
Abstract
The adult mammalian heart has extremely limited cardiac regenerative capacity. Most cardiomyocytes live in a state of permanent cell-cycle arrest and are unable to re-enter the cycle. Cardiomyocytes switch from cell proliferation to a maturation state during neonatal development. Although several signaling pathways are involved in this transition, the molecular mechanisms by which these inputs coordinately regulate cardiomyocyte maturation are not fully understood. Retinoic acid (RA) plays a pivotal role in development, morphogenesis, and regeneration. Despite the importance of RA signaling in embryo heart development, little is known about its function in the early postnatal period. We found that mRNA expression of aldehyde dehydrogenase 1 family member A2 (Aldh1a2), which encodes the key enzyme for synthesizing all-trans retinoic acid (ATRA) and is an important regulator for RA signaling, was transiently upregulated in neonatal mouse ventricles. Single-cell transcriptome analysis and immunohistochemistry revealed that Aldh1a2 expression was enriched in cardiac fibroblasts during the early postnatal period. Administration of ATRA inhibited cardiomyocyte proliferation in cultured neonatal rat cardiomyocytes and human cardiomyocytes. RNA-seq analysis indicated that cell proliferation-related genes were downregulated in prenatal rat ventricular cardiomyocytes treated with ATRA, while cardiomyocyte maturation-related genes were upregulated. These findings suggest that RA signaling derived from cardiac fibroblasts is one of the key regulators of cardiomyocyte proliferation and maturation during neonatal heart development.
Similar content being viewed by others


Introduction
During perinatal heart development, one of the remarkable changes in mammalian cardiomyocytes is the transition from a hyperplastic to hypertrophic state1. Cardiomyocytes withdraw from the cell cycle during the first days after birth, concurrently with further cardiomyocyte maturation2. Despite recent advances showing that several factors are involved in this transition, such as metabolic shift3, oxygen4, and hormones5, the molecular mechanisms involved in these changes are not fully understood.
Retinoic acid (RA), the major active metabolite of vitamin A, is essential for organogenesis, homeostasis, and tissue morphogenesis, where it is generally considered to act in a paracrine manner6. RA is generated in two dehydrogenation reactions by retinol dehydrogenases and retinaldehyde dehydrogenases. ALDH1A2, an isoenzyme of aldehyde dehydrogenases, is an enzyme responsible for RA synthesis during heart development7. RA modulates target gene expression through binding to retinoic acid receptors (RARα, RARβ, RARγ), which form heterodimers with Retinoid X receptors (RXRs)8. RA signaling is known to play a pivotal role in embryo heart development9,10, whereas its function in the early postnatal period remains largely uncharacterized.
Here, we found a spike of peak expression of ALDH1A2 in cardiac fibroblasts during the perinatal period. We observed that treatment of cultured neonatal cardiomyocytes with all-trans retinoic acid (ATRA), an active form of RA, induced a switch from a proliferative to mature state. Our findings suggest that paracrine signals from cardiac fibroblasts modulate cardiomyocyte properties.
Results
Characterization of cell-cycle activity during perinatal heart development
To evaluate cell-cycle activity during perinatal heart development, we performed immunostaining of mouse ventricles for wheat germ agglutinin (WGA) to distinguish cardiomyocytes from non-cardiomyocytes, and for Ki-67, which is expressed in all active phases of the cell cycle including mitotic events (Fig. 1A). The proportions of Ki-67-positive cardiomyocytes were significantly decreased after birth, while there was no significant difference between subendocardium, midwall myocardium, and subepicardium (Fig. 1B). Similarly, the proportions of Ki-67-positive total ventricular cells, including stromal and vascular endothelial cells, were gradually decreased with age (Fig. 1C), suggesting that cell-cycle activity was decreased during the perinatal period throughout the heart.

Characterization of cell-cycle activity during perinatal heart development. (A) Fluorescent images of left ventricles immunostained for Hoechst (blue), Ki-67 (red) and WGA (green) at the indicated stages. Scale bar, 50 µm. (B, C) Left diagrams represent percentage of Ki-67 positive cells (B) in cardiomyocytes (n = 9) and (C) in whole ventricle cells (n = 9), at different pre and postnatal stages of E18.5, P6, P12, and P18. (**p < 0.01, One-way ANOVA with Dunnett’s multiple comparisons test relative to E18.5). Right diagrams represent percentage of Ki-67 positive cells in the subendocardium (red bars, n = 3), midwall myocardium (yellow bars, n = 3), and subepicardium (blue bars, n = 3).
RA signaling suppresses cardiomyocyte proliferation
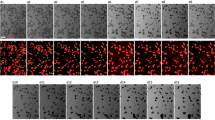
To analyze the function of RA signaling during perinatal heart development, we investigated the mRNA expression levels of multiple retinoid metabolic-related genes at various perinatal stages using RNA-seq analysis on whole-heart tissue (Fig. 2A). Notably, RNA expression of Aldh1a2 revealed dynamic changes from E17.5 to P24, with its highest expression peaking at P6 in the mouse heart. In contrast, the related synthases Rdh10 and Aldh1a3, whose loss is lethal at fetal stages in mouse11, were stable and did not exhibit obvious temporal changes in their expression level (Fig. 2B). We performed a timecourse validation of Aldh1a2 expression in mouse hearts by quantitative real-time PCR (qRT-PCR) and observed a similar pattern of peak and decline, with the highest levels of Aldh1a2 occurring at P6 (Fig. 2C). Analysis of publicly available single-cell transcriptome data of mouse hearts12 showed that Aldh1a2 expression was enriched in Pdgfra-positive cardiac fibroblasts at P2 (Fig. 2D), which was consistent with previous findings13. We confirmed these findings and extended them throughout the perinatal development timecourse by performing immunostaining of ALDH1A2 and PDGFRA in hearts at stages E18.5 through P18. We indeed observed that cells expressing both markers were observed within the subepicardium, midwall myocardium, and subendocardium, indicating that ALDH1A2 is expressed mainly in cardiac fibroblasts during the perinatal stage (Fig. 2E). The proportion of ALDH1A2-positive cells was significantly increased in P6 hearts compared to that in E18.5 (Fig. 2F), while there were no significant changes in the distribution per subepicardium, midwall myocardium, and subendocardium (Fig. 2G). Taken together, our results indicate that a spike in peak expression of ALDH1A2 in cardiac fibroblasts coincides with the timing of a switch from proliferation to maturation in cardiomyocytes. We hypothesized that RA production from cardiac fibroblasts via ALDH1A2 could be involved in this transition. To investigate this possibility, we treated cultured neonatal rat ventricular cardiomyocytes (NRVCMs) with ATRA in combination with 5-ethynyl-2-deoxyuridine (EdU), a marker for DNA synthesis, and found that DNA synthesis in NRVCMs was suppressed in a dose-dependent fashion (Fig. 3A). Additional immunostaining of cultured human cardiac myocytes (HCMs) confirmed the suppression of DNA synthesis by pan-RAR agonist treatment (Fig. 3B). To assess the responsible RAR signaling genes in this context, cultured NRVCMs were treated with pan-RAR agonists (ATRA, EC23), selective RARA agonists (AM580, BMS753), RARB agonist (CD2314), and RARG agonist (BMS961), respectively. Treatment with both pan-RAR agonists and selective RARA agonists suppressed DNA synthesis in cardiomyocytes (Fig. 3C). To further evaluate the effects of ATRA on the mitosis and cytokinesis state of NRVCMs, we performed immunostaining for phosphorylated histone H3 (pH3), a marker for mitosis, or Aurora B kinase, a marker for cytokinesis. Immunocytochemistry analysis revealed that the proportion of pH3 or Aurora B positive NRVCMs was decreased in the ATRA treatment group (Fig. 3D, E), suggesting that augmented RA signaling inhibited the mitosis and cytokinesis of cardiomyocytes.

Short spike of Aldh1a2 expression in neonatal heart. (A, B) RNA-seq-based expression levels of multiple retinoid metabolic genes during murine perinatal heart development (A). The expression levels of Aldh1a2 (orange), Aldh1a3 (green), and Rdh10 (blue) are depicted on the right (B). (n = 2). (C) Quantitative real-time PCR (qRT-PCR) analysis of Aldh1a2 expression in mouse ventricle during heart development. (n = 3; **p < 0.01, One-way ANOVA with Dunnett’s multiple comparisons test relative to eight weeks). (D) Single cell RNA-seq-based Uniform manifold approximation and projection (UMAP) clustering of P2 heart (GSM4644950). Feature Plots of each gene expression showing Aldh1a2 (left panel) and Pdgfra (right panel). (E) Immunofluorescence analysis of ALDH1A2 in mouse heart sections during late-embryonic to neonatal period (green = ALDH1A2, red = PDGFRA, blue = WGA). Note that ALDH1A2 is mainly expressed in cardiac fibroblasts (ALDH1A2+PDGFR+) and not in cardiomyocytes. Higher magnification images are in the insets. Scale bars, 50 μm. (F) Quantitation of ALDH1A2-positive cells at different developmental stages. (n = 3 mice; **p < 0.01, One-way ANOVA with Dunnett’s multiple comparisons test relative to E18.5). (G) Quantitation of ALDH1A2-positive cells in the subendocardium (red bars), midwall myocardium (yellow bars), and subepicardium (blue bars). All data represents mean ± SEM.

RA signaling inhibits cardiomyocyte proliferation. (A) Representative flow cytometry plots of EdU incorporation in cardiac Troponin T (cTnT)-positive cells from ATRA treated cultured NRVCMs and controls at 48 h on the two left plots. Quantitation of EdU-positive and cTnT-positive cells treated with increasing concentrations of ATRA is shown on the right. (n = 4; **p < 0.01, One-way ANOVA with Dunnett’s multiple comparisons test relative to DMSO). (B) Representative fluorescent images of ATRA treated cultured human cardiac myocytes and controls stained for Hoechst (blue), MYH6 (red) and EdU (green). Quantitation of EdU-positive and MYH6-positive cells is shown on the right. (n = 6; *p < 0.05, **p < 0.01, unpaired t test relative to DMSO). Scale bars, 50 μm. (C) Graph showing flow cytometry analysis of 10 µM Pan-RAR agonists (ATRA, EC23), selective RARA agonists (AM580, BMS753), selective RARB agonist (CD2314) or selective RARG agonist (BMS961) treated cultured NRVCMs and controls at 48 h. (n = 4; **p < 0.01, One-way ANOVA with Dunnett’s multiple comparisons test relative to DMSO). (D) Representative images of ATRA treated cultured NRVCMs and controls stained for Hoechst (blue), phosphohistone H3 (pH3) (red) and cTnT (green). Right diagram represents the percentage of pH3- and cTnT-positive cardiomyocytes. (n = 8; *p < 0.05, unpaired t test relative to DMSO). Scale bars, 100 µm. (E) Representative image of cultured NRVCMs stained for Hoechst (blue), Aurora B (red, white arrow) and cTnT (green). Quantitation of Aurora B- and cTnT-positive cardiomyocytes between controls and ATRA-treated NRVCMs is shown in the right. (n = 8; *p < 0.05, unpaired t test relative to DMSO). Scale bar, 50 µm. All data represents mean ± SEM.
Bioinformatic analysis of ATRA-treated prenatal cardiomyocytes
For a comprehensive transcriptional evaluation of the effect of RA signaling on cardiomyocyte cell cycle arrest, we performed RNA-seq analysis of cultured prenatal cardiomyocytes treated with ATRA in vitro. Isolated prenatal rat ventricular cardiomyocytes (PRVCMs) were treated with 10 μM ATRA for 48 h to mimic temporal exposure to RA signaling. Heatmap analysis of a total of 11,692 genes showed that the ATRA-treated cardiomyocyte group (ATRA group) was separated from controls (Control group), with up- and down-regulated genes associated with both groups (Fig. 4A). We identified 606 genes as the down-regulated differentially expressed genes (DEGs) in the ATRA group with log2 fold change < − 1. The gene ontology (GO) terms enriched by the down-regulated DEGs were associated with cell proliferation (Fig. 4B). Gene set enrichment analysis (GSEA) revealed enrichments in the abundance of multiple terms of “growth”, “positive regulation of cell population proliferation”, and “muscle cell proliferation” in the Control group as compared to the ATRA group (Fig. 4C). Heatmap analysis of selected cell cycle regulators showed that many of them were downregulated in the ATRA group compared to the Control group, including c-myc (Myc), proliferating cell nuclear antigen (Pcna), cyclin-dependent kinase1 (Cdk1), Cdk4, and Cdk6 (Fig. 4D)14. Conversely, the regulators of cell cycle progression, such as Wee1 and cyclin dependent kinase inhibitor 1B (Cdkn1b, also known as p27) were upregulated in ATRA-treated cells (Fig. 4E). In addition, Myh6 and myosin light chain 2 (Myl2) expression were increased in the ATRA group, while expression levels of Myh7 and Myl7 were reduced (Fig. 4E), indicative of cardiomyocyte maturation15. Furthermore, qPCR analysis revealed that the ratio of Myh6/Myh7, reliable indicator for cardiomyocyte maturation15, was significantly increased in ATRA-treated NRVCMs (Fig. 4F). The expression of maturation marker genes related to calcium handling (Ryr2, Atp2a2) was also increased by ATRA treatment (Fig. 4F). In addition, the treatment of NRVCMs with ATRA led to an increase in the number of multinucleated cardiomyocytes (Fig. 4G), supporting the idea that ATRA treatment promotes cardiomyocyte maturation.

Bioinformatic analysis of ATRA treated PRVCMs. (A) Heatmap image of gene expression by RNA sequencing analysis illustrating 11,692 genes between prenatal rat ventricular cells which were treated for 48 h with 10 µM ATRA (ATRA) and controls (Control). (B) GO analysis for significantly differentially regulated genes which were downregulated in ATRA treated group. (C) GSEA comparing ATRA treated (red side) and control (blue side) groups. Enrichment plots show the result for the growth, positive regulation of cell population proliferation, and muscle cell proliferation gene sets. Each plot is shown with its normalized enrichment score (NES) and FDR. (D, E) Heatmap images of RNA sequencing analysis for cell cycle regulated genes (D) and cardiac maturation genes (E) between ATRA and Control groups. F. RT-qPCR analysis of the expression ratio of Myh6/Myh7, Ryr2 and Atp2a2 expression in cultured ATRA-treated and DMSO-treated NRVCMs. All data represent mean ± SEM. (n = 7; **p < 0.01, unpaired t test relative to DMSO). (G) Quantitation of multinucleated cardiomyocytes between controls and ATRA-treated NRVCMs is shown in the right. Data represents mean ± SEM. (n = 7; **p < 0.01, unpaired t test relative to DMSO).
Collectively, our results suggest that RA signaling might modulate cardiomyocyte characteristics from proliferation to maturation during early postnatal heart development.
Discussion
The heart undergoes a series of substantial morphological, structural, and functional changes to adapt to body size and cardiac workload during heart development. Understanding the molecular mechanisms of these transitions is an important issue for cardiac regenerative medicine. We propose that dynamic RA production from cardiac fibroblasts by ALDH1A2 is one of the important regulators for cardiomyocyte switching. One important question concerns distinct functional properties of RA signaling in a context-dependent fashion. There are several conflicting studies showing the protective13,16 or harmful17 effects of RA signaling on myocardial ischemia. It is known that ATRA promotes cardiomyocyte proliferation and cardiac regeneration, while our findings appear to show cell cycle exit and cardiomyocyte maturation. One possibility is that different microenvironmental configurations throughout embryonic-to-adult development modulates the effects of RA signaling on cardiomyocytes. Myocardial infarction induces an extreme burst of inflammation with local release of pro-inflammatory mediators along with a hypoxic environment; in this setting RA signaling is observed to lead to activation of cardiomyocyte proliferation16. By comparison, our study emphasizes the developmental progression of cardiomyocyte maturation within physiological parameters of the heart; the homeostatic milieu of cell types and growth factors in this setting might mediate RA signaling responsiveness resulting in cardiomyocyte maturation. In contrast, long-term administration of ATRA leads to cardiac hypertrophy in healthy rats, indicative of cardiotoxic effects18. Another possibility is the consequence of global epigenetic changes during heart development. Indeed, DNA methylation in cardiomyocytes is highly dynamic during postnatal development and is associated with cardiac developmental signaling pathways19. Transcriptional responses to ATRA are associated with differential DNA methylation status in melanoma and breast cancer cells20,21. It has been recently reported that ATRA treatment could enhance the structural and functional maturation of human embryonic stem cell-derived cardiomyocytes22, which is consistent with our findings. In the rodent system, MYH7 is a marker of embryonic cardiomyocytes, while MYH6 is upregulated in mature stages15. Indeed, the ratio of MYH6/MYH7 was increased after ATRA treatment in our experiments (Fig. 4F), supporting the notion that the effect of RA signaling on cardiomyocyte maturation is conserved between species. A key difference between our study and Miao S et al. was the mature state of cardiomyocytes. We have utilized rat ventricular cardiomyocytes isolated from rat heart at P1, while hESC-CMs after day 15 of differentiation show RNA-seq profiles closer to embryonic human hearts at Carnegie stage 14–1623, further supporting that the two studies consider cardiomyocytes at distinct developmental stages.
Cardiac fibroblasts play potent roles in homeostasis, regeneration, and organogenesis through extracellular matrix production and paracrine signaling24. Embryonic cardiac fibroblasts promote cardiomyocyte proliferation in a paracrine fashion25. In contrast, ablation of cardiac fibroblasts during the neonatal period inhibits the cardiomyocyte transition from proliferation to maturation26,27, supporting our findings that RA signaling from cardiac fibroblasts plays a role in this transition. Further studies will be required to confirm the role of cardiac fibroblast-derived RA in cardiomyocyte maturation in vivo. Such analysis would require a combination of inducible and cell type-specific genetic approaches for Aldh1a2 ablation, since global genetic deletion of Aldh1a2 results in embryonic lethality due to cardiac malformations7. As the mechanisms that underlie the loss of regenerative capacity of cardiomyocytes in adult mammals are not fully uncovered, our findings could provide novel insight into new therapeutic approaches in the field of heart regenerative medicine.
Methods
Animals
All experimental protocols were approved by the Animal Ethics Review Board of Nagoya University School of Medicine. All experiments were conducted in accordance with the institutional guidelines and regulations for animal experiments at Nagoya University. This study was reported in accordance with the Animal Research: Reporting of In Vivo Experiments on Animals (ARRIVE) guidelines. C57BL/6 J mice and Wistar rats were purchased from Charles River Laboratories (Yokohama, Japan). Neonatal pups were sacrificed by decapitation, and adult animals were euthanized by isoflurane.
Cell culture
NRVCMs or PRVCMs were enzymatically isolated from 1-to 3-day-old or E18.5 Wistar rat ventricles as previously described28. Briefly, minced ventricle tissue was suspended in 15 mg Trypsin (27250–018, Gibco) / 25 ml Hank's Balanced Salt Solution (HBSS) in 4 °C for 18 h, replaced with 10 mg of Collagenase 2 (LS004176, Worthington Biochemical Corp)/10 ml HBSS solution under continuous agitation, 150 rpm, at 38 °C for 4.5 min five times. Cells were pelleted by centrifugation at 200 g for 5 min and resuspended in culture medium. Cells underwent two pre-platings of 1 h each to minimize fibroblast contamination. Isolated cardiomyocytes were seeded to 70% confluence on laminin-coated (final concentration 38.25 μg/ml, L2020, Sigma) 60 mm culture dishes. Cardiomyocytes were maintained in Dulbecco’s Modified Eagle Medium (12320–032, Gibco) containing 1.5 mM Vitamin B12 (V2876, Sigma), 7% fetal bovine serum and 1% penicillin / streptomycin and incubated for 24 h (37 °C, 5% CO2). After incubation, cardiomyocytes were exposed to 10 µM various RAR agonists, including all-trans retinoic acid (ATRA) (R2625, Sigma), EC23 (SML2404, Sigma), BMS753 (3505, Tocris), AM580 (A3082, Tokyo Chemical Industry), CD2314 (3824, Tocris), BMS961 (3410, Tocris) or Dimethyl sulfoxide (DMSO) as vehicle for 48 h. Proliferating cells were labeled with 5-ethynyl-2-deoxyuridine (EdU) (E10187, Thermo Fisher Scientific) during the last 24 h of the culture period.
Flow cytometry
Cardiomyocytes were fixed and permeabilized with BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (554714, BD Biosciences) at 4 °C for 20 min. EdU was stained by Click-iT™ Plus EdU Alexa Fluor™ 488 Flow Cytometry Assay Kit (C10633, Invitrogen). Cardiomyocytes were incubated with the following primary antibody (4 °C overnight) and secondary antibody (room temperature for 1 h): Cardiac Troponin T (1:400, ab8295, abcam) and Alexa Fluor™ Plus 647(1:400, A32787, Thermo Fisher Scientific). Cardiomyocytes were resuspended in 500 μl Phosphate-Buffered Saline (PBS) and analyzed on FACSCanto (Becton, Dickinson and Company, Franklin Lakes, NJ, USA).
HCMs proliferation assay
HCMs (C-12811, PromoCell) were cultured in Myocyte Basal Medium and Supplement Pack Myocyte cell GM (C-22170, PromoCell) growing to 80% confluence at 37 ℃ and 5% CO2 according to the instruction manual. 10,000 cells / well were seeded into μ-Slide 8 Well Imaging chambers (80821, Ibidi) coated with 38.25 μg/ml laminin. After incubation for 24 h, cells were incubated with ATRA (10 μM) or an equal amount of DMSO as negative control for 48 h. EdU was added to a concentration of 10 µM during the last 24 h of the culture period. After 24 h of EdU labeling, cells were fixed with 4% Paraformaldehyde (PFA) for 15 min, permeabilized with 0.2% Triton-X100 for 10 min, and blocked with 3% Bovine serum albumin blocking buffer for 30 min. Labeled DNA was detected using Click-iT™ EdU Cell Proliferation Kit for Imaging, Alexa Fluor™ 488 dye (C-10337, Invitrogen). Cells were incubated with mouse anti-myosin heavy chain 6 (Myh6) (1:200 MA1-26180, Thermo Fisher Scientific) as primary antibody at 4 °C overnight, followed by goat anti-Mouse IgG (H + L) Alexa Fluor 647 (1:500 A11005, Thermo Fisher Scientific) as secondary antibody. Nuclei were counterstained with Hoechst 33342 (1:1000, H3570, Invitrogen) for 10 min at room temperature. Images were collected using a fluorescence microscope BZ-X710 and BZ-X Analyzer. ImageJ software (version 1.53a) was used to count nuclei.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted using the QIAzol Lysis Reagent (79306, QIAGEN) and miRNeasy Micro Kit (217084, QIAGEN) followed by digestion of DNA using the TURBO DNA-free™ Kit (AM1907, Invitrogen). Reverse transcription for cDNA synthesis was processed using PrimeScript RT reagent Kit (RR037A, TaKaRa). Quantitative real-time PCR was performed using the CFX96 Real-Time PCR Detection System (Bio-Rad, USA) with SYBR Premix Ex Taq™ 2 (RR820A, TaKaRa) or THUNDERBIRD Next SYBR qPCR Mix (QPX-201, TOYOBO). Gapdh was used to normalize gene expression. The following primer pairs were used: mouse Aldh1a2 forward (5’- CACAAGACACGAGCCCATTGGA -3’), mouse Aldh1a2 reverse (5’- GGTTTGATGACCACGGTGTTACC -3’), mouse Gapdh forward (5’-AAAGGGTCATCATCTCCGCC-3’), mouse Gapdh reverse (5’-GCCCTTCCACAATGCCAAAG -3’), rat Myh6 forward (5’- AGAGTGACAGGATGACGGAT-3’), rat Myh6 reverse (5’- TCCTTCACAGTCACCGTCTTG-3’), rat Myh7 forward (5’- AAGTCCTCCCTCAAGCTCCTAAGT-3’), rat Myh7 reverse (5’- TTGCTTTGCCTTTGCCC-3’), rat Ryr2 forward (5’- CAAACAGGGCAGAAGACACC-3’), rat Ryr2 reverse (5’- CTCTGAGGGTGCTCCACCT-3’), rat Atp2a2 forward (5’- CAATCTGGTGACGGATGGTCT-3’), rat Atp2a2 reverse (5’- CCAATAGCCAGGTAACGGAAA-3’), rat Gapdh forward (5’- CAATCTGGTGACGGATGGTCT-3’), rat Gapdh reverse (5’- CCAATAGCCAGGTAACGGAAA-3’).
RNA-sequencing analysis
RNA-seq libraries were prepared as previously described with some modifications29. For cardiac development analysis, ventricles from P3, P6, P12 and P24 mice were minced into several pieces and washed in PBS. Isolated RNA quality was assessed using an Agilent 2200 TapeStation system (Agilent). 1 µg of RNA with a RIN e score of 7 or higher was used for preparation of sequencing libraries with Poly(A) Magnetic Isolation Module (E7490S, New England Bio Labs) and NEBNext Ultra RNA Prep Kit for Illumina (E7530S, New England Bio Labs) according to the manufacturer's instructions. 151-bp pair-end sequencing was performed with a HiSeq 2500 system (Illumina). For PRVCM analysis, cultured PRVCMs were treated with 10 µM ATRA or DMSO for 48 h. Extracted RNA quality was assessed, and 100 ng of RNA were used for library preparation with NEBNext Poly(A) Isolation Module (E7490, New England Bio Labs) and NEBNext Ultra II RNA Prep Kit for Illumina (E7770, New England Bio Labs) according to the manufacturer's instructions. 150-bp pair-end sequencing was performed with a HiSeq X system (Illumina). E17.5 mouse left ventricle gene expression data (GSM2059616, GSM2059617) was also included for further analysis30. Quality of raw sequence data was assessed using FastQC (Version: FastQC 0.10.0). FASTQ files were trimmed for adapters and Phred score (> 20) using TrimGalore! (0.6.4). Trimmed sequenced reads were aligned to the mouse genome assembly (GRCm39 GENCODE vM30) or the Rattus norvegicus genome assembly (mRatBN7.2 Ensembl release 108) using STAR (2.7.3a). Aligned reads were used to quantify mRNA with HTSeq-count (0.11.2). Differential gene expression analysis across samples was performed using DESeq2 package (1.32.0) on protein-coding genes. Genes with mean FPKM > 0.5 across samples were used further analysis. Heat maps were generated with heatmap.3 package (1.1.9) in R. DEGs were selected using false discovery rate (FDR)-adjusted p-value cut-off < 0.05 and absolute log2 fold change > 1. GO analysis was performed with the GO Biological Process of Enrichr using gene lists of the DEGs. GSEA was performed using gseGO from clusterProfiler package (4.22) in R. The results were visualized with the gseaplot2 in the enrichplot package (version 1.14.2) in R. Single-cell RNA-seq dataset of P2 mouse heart (GSE153480) was processed using R (4.1.0) and Seurat (4.3.0) as previously described12,31.
Immunohistochemistry
Mouse ventricle samples were fixed with 4% PFA/PBS and subjected to paraffin embedded blocks. Tissue slices (4 μm) were deparaffinized and retrieved by boiling for 2 min in a pressure cooker with citrate buffer (2.4 g/L sodium citrate, 0.35 g/L citric acid, pH 6.0). Blocking solution was applied and samples were incubated with primary antibodies overnight at 4 °C, followed by secondary antibodies overnight at 4 °C. Nuclear staining was performed with Hoechst 33342 for 10 min at room temperature. Samples were enclosed with Fluoromount-G (0100–01, Southern Biotech), and images were taken on BZ-X710 microscope (Keyence). We classified the myocardium into three layers (subendocardium, midwall myocardium, and subepicardium)32 in short-axis sections of the midventricular region. Antibodies were used as follows: anti-Ki-67 (1:200, ab15580, abcam), platelet-derived growth factor receptor alpha (PDGFRA) (1:200, AF1062, R&D Systems), ALDH1A2 (1:200, ABN420, Sigma Aldrich), wheat germ agglutinin (WGA) (5 μg/mL for 10 min at room temperature, W11261, Invitrogen), Donkey anti-Goat IgG (H + L) 555 (1:400, A21432, Invitrogen), Donkey anti-Rabbit IgG (H + L) 647 (1:400, 20047, Biotium).
Immunocytochemistry
NRVCMs at 80,000 cells / well were cultured on μ-Slide 8 Well Imaging chambers coated with 38.25 μg/ml laminin for 24 h, followed by the treatment with ATRA (10 μM) or an equal amount of DMSO for 48 h. Cells were fixed with 4% PFA/PBS for 15 min, permeabilized with 0.1% Triton-X100 for 10 min, and blocked with 1% Bovine serum albumin blocking buffer for 30 min. Cells were incubated with mouse anti-Cardiac Troponin T (1:400) and rabbit anti-Phospho-Histone H3 (Ser10) (1:800, #3377, Cell Signaling) or rabbit anti-Aurora B (1:100, A5102, Sigma Aldrich) as primary antibody at 4 °C overnight, followed by donkey anti-mouse IgG (H + L) CF 488A (1:500, 20014, Biotium) and donkey anti-rabbit IgG (H + L) CF 555 (1:500, 20038, Biotium) as secondary antibody. Nuclei were counterstained with Hoechst 33342 (1:1000) for 10 min at room temperature. Images were collected using a fluorescence microscope BZ-X710 and BZ-X Analyzer.
Statistical analysis
Statistical Analysis was performed using GraphPad Prism version 7.0. Data are presented as mean ± standard error of mean (SEM). The difference of means between two independent groups was tested for using the unpaired two-tailed Student’s t-test. To test for differences across multiple groups, One-way ANOVA followed by Dunnett’s multiple comparison test was used to analyze three or more variables.
Data availability
All relevant data supporting the results of the present study are included within the article and can be obtained from the corresponding author upon reasonable request. RNA sequencing data is available from the Gene Expression Omnibus (GEO) database: GSE263095 and GSE263096.
References
Karbassi, E. et al. Cardiomyocyte maturation: advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 17, 341–359 (2020).
Maroli, G. & Braun, T. The long and winding road of cardiomyocyte maturation. Cardiovasc. Res. 117, 712–726 (2021).
Chong, D. et al. Neonatal ketone body elevation regulates postnatal heart development by promoting cardiomyocyte mitochondrial maturation and metabolic reprogramming. Cell Discov. 8, 106 (2022).
Puente, B. N. et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 157, 565–579 (2014).
Hirose, K. et al. Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science 364, 184–188 (2019).
Cunningham, T. J. & Duester, G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 16, 110–123 (2015).
Niederreither, K. et al. Embryonic retinoic acid synthesis is essential for heart morphogenesis in the mouse. Development 128, 1019–1031 (2001).
Evans, R. M. & Mangelsdorf, D. J. Nuclear receptors, RXR, and the Big Bang. Cell 157, 255–266 (2014).
Kastner, P. et al. Vitamin A deficiency and mutations of RXRalpha, RXRbeta and RARalpha lead to early differentiation of embryonic ventricular cardiomyocytes. Development 124, 4749–4758 (1997).
Pasutto, F. et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am. J. Hum. Genet. 80, 550–560 (2007).
Kumar, S., Sandell, L. L., Trainor, P. A., Koentgen, F. & Duester, G. Alcohol and aldehyde dehydrogenases: retinoid metabolic effects in mouse knockout models. Biochim Biophys. Acta 1821, 198–205 (2012).
Wang, Z. et al. Cell-type-specific gene regulatory networks underlying murine neonatal heart regeneration at single-cell resolution. Cell Rep. 33, 108472 (2020).
Da Silva, F. et al. Retinoic acid signaling is directly activated in cardiomyocytes and protects mouse hearts from apoptosis after myocardial infarction. Elife 10 (2021).
Mohamed, T. M. A. et al. Regulation of cell cycle to stimulate adult cardiomyocyte proliferation and cardiac regeneration. Cell 173, 104–116 (2018).
Guo, Y. & Pu, W. T. Cardiomyocyte maturation: new phase in development. Circ. Res. 126, 1086–1106 (2020).
Tan, Y. Z. et al. Retinoic acid released from self-assembling peptide activates cardiomyocyte proliferation and enhances repair of infarcted myocardium. Exp. Cell Res. 422, 113440 (2023).
Danzl, K. et al. Early inhibition of endothelial retinoid uptake upon myocardial infarction restores cardiac function and prevents cell, tissue, and animal death. J. Mol. Cell Cardiol. 126, 105–117 (2019).
Silva, R. A. C. et al. Cardiac remodeling induced by all-trans retinoic acid is detrimental in normal rats. Cell Physiol. Biochem. 43, 1449–1459 (2017).
Sim, C. B. et al. Dynamic changes in the cardiac methylome during postnatal development. FASEB J. 29, 1329–1343 (2015).
Fan, J. et al. Silencing and re-expression of retinoic acid receptor beta2 in human melanoma. Pigment Cell Melanoma Res. 23, 419–429 (2010).
Coyle, K.M. et al. DNA methylation predicts the response of triple-negative breast cancers to all-trans retinoic acid. Cancers (Basel) 10 (2018).
Miao, S. et al. Retinoic acid promotes metabolic maturation of human embryonic stem cell-derived cardiomyocytes. Theranostics 10, 9686–9701 (2020).
Meng, Z. et al. Dynamic transcriptome profiling toward understanding the development of the human embryonic heart during different Carnegie stages. FEBS Lett. 594, 4307–4319 (2020).
Plikus, M. V. et al. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 184, 3852–3872 (2021).
Ieda, M. et al. Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev. Cell 16, 233–244 (2009).
Kuwabara, J. T. et al. Regulation of extracellular matrix composition by fibroblasts during perinatal cardiac maturation. J. Mol. Cell Cardiol. 169, 84–95 (2022).
Hortells, L. et al. A specialized population of Periostin-expressing cardiac fibroblasts contributes to postnatal cardiomyocyte maturation and innervation. Proc. Natl. Acad Sci. USA 117, 21469–21479 (2020).
Simmons, W. W., Closs, E. I., Cunningham, J. M., Smith, T. W. & Kelly, R. A. Cytokines and insulin induce cationic amino acid transporter (CAT) expression in cardiac myocytes. Regulation of L-arginine transport and no production by CAT-1, CAT-2A, and CAT-2B. J. Biol. Chem. 271, 11694–11702 (1996).
Kato, K. et al. Pulmonary pericytes regulate lung morphogenesis. Nat. Commun. 9, 2448 (2018).
Liu, X. et al. The complex genetics of hypoplastic left heart syndrome. Nat. Genet. 49, 1152–1159 (2017).
Narita, S. et al. Direct reprogramming of adult adipose-derived regenerative cells toward cardiomyocytes using six transcriptional factors. iScience 25, 104651 (2022).
Streeter, D. D. Jr., Spotnitz, H. M., Patel, D. P., Ross, J. Jr. & Sonnenblick, E. H. Fiber orientation in the canine left ventricle during diastole and systole. Circ. Res. 24, 339–347 (1969).
Acknowledgements
We thank the staff from the Division of Experimental Animals, Nagoya University School of Medicine, for assisting with animal experiments. We acknowledge the Division for Medical Research Engineering, Nagoya University Graduate School of Medicine for the usage of flow cytometry facilities. We would like to thank Yoko Inoue for technical assistance.
Funding
This work was supported, in part, by JST CREST (Grant Number JPMJCR19H4) (to T. M.), JSPS KAKENHI Grant Number JP18H02805, 21H02911 (to T. M.), JP20K17145, JP22K08123 (to K.K.), JP17K09573 (to K.U.), and JP19K17557 (to Y.F.).
Author information
Authors and Affiliations
Contributions
Y.F., K.K., K.U. and S.N. designed the study, performed the experiments, interpreted the results, and wrote the manuscript. Y.O. and Y.S. performed RNA-sequencing. M.T. and T.M. interpreted the data and discussed the research procedures with Y.F., K.K., K.U. and S.N.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fujikawa, Y., Kato, K., Unno, K. et al. Dynamic upregulation of retinoic acid signal in the early postnatal murine heart promotes cardiomyocyte cell cycle exit and maturation. Sci Rep 14, 20222 (2024). https://doi.org/10.1038/s41598-024-70918-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-70918-1
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
