, an overlooked biobased compound. ACS Sustain. Chem. Eng. 6, 3419–3425 (2018).
Luijkx, G. C. A., van Rantwijk, F. & van Bekkum, H. Hydrothermal formation of 1,2,4-benzenetriol from 5-hydroxymethyl-2-furaldehyde and d-fructose. Carbohydr. Res. 242, 131–139 (1993).
Cai, T. et al. Synthesis of renewable C–C cyclic compounds and high-density biofuels using 5-hydromethylfurfural as a reactant. Green Chem. 22, 2468–2473 (2020).
Randolph, C. et al. Biobased chemicals: 1,2,4-benzenetriol, selective deuteration and dimerization to bifunctional aromatic compounds. Org. Process Res. Dev. 22, 1663–1671 (2018).
Hudnall, P. M. in Ullmann’s Encyclopedia of Industrial Chemistry 473–480 (Wiley, 2000).
Krumenacker, L., Costantini, M., Pontal, P. & Sentenac, J. in Kirk‐Othmer Encyclopedia of Chemical Technology (Wiley, 2000).
Fiege, H. et al. in Ullmann’s Encyclopedia of Industrial Chemistry 521–582 (Wiley, 2000).
Li, W., Xie, D. & Frost, J. W. Benzene-free synthesis of catechol: interfacing microbial and chemical catalysis. J. Am. Chem. Soc. 127, 2874–2882 (2005).
Zheng, S., Smit, W., Spannenberg, A., Tin, S. & de Vries, J. G. Synthesis of α-keto aldehydes via selective Cu(i)-catalyzed oxidation of α-hydroxy ketones. Chem. Commun. 58, 4639–4642 (2022).
Wozniak, B., Spannenberg, A., Li, Y., Hinze, S., & de Vries, J. G. Cyclopentanone derivatives from 5-hydroxymethylfurfural via 1-hydroxyhexane-2,5-dione as intermediate. Chem. Sus. Chem. 11, 356–359 (2018).
Wu, W.-P. et al. Selective conversion of 5-hydroxymethylfuraldehyde using Cp*Ir catalysts in aqueous formate buffer solution. Chem. Sus. Chem. 9, 1209–1215 (2016).
Wen, J., Chennamadhavuni, D., Morel, S. R. & Hadden, M. K. Truncated itraconazole analogues exhibiting potent anti-hedgehog activity and improved drug-like properties. ACS Med. Chem. Lett. 10, 1290–1295 (2019).
Romanelli, M. N. et al. Structure−affinity relationships of a unique nicotinic ligand: N-dimethyl-N4-phenylpiperazinium iodide (DMPP). J. Med. Chem. 44, 3946–3955 (2001).
Liu, P. et al. 3,4-difluoropyrrole-, 3,3,4,4-tetrafluoropyrrolidine- and pyrrolidine-based liquid crystals. J. Fluor. Chem. 156, 327–332 (2013).
Grilj, J., Laricheva, E. N., Olivucci, M. & Vauthey, E. Fluorescence of radical ions in liquid solution: Wurster’s Blue as a case study. Angew. Chem. Int. Ed. 50, 4496–4498 (2011).
Starkenmann, C., Niclass, Y., Vuichoud, B., Schweizer, S. & He, X.-F. Occurrence of 2-acetyl-1-pyrroline and its nonvolatile precursors in celtuce (Lactuca sativa L. var. augustana). J. Agric. Food Chem. 67, 11710–11717 (2019).
Abd El-Aty, A. M., Kim, I.-K., Kim, M.-R., Lee, C. & Shim, J.-H. Determination of volatile organic compounds generated from fresh, white and red Panax ginseng (C. A. Meyer) using a direct sample injection technique. Biomed. Chromatogr. 22, 556–562 (2008).
Buntara, T. et al. Caprolactam from renewable resources: catalytic conversion of 5-hydroxymethylfurfural into caprolactone. Angew. Chem. Int. Ed. 50, 7083–7087 (2011).
Xie, Z.-Y., Deng, J. & Fu, Y. W(OTf)6-catalyzed synthesis of γ-lactones by ring contraction of macrolides or ring closing of terminal hydroxyfatty acids in ionic liquid. ChemSusChem 11, 2332–2339 (2018).
Yamazaki, T., Nakai, M. & Kuroki, Y. Process for preparing cyclopentenone derivatives. US patent US3953514A (1974).
Liu, F. et al. Combination of Pd/C and Amberlyst-15 in a single reactor for the acid/hydrogenating catalytic conversion of carbohydrates to 5-hydroxy-2,5-hexanedione. Green Chem. 16, 4110–4114 (2014).
Liu, F. et al. Palladium/carbon dioxide cooperative catalysis for the production of diketone derivatives from carbohydrates. ChemSusChem 7, 2089–2093 (2014).
Acknowledgements
This project was executed as part of the Leibniz Science Campus ComBioCat, with financial support from the Leibniz Association under the auspices of their Strategic Networking Funding Program. We thank the LIKAT analytical department for help. Z.W. was supported by the National Natural Science Foundation of China (22202123).
Funding
Open access funding provided by Leibniz-Institut für Katalyse e.V. (LIKAT Rostock).
Author information
Authors and Affiliations
Contributions
S.Z. conducted the condition optimization, substrate scopes, data analysis and paper preparation. J.G.d.V. wrote the proposal. J.G.d.V. and S.T. directed the project, confirmed the data analysis and revised the paper. B.W. performed initial experiments. F.K. and E.B. provided suggestions regarding data analysis and revised the paper. Z.W. and H.J. carried out DFT computations and revised the paper and supplementary materials. All authors approved the final version for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Sustainability thanks Francesco Mauriello, Marcus Rose, Guoyong Song and Fei Wei for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
1. General remarks. 2. Synthesis of DOH from HMF. 3. Base-catalysed intramolecular aldol condensation of DOH. 4. Proline and derivatives-catalysed intramolecular aldol condensation of DOH. 5. Thiourea catalysed intramolecular aldol condensation of DOH. 6. Condition optimization for 1,4-dipyrrolidinylbenzene (2a) and 4-pyrrolidinylphenol (3a). 7. General procedures for the reactions of DOH and amines. 8. Mechanism studies. 9. Drug molecules containing the N-phenylpiperazinyl moiety. 10. Acid-catalysed intramolecular aldol condensation of DOH. 11. Condition optimization for the conversion of DOA to catechol. 12. Computational details. Appendix I. NMR, GC–MS, IR spectra. References.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zheng, S., Wei, Z., Wozniak, B. et al. Synthesis of valuable benzenoid aromatics from bioderived feedstock. Nat Sustain 6, 1436–1445 (2023). https://doi.org/10.1038/s41893-023-01190-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41893-023-01190-w
This article is cited by
-
A pathway to bio-based aromatics
Nature Sustainability (2023)
