
Abstract
Gliomas are the most common primary brain tumor and are uniformly lethal. Despite significant advancements in understanding the genetic landscape of gliomas, standard-of-care has remained largely unchanged. Subsets of gliomas are defined by gain-of-function mutations in the metabolic genes encoding isocitrate dehydrogenase (IDH). Efforts to exploit mutant IDH activity and/or directly inhibit it with mutant IDH inhibitors have been the focus of over a decade of research. The recently published INDIGO trial, demonstrating the benefit of the mutant IDH inhibitor vorasidenib in patients with low-grade IDH-mutant gliomas, introduces a new era of precision medicine in brain tumors that is poised to change standard-of-care. In this review, we highlight and contextualize the results of the INDIGO trial and introduce key questions whose answers will guide how mutant IDH inhibitors may be used in the clinic. We discuss possible combination therapies with mutant IDH inhibition and future directions for clinical and translational research.
Similar content being viewed by others
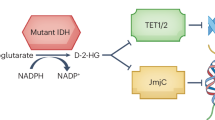


Gliomas are the most common primary malignant brain tumor. Until recently, glioma subclasses had been exclusively defined based on histological subtype and grade (2–4)1. In the past two decades, our understanding of glioma biology has deepened substantially, driven largely by the discovery of predictive and prognostic mutations recurrently observed in gliomas. Mutations in genes encoding isocitrate dehydrogenase (IDH) metabolic enzymes have been described for well over a decade2 and are now formally incorporated into the World Health Organization diagnostic criteria for brain tumors3. IDH mutations are a defining feature of oligodendrogliomas and subtypes of astrocytoma, with the additional presence of 1p19q chromosomal codeletion further distinguishing oligodendrogliomas (1p/19q codeleted) from IDH-mutant astrocytomas (1p/19q intact).
Wild-type IDH enzymes exist in three isoforms, IDH1, IDH2, and IDH34,5. All isoforms convert isocitrate to 2-oxoglutarate (2OG), using either NADP+ (IDH1/2) or NAD+(IDH3) as a cofactor. The conversion of isocitrate to 2OG is a critical step in the Krebs cycle and additionally serves as a cellular source of NADPH/NADH. 2OG also serves as a substrate for families of 2OG-dependent enzymes, including dioxygenases, dehydrogenases, and transaminases4. The majority of IDH mutations in gliomas affect IDH1, the most common of which results in an arginine to histidine substitution (IDH1-R132H)6. The mutant IDH1 (mIDH1) IDH1-R132H protein converts 2OG to the oncometabolite (R)-2-hydroxyglutarate [(R)-2HG)]4,6. Due to the structural similarity between (R)-2HG and 2OG, (R)-2HG competitively inhibits 2OG-dependent enzymes, including 2OG-dependent dioxygenases that affect DNA and histone methylation. Indeed, IDH-mutant gliomas exhibit a distinct hypermethylation pattern called the glioma CpG island methylator phenotype (G-CIMP)7,8,9, underscoring the unique epigenotype of these tumors.
Standard-of-care treatment for IDH-mutant gliomas involves chemotherapy regimens and local therapies that have been used for over 20 years. The recently published INDIGO (Investigating Vorasidenib in Glioma) trial10, demonstrating a clinical benefit from the mIDH1/2 inhibitor vorasidenib, marks a forthcoming shift in this paradigm. In this review, we outline current standard-of-care, examine the INDIGO trial and its implications, and discuss clinical and translational questions that are critical next steps to applying results from INDIGO to management of patients with IDH-mutant glioma.
Current standard-of-care
Standard care for IDH-mutant glioma incorporates surgery, radiation (RT), and/or chemotherapy. Treatment begins with maximal safe resection, which reduces symptoms due to mass effect and allows for tissue sampling for molecular and histopathological analysis11,12,13. Extent of resection is also thought to be prognostic, with increased extent of resection demonstrating an association with improved overall survival12,13,14,15,16. However, due to the highly diffuse nature of gliomas (both IDH-mutant and IDH-wild-type), adjuvant treatment is often required6,17,18.
Among patients with grade 2 IDH-mutant gliomas, adjuvant treatment is determined by risk group, with low-risk patients typically being defined as age ≤40 with gross total resection19,20. Patients deemed low-risk may undergo a “watch and wait” approach, where RT and chemotherapy can be deferred until tumor progression occurs6,21, while high-risk patients (age >40 and/or subtotal resection) often receive adjuvant RT with chemotherapy (PCV [procarbazine, lomustine, and vincristine] or TMZ), though in select cases may be candidates for “watch and wait”19,20,22,23.
For patients with grade 3 IDH-mutant glioma, adjuvant therapy with RT followed by TMZ or PCV is generally used for all patients. Institutional practice varies regarding choice of TMZ or PCV due to (1) an often greater toxicity profile with PCV and (2) lack of modern randomized clinical trial data directly comparing efficacy of TMZ vs. PCV in this setting. Currently, 1p/19q status often informs adjuvant treatment regimen choice in grade 3 IDH-mutant gliomas, with 1p/19q codeleted patients receiving RT + PCV and 1p/19q noncodeleted patients receiving RT + TMZ6,18,24,25,26. This is supported by recent data from the French POLA network demonstrating that among patients with grade 3 oligodendroglioma, RT + PCV was associated with significantly improved 5-year and 10-year overall survival compared to RT + TMZ27. The ongoing and redesigned28 CODEL trial directly compares adjuvant RT + TMZ and RT + PCV among patients with grade 2–3 oligodendroglioma and will provide further insight into this question. For patients with grade 3 IDH-mutant astrocytoma, data from the CATNON trial support use of RT and adjuvant TMZ29.
Given concerns regarding potential long-term toxicities from RT and a desire to reserve RT as an effective salvage option, there is ongoing interest in whether chemotherapy alone (without RT) can be used as upfront treatment in select patients. The ongoing POLCA (NCT02444000) and NOA-18 trials (NCT05331521) are exploring the omission of RT, using PCV (POLCA) or CCNU + TMZ (NOA-18) without concurrent RT.
Following inevitable tumor progression, the options for salvage therapy are largely the same: surgery, RT, and alkylating chemotherapy. As in the upfront setting, each of these therapies have short and long-term toxicity profiles, the latter of which are particularly relevant in this disease where many patients are young at diagnosis and survive long enough to experience long-term toxicities. Importantly, the long disease course of low-grade gliomas can render it challenging to study long-term toxicities from RT. Severity of such toxicities following use of modern radiation techniques is not well-characterized and warrants additional study.
The INDIGO trial
Early-phase studies and the INDIGO trial
Optimal upfront treatment strategies for IDH-mutant glioma have largely centered on use and sequence of the above discussed modalities: surgery, RT, and/or alkylating chemotherapy. While targeted therapies have become standard-of-care in other molecularly-defined cancers (such as EGFR-mutant lung cancers, HER2-positive cancers, and even other IDH-mutant cancers), drugs that target supposed driver mutations in glioma had not demonstrated clinically significant benefits until recently. Indeed, ivosidenib and enasidenib, inhibitors of mIDH1 and mIDH2, respectively, have been approved by the United States Food and Drug Administration for treatment of either IDH-mutant leukemias (enasidenib, ivosidenib) and/or IDH-mutant cholangiocarcinomas (ivosidenib), raising the question as to whether mIDH inhibitors may also demonstrate efficacy in IDH-mutant gliomas.
As such, multiple prospective clinical trials have tested mIDH inhibitors in gliomas. Response assessment in these studies was performed according to Response Assessment in Neuro-Oncology (RANO) criteria, with contrast-enhancing tumors utilizing RANO high-grade glioma guidelines30 and non-enhancing tumors using RANO low-grade glioma guidelines31. Ivosidenib, a selective mIDH1 inhibitor, was tested in a phase I trial that enrolled IDH1-mutant glioma patients who had recurred or not responded to standard-of-care therapy32. Although this phase I study is not powered for efficacy, disease control outcomes were reported as secondary endpoints. Patients with contrast-enhancing tumors (associated with more aggressive disease) had a 0% response rate (0/31) with 54.8% progressing on treatment. In contrast, among patients with non-enhancing tumors, one partial response (1/35, 2.9%) and an overall higher rate of stable disease (30/35, 85.7%) were observed. However, inferring efficacy of mIDH inhibition from these data are limited by the fact that ivosidenib has modest blood-brain barrier penetration33. Subsequent trials have since tested the dual mIDH1/2 inhibitor vorasidenib34, which is more CNS-penetrant and exhibits a significantly higher tumor:plasma ratio in IDH-mutant glioma patients compared to ivosidenib (1.69 vs. 0.10)35. Like ivosidenib, vorasidenib was also first tested in a phase I trial that enrolled patients with recurrent or progressive IDH-mutant gliomas36. While patients with contrast-enhancing disease still responded poorly to vorasidenib (0% response rate and 40% with progressive disease), patients with non-enhancing gliomas exhibited 18.2% (4/22) response rate and 72.7% (16/22) stable disease. When compared directly within the same phase I perioperative study35, vorasidenib demonstrated less variable 2HG suppression compared to ivosidenib and was selected for subsequent phase III testing.
Taken together, the early-phase trials suggested that mIDH inhibitors may be best employed in the earlier, more indolent disease setting, and that the role of mIDH inhibitor monotherapy among patients with advanced, contrast-enhancing disease is limited. The lack of efficacy in the contrast-enhancing disease setting may be due to acquisition of additional drivers in more advanced disease, and/or a “hit-and-run” effect of mIDH37. Nevertheless, these trials set the stage for the INDIGO trial, which aimed to test efficacy of vorasidenib in upfront treatment of low-grade gliomas. The INDIGO trial enrolled patients with residual or recurrent grade 2 IDH-mutant oligodendroglioma or astrocytoma, who had not had any prior therapy other than surgery (1–5 years prior to randomization)10. Residual/recurrent disease was defined as ≥1 target lesion measuring ≥1 cm by ≥1 cm in longest dimensions. Importantly, patients were asymptomatic, not being treated with steroids for symptoms due to glioma, and had either minimal or nonnodular enhancement on magnetic resonance imaging (MRI) scans. Patients were randomized to vorasidenib or placebo control. The primary endpoint of the trial was progression-free survival (PFS), with a secondary endpoint being time to next intervention, defined as initiation of next anticancer treatment.
At a median follow-up of 14.2 months, median PFS was significantly improved with vorasidenib (27.7 months) compared to placebo (11.1 months) (hazard ratio [HR] 0.39, 95% CI 0.27–0.56, p < 0.001). Time to next intervention was also significantly improved with vorasidenib (HR 0.26, 95% CI 0.15–0.43, p < 0.001). Toxicities were mild, with grade ≥3 elevations in alanine aminotransferase (ALT) observed in 9.6% of patients receiving vorasidenib and 0% patients receiving placebo. Frequency of seizures was comparable between vorasidenib and placebo arms, with no clinically meaningful differences in patient-reported health-related quality of life38.
The INDIGO trial represents a step forward in advancing the treatment of IDH-mutant gliomas. In a disease where systemic therapy regimens largely rely on alkylating agent chemotherapies that lack tumor specificity, the INDIGO trial is remarkable in its use of a molecularly-guided targeted therapy and helps transition glioma treatment into the era of precision medicine. Furthermore, the results from INDIGO also clearly demonstrate the role of mIDH and (R)-2HG as drivers in low-grade, non-enhancing IDH-mutant gliomas. These results introduce a new treatment paradigm in which candidates for a “watch and wait” strategy in the pre-INDIGO era (and perhaps others beyond these criteria) may now be considered for mIDH inhibitor monotherapy.
With this said, it is important to acknowledge the limitations of the current data. First, it will be important to clarify whether the current PFS benefit translates to meaningful differences in survival outcomes. Survival data may be difficult to interpret given that 31.9% of patients in the placebo arm crossed over to vorasidenib treatment at progression. If no survival benefit is observed in the vorasidenib arm, it will be difficult to know whether this is a true lack of effect, or whether this is confounded by crossover. In addition, it is not known the extent to which second-line therapies may effectively salvage patients who progress in the control arm. In the absence of survival data, deferring potential toxicities of salvage treatment (including RT) is arguably a clinically meaningful endpoint in and of itself39, though there is a lack of data quantifying the degree of toxicities from brain-directed RT in the modern era. Extrapolating toxicities of RT employed for other brain tumors40,41,42 is confounded by different dose regimens, treatment fields, and patient demographics. Second, the INDIGO trial demonstrated a PFS benefit of vorasidenib compared to patients treated with placebo, though it is unknown how efficacy of vorasidenib compares to RT + PCV or RT + TMZ, which is often used adjuvantly as standard-of-care among patients who do not undergo observation after surgery6,18.
Other mutant IDH inhibitors
In addition to vorasidenib and ivosidenib, additional mIDH inhibitors have demonstrated ability to reduce brain tumor levels of 2HG and may be similarly studied in future phase 3 trials. The brain-penetrant mIDH1 inhibitor olutasidenib showed stable disease as a best response in 40% of recurrent/progressive IDH1-mutant glioma patients in a phase Ib/II trial43. Results from an ongoing phase I clinical trial investigating safusidenib (DS-1001b), another brain-penetrant mIDH1 inhibitor, demonstrated objective response rates of 17.1% and 33.3% in contrast-enhancing and non-enhancing tumors, respectively44. Safusidenib is currently undergoing additional clinical testing in the upfront setting (NCT04458272) and in the recurrent/progressive setting (NCT05303519). The mIDH1 inhibitor BAY143603245 was well-tolerated and demonstrated a response rate (complete or partial) of 11.4% in patients with IDH-mutant low-grade glioma in a phase I trial46. Further randomized clinical trials may lead to inclusion of these drugs as additional options to treat patients with IDH-mutant glioma.
Response heterogeneity and resistance to mutant IDH inhibition
As with any impactful clinical study, the INDIGO trial prompts many follow-up questions that will shape how mIDH inhibitors are used to treat gliomas. First, who should receive mIDH inhibitor therapy? The INDIGO trial enrolled a highly selected patient population with a very favorable disease profile: non-enhancing, grade 2 tumors (52.0% with 1p/19q codeletion) with no prior RT or chemotherapy and who would have otherwise been candidates for “watch and wait.” However, it is unclear whether patients who do not meet these narrow eligibility criteria will also benefit from mIDH inhibitors. This is especially relevant given that mIDH inhibition is clearly not an efficacious treatment strategy in all IDH-mutant glioma patients. Patients with aggressive, contrast-enhancing disease do not appear to benefit from vorasidenib36. How should mIDH inhibitors be used in the many patients whose disease risk profile falls between INDIGO enrollment criteria and contrast-enhancing disease? What clinical or molecular biomarkers may help identify patients who are most likely to respond and benefit from mIDH inhibitors? Existing clinical data suggest that grade 2 and grade 3 patients may have similar clinical outcomes, as evidenced by Reuss et al., who reported median overall survival times for patients with grade 2 and grade 3 IDH-mutant glioma across three separate clinical cohorts as 10.9 years and 9.3 years, respectively47. This observation was also corroborated in an independent Swedish cohort of patients, where comparable overall survival was observed among grade 2 and grade 3 IDH-mutant glioma patients without known CDKN2A/B deletions (11.4 years and 10.9 years, respectively)48. These data may support use of mIDH inhibitors in patients with grade 3 disease, though it is difficult to assess in the absence of randomized clinical data. Whether comparable survival between patients with grade 2 and grade 3 IDH-mutant tumors will translate to similar clinical benefit from mIDH inhibitor therapy is unknown. Furthermore, it is unclear whether vorasidenib monotherapy improves outcomes compared to adjuvant chemotherapy and RT, or whether combining mIDH inhibitors with adjuvant RT+chemotherapy would be superior to RT+chemotherapy alone.
Part of the difficulty in identifying patients who will respond to mIDH inhibitor treatment stems from the fact that it is unknown which of the many downstream effects of mIDH are reversed with mIDH inhibition, whether that reversibility changes over the course of the glioma life cycle, and whether additional driver mutations predominate in later stage disease. Few preclinical models exist that can be used to study oncogenic mechanisms of IDH mutations in the context of grade 2–3 disease49,50. The indolent nature of low-grade IDH-mutant gliomas renders these tumors difficult to generate patient-derived cell lines that proliferate in vitro or in xenograft models, and engineered approaches that utilize exogenous mutant IDH expression are limited in their ability to fully capture the genetic and epigenetic landscape observed in human tumors. As the microenvironmental effects of mutant IDH are becoming more understood, it also increases the importance and presents challenges for researchers to develop in vivo models generated in immune-intact (ideally, native immune) systems. Recent advances such as patient-derived organoids51, genetically engineered models of IDH-mutant glioma52,53,54,55, and assessment of samples acquired from patients treated with mIDH inhibitors56 may be helpful in this regard. Analysis from patients in the INDIGO trial may also provide insight in identifying molecular changes that correlate with response or resistance to vorasidenib.
A related question to understanding mechanisms of sensitivity is how IDH-mutant gliomas acquire resistance to mIDH inhibition (Fig. 1). Given the 28% progression rate reported in vorasidenib-treated patients enrolled in INDIGO, clinicians will increasingly need to decide how to employ salvage therapy for patients who have progressed on mIDH inhibitors. Part of this challenge is a lack of current understanding as to how prolonged mIDH inhibition alters the biology of IDH-mutant gliomas by time of progression. Do patients progress while on vorasidenib with high (R)-2HG, rendering them potential candidates for synthetic lethal strategies that exploit ongoing presence of (R)-2HG? Or do patients progress on vorasidenib with low (R)-2HG, indicating that salvage combination treatments that do not rely on elevated (R)-2HG for efficacy should be prioritized? It may also be the case that tumors progress during vorasidenib treatment with low (R)-2HG, but revert to a high (R)-2HG state upon cessation of mIDH inhibitor treatment. If so, then the kinetics of (R)-2HG re-accumulation in this setting are especially important in dictating choice and timing of salvage therapies. Given associations between IDH mutations and DNA damage deficits57, how do (R)-2HG levels impact efficacy of common salvage therapies such as RT?

Left: Role of mutant IDH in untreated glioma tumors. Collateral vulnerabilities include druggable synthetic lethal targets of mutant IDH. Middle: Schematic of mutant IDH inhibitor treatment in IDH-mutant gliomas responsive to mutant IDH inhibition. It is unknown whether strategies that target collateral vulnerabilities conferred by mutant IDH may still be utilized effectively in these settings. Right: IDH-mutant gliomas with de novo or acquired resistance to mutant IDH inhibitors. As in mutant IDH inhibitor-sensitive IDH-mutant gliomas, tumors resistant to mutant IDH inhibition may be candidates for therapies that target collateral vulnerabilities, depending on the mechanism of action.
Understanding these mechanisms of resistance are not only important for guidance of existing salvage treatment options, but may also reveal novel druggable targets for second-line therapies. If acquired resistance is driven by secondary IDH mutations that allow tumors to maintain (R)-2HG production (Fig. 1), drugs that block canonical and acquired IDH mutations may be beneficial, akin to secondary T790M EGFR mutations observed following tyrosine kinase inhibitor (TKI) treatment in EGFR-mutant lung cancer58. Such “(R)-2HG-restoring” mechanisms of resistance have been described in IDH-mutant leukemia patients treated with ivosidenib or enasidenib and in IDH-mutant cholangiocarcinomas59,60,61,62,63,64. However, it is also possible that resistance to mIDH inhibition in glioma may be mediated through (R)-2HG-independent mechanisms. Prolonged mIDH inhibition may select for silencing or activation of one or more downstream (R)-2HG effectors, akin to MET-amplification as an alternative mechanism of resistance to TKIs in EGFR-mutant lung cancer65. These are critical questions to address, particularly as the population of mIDH inhibitor-resistant IDH-mutant glioma patients is expected to substantially increase as vorasidenib (and perhaps other mIDH inhibitors) becomes increasingly utilized in the clinic.
Additional emerging treatment strategies for IDH-mutant gliomas
Unfortunately, despite resection and standard-of-care chemotherapy and RT, progression is inevitable, which is likely to remain true even as longer-term data from the INDIGO trial are reported. Several ongoing efforts to translate novel therapeutic strategies from preclinical work are outlined below and continue to be relevant. There is additionally a pressing need to understand how these therapies may be best employed either with concurrent mIDH inhibitor treatment, or as salvage in the mIDH inhibitor-resistant setting.
PARP inhibitors
(R)-2HG has been shown to impair homology-dependent DNA repair due to inhibition of 2OG-dependent enzymes involved in homologous recombination66,67. IDH mutations are thus thought to confer a “BRCA-like” defect that renders IDH-mutant tumors sensitive to poly(ADP-ribose) polymerase (PARP) inhibitors. Of note, this mechanistic model has been recently challenged by data suggesting that sensitivity of IDH-mutant tumors to PARP inhibition may instead be due to mIDH-dependent heterochromatin formation and resultant replication stress68. Sensitivity to PARP inhibitors may be further enhanced in IDH-mutant gliomas by impairments in NAD+ metabolism69,70. Currently, PARP inhibitors are being tested in early-phase clinical trials in IDH-mutant gliomas6. Results thus far from monotherapy testing in the “Using Olaparib in Recurrent IDH-mutant Glioma” (OLAGLI) trial have demonstrated limited success with olaparib alone71, prompting interest in results from ongoing trials using PARP inhibitors in combination with agents such as TMZ (NCT03914742) or immunotherapy (NCT03991832)6. Importantly, depletion of (R)-2HG with IDH inhibitor treatment reversed sensitivity to PARP inhibitors67, suggesting that PARP inhibitors may have decreased efficacy when used in combination with mIDH inhibitors or in tumors that progress on mIDH inhibitors with low (R)-2HG.
CDK inhibitors
Homozygous deletion of the CDKN2A/B tumor suppressor confers a particularly poor prognosis in IDH-mutant gliomas72 and is now formally incorporated into WHO diagnostic criteria3. The p16INK4a protein encoded by CDKN2A normally functions to inhibit cyclin-dependent kinases 4 and 6 (CDK4/6). Loss of CDKN2A therefore increases CDK4/6 activity and can cause dysregulation in downstream targets of CDK4/6, including the tumor suppressor Rb. CDK4/6 inhibition is thus an appealing strategy in CDKN2A/B-deleted tumors and has been tested in other solid tumors with CDKN2A alterations73,74. Phase II trials are currently underway testing the CDK4/6 inhibitors palbociclib (NCT02530320) and abemaciclib (NCT03220646) in IDH-mutant oligodendrogliomas.
Demethylating agents
Given that (R)-2HG competitively inhibits 2OG-dependent enzymes, many of which contribute to DNA and histone demethylation, there has been interest in whether methyltransferase inhibitors may be useful in treating IDH-mutant gliomas. Indeed, preclinical data show that the DNA methyltransferase inhibitor 5-azacytidine can reverse upregulation of the PDGFRA oncogene driven by IDH mutations75. One of the challenges associated with this strategy is that DNA hypermethylation patterns caused by mIDH7,8,9 include silencing of many genes, and it is not known (1) which of the locus-specific hypermethylation events are reversible, and (2) whether reversal of hypermethylation will be effective in halting tumor growth in patients, as it is in mouse models76,77. Results from ongoing trials testing the DNA methyltransferase inhibitors decitabine and 5-azacytidine in recurrent IDH-mutant gliomas (NCT03922555 and NCT03666559, respectively) will be helpful in this regard.
Metabolic pathway inhibitors
Additional therapeutic strategies in various stages of clinical translation have leveraged the metabolic dependencies conferred by mIDH to selectively target IDH-mutant gliomas. Glutaminase inhibitors have been shown to exploit metabolic defects conferred by IDH oncogenes in glioma78,79. (R)-2HG inhibits 2OG-dependent branched chain transaminases BCAT1/2 in IDH-mutant gliomas, creating a dependency on glutaminase to maintain glutathione pools. Glutaminase inhibitors thus exploit this defect and effectively decrease glutathione pools in IDH-mutant gliomas, increasing sensitivity to RT and oxidative stress. A phase Ib trial using the glutaminase inhibitor teleglenastat (CB-839) in combination with RT in IDH-mutant grade 2–3 astrocytomas demonstrated acceptable safety profiles80,81. Importantly, redox stress induced by mIDH was rescued with mIDH inhibitor treatment78, suggesting that concurrent mIDH inhibitor treatment may antagonize teleglenastat/RT efficacy.
Work from Tateishi et al.70 and Nagashima et al.82 have similarly identified metabolic vulnerabilities conferred by mIDH that can be therapeutically targeted. Specifically, mIDH decreases NAD+ pools, creating a dependency that renders IDH-mutant gliomas sensitive to drugs that further deplete NAD+. Such strategies include inhibitors of nicotinamide phosphoribosyltransferase (NAMPT)70 or combination of poly(ADP-ribose) glycohydrolase (PARG) inhibition and TMZ82. Clinical studies of NAMPT inhibitors have been limited by toxicity83,84, but PARG inhibitors are currently in clinical development that could be tested for IDH-mutant glioma therapy in future trials.
More recently, several brain tumor types including IDH-mutant gliomas have been found to be sensitive to inhibitors of dihydroorotate dehydrogenase (DHODH), an enzyme in the de novo pyrimidine synthesis pathway53,85,86. IDH-mutant gliomas are hyperdependent on de novo pyrimidine synthesis, and treatment with the brain-penetrant DHODH inhibitor orludodstat (BAY2402234) induces replication stress and DNA damage53. Clinical trials are forthcoming to test orludodstat in IDH-mutant glioma patients.
Immunotherapy
(R)-2HG has been shown to suppress the immune microenvironment through multiple mechanisms, including altered leukocyte chemotaxis87 and suppression of T cell function in both in IDH-mutant glioma88,89,90 and IDH-mutant cholangiocarcinoma91. These findings raise the question as to whether mIDH-driven immunosuppression can be reversed with mIDH inhibitor treatment, supported by data suggesting that immune activation by ivosidenib is a critical mechanism of action in IDH-mutant cholangiocarcinoma91. mIDH inhibitor treatment may therefore enhance efficacy of immune checkpoint blockade if used concurrently in IDH-mutant glioma, raising potential combination therapy strategies. Indeed, a phase II trial testing combination nivolumab and ivosidenib treatment in patients with advanced IDH-mutant solid tumors is underway (NCT04056910), as well as a phase I trial testing combination pembrolizumab and vorasidenib treatment in patients with grades 2 and 3 IDH-mutant glioma (NCT05484622).
In addition to immune checkpoint blockade, vaccines for both IDH-mutant and IDH-wild-type gliomas have presented exciting avenues for immunotherapy in brain tumors. A recent phase I clinical trial demonstrated promising results from a peptide vaccine specific for the IDH1-R132H mutant protein92. Among the 33 patients enrolled, 93.3% (30/32) displayed an immune response induced by the IDH1-R132H vaccine across multiple HLA alleles, with an overall response rate of 84.4% (per RANO criteria, including stable disease) and acceptable safety profiles. Because mIDH inhibitors have been shown to reverse (R)-2HG-mediated T cell suppression, it is plausible that they could augment immunogenicity of IDH1-R132H vaccines. This rational combination therapeutic strategy may be tested in future studies.
Future clinical and translational directions
Results from the INDIGO trial are poised to establish a new standard in the treatment of IDH-mutant glioma, a significant advance in a field that is in desperate need of effective clinical therapies. IDH-mutant gliomas are the most recent IDH-mutant cancers to demonstrate clinical benefit from mIDH inhibitor treatment (following IDH-mutant leukemia and IDH-mutant cholangiocarcinoma), and these results reflect the culmination of more than a decade of basic, translational, and clinical research efforts. The INDIGO trial results also underscore the importance of several questions: (1) what are the molecular mechanisms that mediate sensitivity of gliomas to mIDH inhibition? (2) what biomarkers predict who will respond to mIDH inhibitors? (3) how do tumors acquire resistance after treatment with mIDH inhibitors? (4) how should mIDH inhibitors be used in combination with existing and emerging treatments for IDH-mutant gliomas?
Addressing these questions requires a deeper understanding of IDH-mutant glioma biology. Mechanisms underlying sensitivity and resistance to mIDH inhibitor treatment are likely to be closely related to the downstream oncogenic targets of mIDH, which are not fully understood. Thoughtful combination treatment strategies necessitate an understanding of how new and existing therapies for IDH-mutant glioma exert their efficacy, allowing for rational integration of these treatments with mIDH inhibitors. Additional clinical (and molecular) data from patients treated with mIDH inhibitors will also be useful in addressing these questions. In addition, preclinical data using glioma models responsive to mIDH inhibitor therapy will serve as important systems for mechanistic investigation. Collectively, these efforts will refine how mIDH inhibitors should be optimally used in order to maximize clinical benefit for IDH-mutant glioma patients.
Data availability
There are no original data presented in this review article. All data discussed in this article can be found in the References section.
References
Louis, D. N. et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 114, 97–109 (2007).
Yan, Hai et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773 (2009).
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 23, 1231–1251 (2021).
Losman, J.-A. & Kaelin, W. G. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 27, 836–852 (2013).
Pirozzi, C. J. & Yan, H. The implications of IDH mutations for cancer development and therapy. Nat. Rev. Clin. Oncol. 18, 645–661 (2021).
Miller, J. J. et al. Isocitrate dehydrogenase (IDH) mutant gliomas: a Society for Neuro-Oncology (SNO) consensus review on diagnosis, management, and future directions. Neuro Oncol. 25, 4–25 (2023).
Noushmehr, H. et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17, 510–522 (2010).
Malta, T. M. et al. Glioma CpG island methylator phenotype (G-CIMP): biological and clinical implications. Neuro Oncol. 20, 608–620 (2018).
Turcan, S. et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483 (2012).
Mellinghoff, I. K. et al. Vorasidenib in IDH1- or IDH2-mutant low-grade glioma. N. Engl. J. Med. 389, 589–601 (2023).
Jakola, A. S. et al. The impact of resection in IDH-mutant WHO grade 2 gliomas: a retrospective population-based parallel cohort study. J. Neurosurg. https://doi.org/10.3171/2022.1.JNS212514 (2022).
Wijnenga, M. M. J. et al. The impact of surgery in molecularly defined low-grade glioma: an integrated clinical, radiological, and molecular analysis. Neuro Oncol. 20, 103–112 (2018).
Jakola, A. S. et al. Comparison of a strategy favoring early surgical resection vs a strategy favoring watchful waiting in low-grade gliomas. JAMA 308, 1881–1888 (2012).
Smith, J. S. et al. Role of extent of resection in the long-term outcome of low-grade hemispheric gliomas. J. Clin. Oncol. 26, 1338–1345 (2008).
Choi, J. et al. Extent of resection and molecular pathologic subtype are potent prognostic factors of adult WHO grade II glioma. Sci. Rep. 10, 2086 (2020).
Jakola, A. S. et al. Surgical resection versus watchful waiting in low-grade gliomas. Ann. Oncol. 28, 1942–1948 (2017).
Karim, A. B. et al. A randomized trial on dose-response in radiation therapy of low-grade cerebral glioma: European Organization for Research and Treatment of Cancer (EORTC) Study 22844. Int. J. Radiat. Oncol. Biol. Phys. 36, 549–556 (1996).
Weller, M. et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 18, 170–186 (2021).
Buckner, J. C. et al. Radiation plus procarbazine, CCNU, and vincristine in low-grade glioma. N. Engl. J. Med. 374, 1344–1355 (2016).
Nabors, L. B. et al. NCCN Clinical Practice Guidelines in Oncology Central Nervous System Cancers. National Comprehensive Cancer Network Central Nervous System Cancers. https://www.nccn.org/professionals/physician_gls/pdf/cns.pdf (2023).
van den Bent, M. J. et al. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial. Lancet 366, 985–990 (2005).
Nabors, B., Portnow, J., Hattangadi-Gluth, J. & Horbinski, C. NCCN CNS tumor guidelines update for 2023. Neuro Oncol. 25, 2114–2116 (2023).
Shaw, E. G. et al. Randomized trial of radiation therapy plus procarbazine, lomustine, and vincristine chemotherapy for supratentorial adult low-grade glioma: initial results of RTOG 9802. J. Clin. Oncol. 30, 3065–3070 (2012).
Lassman, A. B. et al. Joint final report of EORTC 26951 and RTOG 9402: phase III trials with procarbazine, lomustine, and vincristine chemotherapy for anaplastic oligodendroglial tumors. J. Clin. Oncol. 40, 2539–2545 (2022).
van den Bent, M. J. et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J. Clin. Oncol. 31, 344–350 (2013).
Cairncross, G. et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J. Clin. Oncol. 31, 337–343 (2013).
Kacimi, S. et al. KS02.5. A overall survival associated with first-line PCV or temozolomide in combination with radiotherapy in patients with IDH-mutant, 1p/19q codeleted, grade 3 oligodendroglioma: analysis from the POLA cohort. Neuro Oncol. 25, ii4 (2023).
Jaeckle, K. A. et al. CODEL: phase III study of RT, RT+ TMZ, or TMZ for newly diagnosed 1p/19q codeleted oligodendroglioma. Analysis from the initial study design. Neuro Oncol. 23, 457–467 (2020).
van den Bent, M. J. et al. Adjuvant and concurrent temozolomide for 1p/19q non-co-deleted anaplastic glioma (CATNON; EORTC study 26053-22054): second interim analysis of a randomised, open-label, phase 3 study. Lancet Oncol. 22, 813–823 (2021).
Wen, P. Y. et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J. Clin. Oncol. 28, 1963–1972 (2010).
Bent et al. Response assessment in neuro-oncology (a report of the RANO group): assessment of outcome in trials of diffuse low-grade gliomas. Lancet Oncol. 12, 583–593 (2011).
Mellinghoff, I. K. et al. Ivosidenib in isocitrate dehydrogenase 1–mutated advanced glioma. J. Clin. Oncol. 38, 3398–3406 (2020).
Popovici-Muller, J. et al. Discovery of AG-120 (Ivosidenib): a first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med. Chem. Lett. 9, 300–305 (2018).
Konteatis, Z. et al. Vorasidenib (AG-881): a first-in-class, brain-penetrant dual inhibitor of mutant IDH1 and 2 for treatment of glioma. ACS Med. Chem. Lett. 11, 101–107 (2020).
Mellinghoff, I. K. et al. Vorasidenib and ivosidenib in IDH1-mutant low-grade glioma: a randomized, perioperative phase 1 trial. Nat. Med. 29, 615–622 (2023).
Mellinghoff, I. K. et al. Vorasidenib, a dual inhibitor of mutant IDH1/2, in recurrent or progressive glioma; results of a first-in-human phase I trial. Clin. Cancer Res. 27, 4491–4499 (2021).
Johannessen, T.-C. A. et al. Rapid Conversion of Mutant IDH1 from Driver to Passenger in a Model of Human Gliomagenesis. Mol. Cancer Res. 14, 976–983 (2016).
Peters, K. et al. QOL-26. A randomized, double-blind phase 3 study of vorasidenib vs placebo in patients with mutant IDH1/2diffuse glioma (INDIGO): analysis of health-related quality of life, neurocognition and seizures. Neuro Oncol. 25, v254–v255 (2023).
Schiff, D. Headway against brain tumors with molecular targeting of IDH-mutant gliomas. N. Engl. J. Med. 389, 653–654 (2023).
Saraf, A. et al. Long-term outcomes and late toxicity of adult medulloblastoma treated with combined modality therapy: a contemporary single-institution experience. Neuro Oncol. 24, 2180–2189 (2022).
Brown, P. D. et al. Hippocampal avoidance during whole-brain radiotherapy plus memantine for patients with brain metastases: phase III trial NRG oncology CC001. J. Clin. Oncol. 38, 1019–1029 (2020).
Brown, P. D. et al. Memantine for the prevention of cognitive dysfunction in patients receiving whole-brain radiotherapy: a randomized, double-blind, placebo-controlled trial. Neuro Oncol. 15, 1429–1437 (2013).
de la Fuente, M. I. et al. Olutasidenib (FT-2102) in patients with relapsed or refractory IDH1-mutant glioma: a multicenter, open-label, phase Ib/II trial. Neuro Oncol. 25, 146–156 (2022).
Natsume, A. et al. The first-in-human phase I study of a brain-penetrant mutant IDH1 inhibitor DS-1001 in patients with recurrent or progressive IDH1-mutant gliomas. Neuro Oncol. 25, 326–336 (2023).
Pusch, S. et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. 133, 629–644 (2017).
Wick, A. et al. Phase I assessment of safety and therapeutic activity of BAY1436032 in patients with IDH1-mutant solid tumors. Clin. Cancer Res. 27, 2723–2733 (2021).
Reuss, D. E. et al. IDH mutant diffuse and anaplastic astrocytomas have similar age at presentation and little difference in survival: a grading problem for WHO. Acta Neuropathol. 129, 867–873 (2015).
Carstam, L. et al. WHO grade loses its prognostic value in molecularly defined diffuse lower-grade gliomas. Front Oncol. 11, 803975 (2021).
Dasgupta, P., Balasubramanyian, V., de Groot, J. F. & Majd, N. K. Preclinical models of low-grade gliomas. Cancers 15, 596 (2023).
Hicks, W. H. et al. Contemporary mouse models in glioma research. Cells 10, 712 (2021).
Abdullah, K. G. et al. Establishment of patient-derived organoid models of lower-grade glioma. Neuro Oncol. 24, 612–623 (2022).
Yanchus, C. et al. A noncoding single-nucleotide polymorphism at 8q24 drives IDH1-mutant glioma formation. Science 378, 68–78 (2022).
Shi, D. D. et al. De novo pyrimidine synthesis is a targetable vulnerability in IDH mutant glioma. Cancer Cell 40, 939–956.e16 (2022).
Shi, D. D. et al. Protocol to establish a genetically engineered mouse model of IDH1-mutant astrocytoma. STAR Protoc. 4, 102281 (2023).
Laugesen, E. et al. MODL-47. A new mouse model of IDH mutated gliomas identifies tumor cells of origin and determinants of sensitivity to IDH inhibitors. Neuro Oncol. 25, v309 (2023).
Spitzer, A. et al. Mutant IDH inhibitors induce lineage differentiation in IDH-mutant oligodendroglioma. Cancer Cell https://doi.org/10.1016/j.ccell.2024.03.008 (2024).
Shi, D. D., Anand, S., Abdullah, K. G. & McBrayer, S. K. DNA damage in IDH-mutant gliomas: mechanisms and clinical implications. J. Neuro. Oncol. 162, 515–523 (2023).
Ma, C., Wei, S. & Song, Y. T790M and acquired resistance of EGFR TKI: a literature review of clinical reports. J. Thorac. Dis. 3, 10–18 (2011).
Intlekofer, A. M. et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 559, 125–129 (2018).
Choe, S. et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 4, 1894–1905 (2020).
Reinbold, R. et al. Resistance to the isocitrate dehydrogenase 1 mutant inhibitor ivosidenib can be overcome by alternative dimer-interface binding inhibitors. Nat. Commun. 13, 4785 (2022).
Oltvai, Z. N. et al. Assessing acquired resistance to IDH1 inhibitor therapy by full-exon IDH1 sequencing and structural modeling. Cold Spring Harb. Mol. Case Stud. 7, a006007 (2021).
Cleary, J. M. et al. Secondary IDH1 resistance mutations and oncogenic IDH2 mutations cause acquired resistance to ivosidenib in cholangiocarcinoma. npj Precis. Onc. 6, 61 (2022).
Harding, J. J. et al. Isoform switching as a mechanism of acquired resistance to mutant isocitrate dehydrogenase inhibition. Cancer Discov. 8, 1540–1547 (2018).
Coleman, N. et al. Beyond epidermal growth factor receptor: MET amplification as a general resistance driver to targeted therapy in oncogene-driven non-small-cell lung cancer. ESMO Open 6, 100319 (2021).
Sulkowski, P. L. et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 582, 586–591 (2020).
Sulkowski, P. L. et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 9, eaal2463 (2017).
Schvartzman, J.-M. et al. Oncogenic IDH mutations increase heterochromatin-related replication stress without impacting tumor mutation burden. Mol Cell 83, 2347–2356.e8 (2023).
Lu, Y. et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res 77, 1709–1718 (2017).
Tateishi, K. et al. Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell 28, 773–784 (2015).
Ducray, F. et al. Olaparib in recurrent IDH-mutant high-grade glioma (OLAGLI). J. Clin. Oncol. 39, 2007–2007 (2021).
Varn, F. S. et al. Glioma progression is shaped by genetic evolution and microenvironment interactions. Cell 185, 2184–2199.e16 (2022).
Al Baghdadi, T. et al. Palbociclib in patients with pancreatic and biliary cancer with CDKN2A alterations: results from the targeted agent and profiling utilization registry study. JCO Precis Oncol. 3, 1–8 (2019).
Ahn, E. R. et al. Palbociclib in patients with non-small-cell lung cancer With CDKN2A alterations: results from the targeted agent and profiling utilization registry study. JCO Precis Oncol. https://doi.org/10.1200/PO.20.00037 (2020).
Flavahan, W. A. et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114 (2016).
Turcan, S. et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet 50, 62–72 (2018).
Yamashita, A. S. et al. Demethylation and epigenetic modification with 5-azacytidine reduces IDH1 mutant glioma growth in combination with temozolomide. Neuro Oncol. 21, 189–200 (2019).
McBrayer, S. K. et al. Transaminase inhibition by 2-hydroxyglutarate impairs glutamate biosynthesis and redox homeostasis in glioma. Cell 175, 101–116.e25 (2018).
Seltzer, M. J. et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 70, 8981–8987 (2010).
Kizilbash, S. et al. CTNI-23. Preliminary safety and pharmacokinetics data for a phase 1B trial of telaglenastat in combination with radiation therapy and temozolomide in patients with IDH-mutant grade 2/3 astrocytoma (NCI-10218). Neuro Oncol. 24, vii75 (2022).
Kizilbash, S. H. et al. A phase Ib trial of CB-839 (telaglenastat) in combination with radiation therapy and temozolomide in patients with IDH-mutated diffuse astrocytoma and anaplastic astrocytoma (NCT03528642). J. Clin. Oncol. 37, TPS2075 (2019).
Nagashima, H. et al. Poly(ADP-ribose) glycohydrolase inhibition sequesters NAD+ to potentiate the metabolic lethality of alkylating chemotherapy in IDH mutant tumor cells. Cancer Discov. 10, 1672–1689 (2020).
Goldinger, S. M. et al. Efficacy and safety of APO866 in patients with refractory or relapsed cutaneous T-cell lymphoma: a phase 2 clinical trial. JAMA Dermatol. 152, 837–839 (2016).
Wei, Y., Xiang, H. & Zhang, W. Review of various NAMPT inhibitors for the treatment of cancer. Front Pharm. 13, 970553 (2022).
Gwynne, W. D. et al. Cancer-selective metabolic vulnerabilities in MYC-amplified medulloblastoma. Cancer Cell 40, 1488–1502.e7 (2022).
Pal, S. et al. A druggable addiction to de novo pyrimidine biosynthesis in diffuse midline glioma. Cancer Cell 40, 957–972.e10 (2022).
Amankulor, N. M. et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 31, 774–786 (2017).
Bunse, L. et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 24, 1192–1203 (2018).
Notarangelo, G. et al. Oncometabolite d-2HG alters T cell metabolism to impair CD8+ T cell function. Science 377, 1519–1529 (2022).
Kohanbash, G. et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Invest 127, 1425–1437 (2017).
Wu, M.-J. et al. Mutant IDH inhibits IFNγ-TET2 signaling to promote immunoevasion and tumor maintenance in cholangiocarcinoma. Cancer Discov. 12, 812–835 (2022).
Platten, M. et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 592, 463–468 (2021).
Acknowledgements
This study was funded by K12CA0903354. The funder played no role in the conception or writing of this manuscript.
Author information
Authors and Affiliations
Contributions
D.D.S., S.K.M. and K.A.A. conceptualized the manuscript. D.D.S., M.D.L., and A.C.-Y.T. drafted the manuscript. S.K.M. and K.A.A. edited the manuscript. M.D.L. and A.C.-Y.T. contributed equally to this work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing non-financial interests but the following competing financial interests: S.K.M. has consulted for Agios Pharmaceuticals. D.D.S., M.D.L., A.C.-Y.T. and K.A.A. declare no competing financial or non-financial interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, M.D., Tsai, A.CY., Abdullah, K.G. et al. Treatment of IDH-mutant glioma in the INDIGO era. npj Precis. Onc. 8, 149 (2024). https://doi.org/10.1038/s41698-024-00646-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-024-00646-2
