
Abstract
Morphine has been suggested to affect cancer cell dynamics and decrease survival rates in lung cancer patients at specific doses, but the precise mechanisms poorly understood. In this study, we aimed to investigate the molecular mechanisms by which morphine modulates the malignant characteristics of non-small cell lung cancer. Cell proliferation was assessed via the Cell Counting Kit-8 assay, and cell migration and invasion were examined via wound healing and Transwell assays. We employed immunofluorescence staining to evaluate E-cadherin expression in A549 and Lewis lung cancer (LLC) cell lines and immunohistochemistry to evaluate E-cadherin expression in nude mice tumours. Additionally, the in vivo effects of morphine on lung cancer progression were explored in a xenograft tumour experiments, in which naloxone was used as a morphine antagonist. Western blot analysis was performed to detect E-cadherin, phosphorylated mTOR (p-mTOR), mTOR, phosphorylated AKT (p-AKT), AKT, phosphorylated PI3K (p-PI3K), and PI3K protein levels in A549 and LLC cells as well as in tumour samples. Morphine (10 µM) significantly increased the proliferation of A549 and LLC cells in vitro (p < 0.05). It also enhanced the migratory and invasive capacities of these cell lines (p < 0.01). Mechanistically, morphine treatment (10 µM) led to a reduction in the expression of E-cadherin, and an increase in the phosphorylation of PI3K, AKT, and mTOR in A549 and LLC cells (p < 0.01). Morphine treatment (1.5 mg/kg) also reduced E-cadherin expression in xenograft tumours and promoted tumour growth in vivo (p < 0.05). This effect was reversed by naloxone (0.1 mg/kg). The results demonstrated that morphine stimulates the malignant proliferation of A549 and LLC cell lines and promotes xenograft tumour growth. Perhaps by specifically targeting MOR, morphine triggers a signalling cascade that activates the PI3K/AKT/mTOR pathway while inhibiting the EMT marker E-cadherin, which may consequently promote the progression of lung cancer.
Similar content being viewed by others


Lung cancer ranks first among cancers’ number of new deaths (1 796 144, 18.0%) and ranks second among cancers’ number of new cases (2 206 771, 11.4%)1. According to the World Health Organization, the number of lung cancer cases worldwide is expected to increase notably, primarily due to industrial and environmental pollutants2. There are two main subtypes of lung cancer: small cell lung cancer (SCLC), which accounts for approximately 15% of cases, and non-small cell lung cancer (NSCLC), which accounts for approximately 85% of cases. Despite advancements, the prognosis for advanced-stage patients remains bleak, often marked by debilitating pain necessitating treatment with opioids, including morphine or fentanyl3. These agents function by activating the mu opioid receptor (MOR), a pivotal member of the opioid receptor family expressed across various cancer types.
Morphine, which is commonly prescribed for postoperative care and pain management in advanced lung cancer patients, has been the focus of recent research. Studies suggest that opioids may influence tumour progression, resistance to anticancer therapies, and patient outcomes by directly affecting cancer cell viability and migration4,5. Additionally, emerging research highlights the potential of morphine to induce angiogenesis in breast tumours6, which might negatively affect prognosis7. However, the precise mechanisms by which morphine affects lung cancer cells and prognosis remain elusive. Our study aimed to investigate the effects of morphine administration on the progression of lung cancer and the possible underlying molecular mechanism in cell lines and experimental animal models.
The epithelial‒mesenchymal transition (EMT) is a dynamic genetic phenomenon that enables the transformation of polarized epithelial cells into mesenchymal cells8. In cancer, EMT is important because of its association with tumour cell plasticity and its role in conferring resistance to conventional targeted therapies. Morphine is used for analgesia in patients with advanced lung cancer, which can effectively reduce the progression of inflammation in cancer patients. Studies have shown that in the inflammatory tumour microenvironment, fibroblasts, macrophages, granulocytes and lymphocytes produce inflammatory mediators, some of which can induce EMT9,10. EMT involves substantial morphological changes, characterized by a decrease in the expression of epithelial markers such as E-cadherin and β-catenin, and an increase in mesenchymal protein expression. This process includes extensive cytoskeletal remodelling, which increases the invasive and proliferative capacities of epithelial cells, ultimately promoting tumour metastasis and the adoption of a mesenchymal phenotype. Notably, E-cadherins, a family of Ca2+-dependent adhesive proteins, play pivotal roles in modulating cell‒cell interactions10.
Overall, this study aimed to determine whether morphine promotes the proliferation of A549 human non-small cell lung cancer cells and Lewis lung cancer (LLC) cells and the growth of xenograft tumours. Additionally, we explored the potential influence of morphine on malignant behaviour and elucidated the role of the PI3K/Akt/mTOR pathway in regulating the expression of the EMT marker E-cadherin induced by morphine. This investigation employs a comprehensive approach that includes both cell lines and experimental animal models.
Materials and methods
Ethical review
The animal experimental protocol of this study was approved by the Ethics Committee for the Use of Laboratory Animals of Zhejiang Cancer Hospital (Record No. 2022-07-099), and all methods were performed in accordance with the ARRIVE guidelines and the American Veterinary Medical Association (AVMA) Guidelines for Animal Euthanasia (2020).
Experimental animals
Female nude mice aged 4–6 weeks and weighing 16–18 g were purchased from Hangzhou Hangsi Biotechnology Company. The mice were housed at the Animal Centre of the Institute of Basic Medical Sciences and Oncology, Chinese Academy of Sciences, Hangzhou, China, under specific-pathogen-free (SPF) conditions: temperature of 20–26 °C, humidity of 40–60%, and a light‒dark cycle of 12 h. Bedding was changed daily, and the mice were given free access to water and food. The animals were euthanized with carbon dioxide at the desired time point or at the end of the experiment as required; for this step, the animals were placed in a closed box, and 100% carbon dioxide was introduced at a fill rate of approximately 20% of the box volume per minute.
Materials
Morphine was obtained from Sigma (USA). Various kits, reagents, and proteins, including a BCA protein assay kit (Sigma–Aldrich); RPMI-1640 (HyClone, USA); DMEM (HyClone, USA); foetal bovine serum (Gibco, USA); trypsin (Gibco, USA); Cell Counting Kit-8 (CCK-8) (TargetMol, USA); methylnaltrexone bromide (TargetMol, USA); antibodies targeting phospho-AKT (A17909; ABclonal, China), AKT (A11016; ABclonal, China), phospho-PI3K p85 (AP0427; ABclonal, China), PI3K p85 (A4992, ABclonal, China), phospho-mTOR (A2445; Proteintech, China), mTOR (AP0094, ABclonal, China), E-cadherin (A18135; ABclonal, China), and β-actin (ab9385; Abcam, UK); and Matrigel (Corning, USA), were used in this study.
Cell culture
A549 and LLC cells were authenticated by the National Center for Biotechnology Information. A549 cells were cultured in RPMI-1640 medium supplemented with 10% foetal bovine serum, 100 U/ml penicillin, and 100 mg/ml streptomycin at 37 °C in a 5% CO2 atmosphere. LLC cells were cultured in DMEM supplemented with 10% foetal bovine serum, 100 U/ml penicillin, and 100 mg/ml streptomycin and cultured under the same CO2 and temperature conditions. Morphine was dissolved in normal DPBS and subsequently diluted to different concentrations. Naloxone followed a similar dissolution pattern in DPBS and was then diluted to different concentrations. Additionally, the cells were pretreated with normal saline (NS) or 1 µM, 10 µM, or 100 µM morphine to investigate the underlying mechanisms driving morphine-induced malignant biological behaviours. The cells in the normal control groups were cultured in RPMI-1640 medium following standard protocols.
Determination of cell viability
A549 and LLC cells were seeded in 96-well plates, with each well containing 2 × 103 cells. Six replicate wells were designated for each group. After a 48-h incubation at 37 °C, the supernatant was removed, and 10 µl of CCK-8 reagent was added to each well. Following a 1-h incubation at 37 °C in the dark, optical density (OD) measurements at 450 nm were taken via a Thermo Fisher instrument to assess cellular viability. Each experiment was replicated three times to ensure robust statistical validation.
Colony formation analysis
The cells were seeded in six-well plates and allowed to grow undisturbed for 7 days. After incubation, the cells were fixed with 4% paraformaldehyde for 30 min and rinsed with phosphate-buffered saline (PBS). Colonies were stained with a 5% crystal violet solution for 15 min and analysed via ImageJ software. Colonies containing more than 50 cells were meticulously counted. Each experimental group included three replicates to ensure robust statistical validation.
5-Ethynyl-20-deoxyuridine (EdU) staining
A549 cell proliferation was assessed via the BeyoClickTM EdU Cell Proliferation Kit (C0075S; Beyotime, China). A549 cells were first washed twice with PBS after they were allowed to proliferate for 48 h. The samples were subsequently incubated in an EdU working solution (10 µM) for 2 h at 37 °C under light-shielded conditions. After being incubated with EdU, the cells were rinsed twice with PBS and fixed in 4% paraformaldehyde for 15 min. Next, the samples were permeabilized with 0.1% Triton X-100 for 15 min, followed by three cycles of rinses with PBS. The cells were then stained with Hoechst for 10 min and rinsed three times with PBS. A fluorescence microscope (Evos flauto, Life Technologies, United States) at 100 × magnification was used to observe red fluorescence, indicating that the cells underwent DNA replication during the incubation period, whereas blue fluorescence indicated the nucleus.
Immunohistochemistry (IHC)
For IHC analyses, sections of lung cancer tissue from female nude mice were subjected to a systematic process of deparaffinization and alcohol rehydration, followed by antigen retrieval via sodium citrate buffer. The tumour sections were then subjected to a blocking step with 5% normal goat serum, 0.1% Triton X-100, and 3% H2O2 in PBS for 60 min at room temperature. For IHC, the sections were incubated overnight at 4 °C with the appropriate primary antibody and then with a horseradish peroxidase (HRP)-conjugated secondary antibody that releases 3,3′-diaminobenzidine tetrahydrochloride (DAB) upon binding to the primary antibody for visualization. Hoechst nuclear staining was performed, and images were obtained under a Nikon microscope.
Wound healing assay
In accordance with established protocols, the wound-healing assay was performed to clarify the effects on cell migration. A549 or LLC cells (5 × 105 cells/well) were cultured in 6-well plates until they reached 95% confluence. A precise “wound” was then created by gently scratching the cell monolayers with a 200-µL pipette tip. The cells were subsequently treated with 10 µM morphine in serum-free medium for 24 h. Images of the wound area at 0 and 24 h were acquired via an Olympus microscope (Tokyo, Japan). Quantitative analysis of the migration distance was performed via ImageJ software. Each experimental run was repeated a minimum of three times to ensure the robustness and consistency of the outcomes.
Transwell assay
Following established methods, Transwell assays were utilized as a platform to explore the impact of morphine on the migratory and invasive properties of A549 or LLC cells. The cells (3 × 104) were seeded in 100 µL of serum-free medium into dedicated chambers placed in a 24-well culture dish, with some chambers featuring uncoated membranes and others coated with Matrigel. Following a 24-h incubation period, the chambers were carefully removed for cleaning, and the cells on the lower surface were fixed with methanol and stained with crystal violet. The migrated cells were subsequently photographed under an Olympus microscope (Tokyo, Japan) at 100 × magnification; five random fields were photographed at 400 × magnification; and these images were used to quantify cell migration. To ensure the reliability and consistency of the results, each experiment was repeated more than three times.
Western blot analysis
Following established methods, Western blot analysis commenced with the treatment of A549 and LLC cells with morphine for 48 h. After treatment, the samples were thoroughly washed with cold PBS, followed by lysis with a specialized lysis solution. After a 30-min incubation at 4 °C and centrifugation at 12,000 rpm for 15 min, the resulting supernatants were carefully collected. The protein concentration in each sample was then assessed via a BCA protein assay kit (Sigma‒Aldrich) following the manufacturer’s instructions. Homogenized cell supernatants containing 40 µg protein equivalent per sample were prepared and boiled briefly at 100 °C for 5 min in sample buffer (P0015L, Beyotime, China). The proteins were subsequently separated on a 10% SDS‒PAGE gel and transferred onto PVDF membranes. After transfer, the membranes were blocked with 5% BSA solution in TBST to prevent nonspecific binding and subsequently incubated with the primary antibody solution following the manufacturer’s protocol. After several washes with TBST to eliminate unbound primary antibody, the membranes were incubated with the secondary antibody for 1 h at room temperature. The protein bands were visualized via an enhanced chemiluminescence (ECL) system. Each experimental was performed at least three times to guarantee the accuracy and consistency of the results.
Immunofluorescence staining
Immunofluorescence assays were performed following established protocols. Cells cultured on coverslips in 24-well plates were first fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and then blocked with normal goat serum. Next, the cells were incubated overnight at 4 °C with a primary E-cadherin antibody. They were then incubated with Cy3-conjugated goat anti-rabbit IgG as a secondary antibody, and DAPI nuclear staining was performed. Imaging was performed using a confocal microscope (Olympus, Tokyo, Japan). Each experimental setup was repeated more than three times to ensure the accuracy and reliability of the results.
Tumour xenograft experiments
Young female nude mice, aged 4–6 weeks, were randomly assigned to either the control or treatment group (n = 5). Each mouse was then injected with 7 × 106 cells suspended in 100 µl of PBS subcutaneously on their backs. The treatment group was administered morphine at a dosage of 1.5 mg/kg every other day for a total of 3 weeks on the basis of our preliminary research. Moreover, the control group received injections of NS according to the same schedule. Tumour growth was closely monitored each week over a 4-week period, and tumour size was measured via a ruler after treatment. After 28 days, the mice were euthanized, and their tumour tissues were meticulously excised and weighed. The tumour volume was calculated via the following formula: volume (mm3) = 1/2 × (length × width × width).
Statistical analysis
The results are presented as the mean ± standard error of the mean. Statistical analysis included paired and unpaired Student’s t tests, followed by Tukey’s post hoc test and multiple comparisons where appropriate. Furthermore, for normally distributed data, one-way analysis of variance (ANOVA) was utilized, and the results are presented as the means ± standard errors. Statistical significance was indicated by a significance threshold of p < 0.05. Statistical calculations and data visualization were performed via GraphPad Prism software (version 9, GraphPad Software, San Diego, CA, USA).
Results
Morphine augments A549 cell proliferation, migration, and invasion
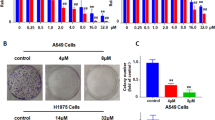
Following the established protocols, we observed a substantial increase in A549 and LLC cell proliferation after 48 h of exposure to morphine, particularly after 10 µM morphine treatment (Fig. 1a). The EdU and colony formation assays results provided additional evidence of the promoting effect of 10 µM morphine on the proliferation of tumour cells (Fig. 1b, c). Moreover, scratch assays and Transwell assays validated the enhancements in migration and invasion induced by 10 µM morphine in A549 cells, as demonstrated by a significant increase in migration and invasion after 48 h of 10 µM morphine treatment (Fig. 1d, e).

Morphine promotes the proliferation and migration of A549 and LLC cells. (a) A549 and LLC cells were incubated with different concentrations of morphine. A significant increase in the percentage of viable cells was observed at a concentration of 10 µM morphine. (b) Proliferation was assessed by an EdU assay after A549 cells were preincubated with normal medium or 10 µM morphine for 48 h. Scale bar = 500 µm. (c) Representative images of A549 cell colony formation after treatment with 10 µM morphine for 7 days. (d) In the scratch wound healing assay, confluent A549 cells were preincubated with either 0 µM or 10 µM morphine for 48 h. Wound closure progress was monitored at 0, 24, and 48 h. (e) In the Transwell invasion assay of A549 and LLC cells, a notable increase in the number of invading cells was observed in the 10 µM morphine group compared with the control group. Representative images depict A549 cell colony formation after treatment with either normal medium or 10 µM morphine for 7 days. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Morphine decreases E-cadherin expression and activates the PI3K/AKT/mTOR signalling pathway related proteins
Our immunofluorescence results revealed that the expression of E-cadherin was significantly reduced after pretreatment of A549 cells and LLC cells with 10 µM morphine (Fig. 2a, b). Western blot analysis shows 10 µM morphine decreases E-cadherin expression (Fig. 2c, d). To investigate whether morphine-induced cell proliferation is associated with the PI3K/Akt/mTOR signalling pathway, we conducted Western blot analysis. Compared with the control group, the morphine-treated group presented increased phosphorylation of AKT and activation of PI3K and mTOR (Fig. 2c, d). These results suggest that morphine may activate the PI3K/AKT/mTOR signalling pathway, consistent with previous studies on malignant cellular processes. This activation indicates a potential mechanism by which morphine promotes cell proliferation, migration, and invasion.

Morphine downregulates the expression of E-cadherin and activates the PI3K/AKT/mTOR signalling pathway. (a) Images and (b) quantification of E-cadherin immunoreactivity in A549 and LLC cells with 10 µM morphine. (c) Images and (d) quantification of Western blot showing the expression of E-cadherin, p-mTOR, mTOR, p-AKT, AKT, p-PI3K and PI3K in A549 and LLC cells after treatment with 10 µM morphine.
Morphine promotes the growth of lung cancer tumours by decreasing E-cadherin expression through the mu opioid receptor (MOR)
Given the significant role of E-cadherin in initiating lung cancer metastasis and the correlation between MOR and cell metastasis ability, we detected E-cadherin and PI3K/AKT/mTOR signalling pathway-related proteins in tumour samples of nude mice and antagonized morphine with naloxone. Our investigation revealed that exposure to morphine (1.5 mg/kg) led to a reduction in E-cadherin levels and promote lung cancer tumour cells proliferation in tumour samples. However, this effect was reversed by treatment with naloxone (a competitive opioid receptor antagonist) (Fig. 3a–e).

Morphine downregulates E-cadherin expression in tumour samples via MOR. (a) Immunohistochemical analysis of in situ E-cadherin expression in tumour samples collected from female nude mice tumours (aged 4–6 weeks, n = 5). (b) Ki67-stained tissues demonstrated that morphine(1.5 mg/kg) promoted the proliferation of solid tumour cells, an effect that could be reversed by naloxone(0.1 mg/kg). (c) Images and (d) quantification of Western blot showing the expression of E-cadherin and p-mTOR, mTOR, p-AKT, AKT, p-PI3K and PI3K in tumour samples following treatment with 1.5 mg/kg morphine. (e) Nude mice (n = 5) were euthanized at the end of the experiment (Day 28), and images of the dissected tumours from five representative female nude are shown. Scale bar: 1 cm. (f, g) Subcutaneous xenograft tumour weight (g) and tumour volume (cm3) were measured at the end of the experiment. The statistical data for tumour weight and volume represent the average ± SD of five independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
The enhancement of tumour growth and E-cadherin downregulation induced by morphine may occur through PI3K/AKT/mTOR pathway activation in nude mice
The immunohistochemistry results revealed a notable decrease in E-cadherin expression and an increase in Ki67 expression in lung cancer tissue samples from morphine-treated mice (Fig. 3a, b). Western blot analysis revealed decreases E-cadherin expression and increased activation of p-mTOR, p-AKT, p-PI3K in the mice treated with morphine compared with the control mice (Fig. 3c, d). For the tumour formation experiments, female nude mice were divided into three groups and received subcutaneous injections of morphine (1.5 mg/kg), NS, or naloxone (0.1 mg/kg), a competitive opioid receptor antagonist, on the 7th day. By the 28th day, the tumours in the morphine-treated groups were significantly larger than those in the other groups (Fig. 3e–g).
Discussion
A large proportion of patients (66%) with advanced lung cancer experience pain, often severe, necessitating the use of strong opioids like morphine11. Estimates suggest that around 50–90% of advanced cancer patients require opioid analgesics12. The use of morphine, a potent µ-opioid receptor (MOR) activator, has been suggested to affect cancer cell dynamics and decrease survival rates in lung cancer patients at specific doses.
The effects of morphine, a widely used drug for postoperative analgesia in patients with various tumours, on tumours are still controversial. Many studies have demonstrated that morphine can inhibit the growth of various human cancer cell lines, including breast, stomach, lung and prostate cancer cell lines13,14,15. However, other studies have indicated that morphine can stimulate the growth of tumour cells in the body, a phenomenon also observed in in vitro experiments16,17,18. Our study revealed that morphine (10 µM or 1.5 mg/kg) significantly increase the proliferation, invasion, and metastasis of non-small cell lung cancer cells (A549, LLC) and promotes the growth of lung cancer tumours by downregulating the expression of E-cadherin. These contrasting conclusions could be attributed to factors such as the dosage of morphine administered, the route of administration, or the steady-state plasma concentration achieved.
In this study, 10 µM or morphine was shown to enhance the malignant behaviour of lung cancer cells, whereas 100 µM morphine did not have the same effect. This difference could be attributed to the greater concentration of morphine inhibiting cell cycle progression from the G (1) to the S phase19 Studies both in vitro and in vivo have indicated that low daily doses of morphine and tumour enhancement occur after a single analgesic application of morphine, whereas tumour suppression occurs after long-term high doses of morphine20,21,22,23.
Our study elucidates the potential pathways by which morphine influences lung cancer progression, offering insights with implications for patient survival. Importantly, the enhanced progression of lung cancer induced by morphine was alleviated by naloxone, indicating the potential involvement of opioid receptors. Previous retrospective analyses have suggested an association between increased opioid doses postoperatively and increased recurrence rates in patients with NSCLC within 5 years24. Additionally, the influence of morphine on mast cell activation, cytokine elevation, and angiogenesis modulation underscores its multifaceted involvement in cancer progression.
We found that morphine triggered a series of events that lead to increased tumour growth, migration, and invasion, coupled with the downregulation of E-cadherin expression, and that these effects may be induced via activation of the PI3K/AKT/mTOR pathway. This effect was reversed by naloxone. Our results highlight the contribution of morphine to tumour growth and reduced overall survival in murine models, possibly mediated by its interaction with MOR. Tripolt’s study demonstrated that opioids promote breast cancer metastasis by engaging the δ-opioid receptor and causing the downregulation of E-cadherin25. Liu13 showed that morphine promotes the malignant biological behaviour of H460 cells by activating the MOR and Src/mTOR signalling pathways. Clinical trials have shown that opioid use is correlated with poor clinical outcomes in immune checkpoint inhibitor-treated NSCLC patients26,27. These studies yield results that are consistent with those of our research. Our study not only sheds light on the impact of morphine on NSCLC cell behaviour but also highlights the therapeutic potential of MOR antagonists in the management of lung cancer.
Although our study provides valuable information, it has some limitations. First, owing to time and resource constraints, in the cell-related experiments, we were unable to generate the dose–response (proliferation) or IC50 (cytotoxicity) curves for A549 and LLC cells treated with morphine. Instead, we solely evaluated the effects of morphine on these lung cancer cells through CCK-8 experiments. Second, we assessed the activation of relevant pathways and the levels of EMT-related markers at the protein level, but we did not assess RNA levels. Third, we speculated that the reduced expression of E-cadherin in tumour cells pretreated with morphine and in mouse tumour tissues may be correlated with the PI3K signalling pathway. However, we did not conduct additional experiments to establish a direct relationship between the two. Fourth, we did not conduct reverse verification of the relationship between the PI3K/Akt/mTOR pathway and E-cadherin downregulation, which is a consideration for our future experiments. Fifth, we assessed E-cadherin expression in the tumour tissues of xenografted mice through immunohistochemical experiments, but we did not investigate the expression of MOR and other EMT-related markers in mouse tumour tissues. Exploring the relationships among MOR, E-cadherin, and other relevant markers in tumour tissues could offer valuable insights into the impact of morphine on the EMT process in tumorigenesis. Finally, owing to insufficient time and sample size, we did not obtain relevant immunohistochemical data for MOR and E-cadherin in human tumour tissues. We will do this in future studies to better understand the effects of morphine on tumours. Figure 4 illustrates the hypothesis of this topic (Fig. 4).

Schematic model illustrating the potential pathway associated with morphine-induced malignant biological behaviour in A549 cells. Morphine acts on an MOR located on the A549 cell membrane, leading to increased levels of phospho-PI3K and phospho-AKT. The activation of the PI3K/AKT pathway further activates mTOR, may be leading to the downregulation of E-cadherin, which induces cell proliferation, migration and invasion.
Conclusions
In summary, our in vivo and in vitro experiments unequivocally demonstrate that morphine promotes the malignant behaviours of A549 and LLC non-small cell lung cancer. Morphine may function by binding to MOR, initiating a sequence of events that trigger the activation of the PI3K/AKT/mTOR pathways. This activation, in a cascading manner, may lead to the downregulation of E-cadherin, ultimately contributing to the progression of lung cancer. The results indicate that the impact of opioid dosage on tumour progression should be taken into account when managing pain in patients with lung cancer. We will conduct more extensive studies in the future to further validate this conclusion and investigate the detailed molecular mechanisms by which morphine promotes tumour growth.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author upon reasonable request.
References
Rebecca, L. S. et al. Cancer statistics, 2021. CA Cancer J. Clin. 71, 7–33 (2021).
Hyuna, S. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Smith, T. J. et al. Randomized clinical trial of an implantable drug delivery system compared with comprehensive medical management for refractory cancer pain: Impact on pain, drug-related toxicity, and survival. J. Clin. Oncol. 20, 4040–4049 (2002).
Afsharimani, B. et al. Comparison and analysis of the animal models used to study the effect of morphine on tumour growth and metastasis. Br. J. Pharmacol. 172, 251–259 (2015).
Díaz-Cambronero, O. et al. Mu opioid receptor 1 (MOR-1) expression in colorectal cancer and oncological long-term outcomes: A five-year retrospective longitudinal cohort study. Cancers (Basel) 12, 134. https://doi.org/10.3390/cancers12010134 (2020).
Li, Y. et al. The mu-opioid receptor (MOR) promotes tumor initiation in hepatocellular carcinoma. Cancer Lett. 453, 1–9 (2019).
Nguyen, J. et al. Morphine stimulates cancer progression and mast cell activation and impairs survival in transgenic mice with breast cancer. Br. J. Anaesth. 113(S1), i4–i13. https://doi.org/10.1093/bja/aeu090 (2014).
Ramundo, V. et al. The epithelial-to-mesenchymal transition (EMT) in the development and metastasis of malignant pleural mesothelioma. Int. J. Mol. Sci. 22, 12216. https://doi.org/10.3390/ijms222212216 (2021).
Ribatti, D. et al. Epithelial–mesenchymal transition in cancer: A historical overview. Transl. Oncol. 13, 100773. https://doi.org/10.1016/j.tranon (2020).
Odarenko, K. V. et al. The nexus of inflammation-induced epithelial–mesenchymal transition and lung cancer progression: A roadmap to pentacyclic triterpenoid-based therapies. Int. J. Mol. Sci. 24, 17325. https://doi.org/10.3390/ijms242417325 (2023).
Christina, A. S. et al. Opioid analgesics for nociceptive cancer pain: A comprehensive review. CA Cancer J. Clin. 74, 286–313 (2024).
Mercadante, S. Cancer pain treatment strategies in patients with cancer. Drugs 82, 1357–1366 (2022).
Liu, X. et al. Morphine promotes the malignant biological behavior of non-small cell lung cancer cells through the MOR/Src/mTOR pathway. Cancer Cell Int. 21, 622. https://doi.org/10.1186/s12935-021-02334-8 (2021).
Ge, Z. H. et al. Morphine improved the antitumor effects on MCF-7 cells in combination with 5-Fluorouracil. Biomed. Pharmacother. 68, 299–305 (2014).
Kampa, M. et al. Opioid alkaloids and casomorphin peptides decrease the proliferation of prostatic cancer cell lines (LNCaP, PC3 and DU145) through a partial interaction with opioid receptors. Eur. J. Pharmacol. 335(2–3), 255–265 (1997).
Qin, Y. et al. Exogenous morphine inhibits human gastric cancer MGC-803 cell growth by cell cycle arrest and apoptosis induction. Asian Pac. J. Cancer Prev. 13, 1377–1382 (2012).
Sabrina, B. et al. Morphine promotes tumor angiogenesis and increases breast cancer progression. Biomed. Res. Int. 2015, 161508. https://doi.org/10.1155/2015/161508 (2015).
Kalpna, G. et al. Morphine stimulates angiogenesis by activating proangiogenic and survival-promoting signaling and promotes breast tumor growth. Cancer Res. 62, 4491–4498 (2002).
Tegeder, I. et al. G protein-independent G1 cell cycle block and apoptosis with morphine in adenocarcinoma cells: Involvement of p53 phosphorylation. Cancer Res. 63, 1846–1852 (2003).
Niu, D. G. et al. Morphine promotes cancer stem cell properties, contributing to chemoresistance in breast cancer. Oncotarget 6, 3963–3976 (2015).
Harimaya, Y. et al. Potential ability of morphine to inhibit the adhesion, invasion and metastasis of metastatic colon 26–L5 carcinoma cells. Cancer Lett. 187, 121–127 (2002).
Sasamura, T. et al. Morphine analgesia suppresses tumor growth and metastasis in a mouse model of cancer pain produced by orthotopic tumor inoculation. Eur. J. Pharmacol. 441, 185–191 (2002).
Zong, J. & Pollack, G. M. Morphine antinociception is enhanced in mdr1a gene-deficient mice. Pharm. Res. 17, 749–753 (2000).
Maher, D. P. et al. Association of increased postoperative opioid administration with non-small-cell lung cancer recurrence: A retrospective analysis. Br. J. Anaesth. 113(Suppl 1), i88–i94. https://doi.org/10.1093/bja/aeu192 (2014).
Tripolt, S. et al. Opioids drive breast cancer metastasis through the δ-opioid receptor and oncogenic STAT3. Neoplasia 23, 270–279 (2021).
Guo, H. et al. A novel investigation into the negative impact of opioid use on the efficacy of immune checkpoint inhibitors in advanced non-small cell lung cancer patients. Int. Immunopharmacol. 129, 111611. https://doi.org/10.1016/j.intimp.2024.111611 (2024).
Young, P. et al. Impact of opioid use on duration of therapy and overall survival for patients with advanced non-small cell lung cancer treated with immune checkpoint inhibitors. Curr. Oncol. 31, 260–273 (2024).
Acknowledgements
The authors acknowledge the use of OpenAI’s GPT-4 language model for refining the English language of this paper.
Funding
This work was supported by the Zhejiang Provincial Health Science and Technology Program Young Innovative Talents Project [grant number 2022RC015]; Zhejiang Province Traditional Chinese Medicine Science and Technology Project Foundation [grant number 2022ZA006]; Zhejiang Traditional Chinese Medicine Science and Technology Plan [grant number 2023ZL302]; and the National Natural Science Foundation of China [grant number 82160229].
Author information
Authors and Affiliations
Contributions
G.F.L.: project administration; W.F.Y. and K.J.X.: study conception and design; revision of the manuscript for important intellectual content; approval of the final version to be published. Y.X.Z. and L.L.T.: acquisition of data; analysis of data; drafting of the manuscript; approval of the final version to be published. J.Y.C. and C.Z.: writing—review and editing. H.Z.X., L.L.T. and K.J.X.: funding acquisition and supervision. All the authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gu, F., Zhou, Y., Tian, L. et al. Morphine promotes non-small cell lung cancer progression by downregulating E-cadherin via the PI3K/AKT/mTOR pathway. Sci Rep 14, 21130 (2024). https://doi.org/10.1038/s41598-024-72198-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-72198-1
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
