
Abstract
Mammalian orthoreoviruses (MRVs), belonging to the genus Orthoreovirus in the family Spinareoviridae, possess a double-stranded RNA segmented genome. Due to the segmented nature of their genome, MRVs are prone to gene reassortment, which allows for evolutionary diversification. Recently, a genotyping system for each MRV gene segment was proposed based on nucleotide differences. In the present study, MRVs were isolated from the fecal samples of Japanese Black cattle kept on a farm in Japan. Complete genome sequencing and analysis of 41 MRV isolates revealed that these MRVs shared almost identical sequences in the L1, L2, L3, S3, and S4 gene segments, while two different sequences were found in the S1, M1, M2, M3, and S2 gene segments. By plaque cloning, at least six genetic constellation patterns were identified, indicating the occurrence of multiple inter- (S1 and M2) and intra- (M1, M3, and S2) reassortment events. This paper represents the first report describing multiple reassortant MRVs on a single cattle farm. These MRV gene segments exhibited sequence similarity to those of MRVs isolated from cattle in the U.S. and China, rather than to MRVs previously isolated in Japan. Genotypes consisting solely of bovine MRVs were observed in the L1, M1, and M2 segments, suggesting that they might have evolved within the cattle population.
Similar content being viewed by others


Introduction
Mammalian orthoreoviruses (MRVs) are members of the genus Orthoreovirus, along with Avian orthoreovirus, Nelson Bay orthoreovirus, Baboon orthoreovirus, Reptilian orthoreovirus, Mahlapitsi orthoreovirus, Piscine orthoreovirus, Broome orthoreovirus, Neoavian orthoreovirus, and Testudine orthoreovirus within the family Spinareoviridae, order Reovirales (https://ictv.global/report/chapter/spinareoviridae/spinareoviridae/orthoreovirus as of June 2024). MRV is an icosahedral, non-enveloped, double-stranded RNA virus. Their virions have a diameter of 60–80 nm and contain ten gene segments consisting of three large (L1, L2, and L3), three medium (M1, M2, and M3), and four small (S1, S2, S3, and S4) genes. These ten gene segments encode eight structural proteins (λ1, λ2, λ3, µ1, µ2, σ1, σ2, and σ3) and four nonstructural proteins (µNS, µNSC, σNS, and σ1s)1. The σ1 protein is a cell attachment protein and a serotype-specific antigen of MRV, recognized by neutralizing antibodies2,3. Based on antigenicity in neutralization, hemagglutination inhibition tests, and the genetic relationship of the S1 gene, MRVs are currently classified into four serotypes: MRV1-4, whose representative prototype strains are Lang (T1L), Jones (T2J), Dearing (T3D), Abney (T3A), and Ndelle (T4N)4,5. Due to their discrete segmented genes, MRV genomes have the capacity for genetic reassortment during co-infection with different MRV strains6. Recently, a genotyping system for each MRV gene segment was proposed based on phylogenetic relationships and nucleotide sequence conservation7.
MRVs spread mainly by respiratory or fecal–oral routes and are widely distributed throughout the world. MRVs infect a broad range of animal species, such as humans8,9, pigs10,11,12,13,14,15,16,17,18, horses19, deer20, chamois21, dogs22,23, cats24,25, bats26,27,28,29,30, rodents31,32, minks33,34, shrews35, and masked palm civets36. MRVs have also been found in wastewater37,38. Most cases of MRV infection appear to be asymptomatic; however, sporadic cases with respiratory disorders and gastroenteritis have been reported in humans and pigs9,10,13,14,15,16,17,39. Recently, more severe cases showing respiratory and neurological diseases have been reported in humans39,40,41,42,43. In cattle, although the clinical signs of MRV infection are unclear, several bovine MRV strains have been isolated from fecal samples44. However, at present, only three complete genome sequences of bovine MRV strains are available45. In the present study, while investigating enteric viruses in Japanese Black cattle on a farm, we isolated MRVs from fecal samples of cattle. These MRVs were found to be one of the most dominant strains of bovine MRVs and prevalent in specific areas. Furthermore, multiple inter- and intra-reassortment events were identified in these viruses.
Results
Isolation and complete genome sequencing of MRVs from fecal samples of cattle
Forty-one fecal samples were collected from four diarrheal and 37 non-diarrheal Japanese Black cattle (including three calves with fever) kept on a farm located on the main island of Japan. These samples were tested by reverse transcription-polymerase chain reaction (RT-PCR) for rotavirus A, bovine coronavirus, bovine torovirus, and bovine viral diarrhea virus, resulting in 10, 1, 1, and 1 positive samples, respectively (Supplementary Table 1). All samples were subjected to virus isolation using a monkey kidney epithelial cell line (MA104). After two or three blind passages, cytopathic effects (CPE) were observed in MA104 cells two to four days after inoculation from all 41 samples. Supernatants of cell cultures exhibiting CPE were subjected to deep sequencing, and the nearly complete genome sequences of the 10 MRV segments from 41 strains [MRV/Cattle/HH-xx/2021/JPN (HH strain)] were determined. Twelve isolates had a single near-complete gene sequence in each of the 10 MRV gene segments, while the remaining 29 isolates were found to possess two different near-complete or partial sequences in any of the S1, M1, M2, M3, and S2 gene segments, indicating co-infection of multiple MRV strains (Table 1). The nucleotide sequences of these MRV strains obtained in this study were deposited in the GenBank/EMBL/DDBJ under accession numbers LC818341–LC818808 (Supplementary Table 2).
Phylogenetic analysis
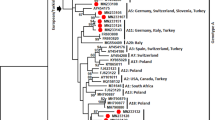
Phylogenetic analysis was performed using nearly complete S1 gene sequences of HH strains and MRV S1 gene sequences obtained from the GenBank/EMBL/DDBJ databases. The S1 gene sequences of HH strains were divided into T1 (25 samples) and T3 (35 samples) genotypes, and 19 strains were found to possess both T1 and T3 sequences. The sequence of the HH strain classified as T1 showed 94.84–94.18% nucleotide identity to that of human and bat MRVs and Chinese bovine MRV YNSZ/V207/2016, forming a cluster within T1(1). On the other hand, all T3 sequences of the HH strain branched in close relation to the Chinese bovine MRV GXLZ2301/2023, showing 92.55% nucleotide identity and forming T3(2) clusters with human and bat MRVs (Fig. 1A, Table 2). Phylogenetic analysis of the M2 gene showed that HH strains were also divided into two groups. Twenty-eight M2 gene sequences of HH strains branched together with US bovine MRV 00304/2014, showing 94.67–94.71% nucleotide identity, forming a cluster of bovine-only genotype 4. On the other hand, 25 HH strain M2 gene sequences formed a cluster with Chinese bovine MRV GXLZ2301/2023, showing 91.52–92.61% nucleotide identity within genotype 2. This genotype 2 bovine MRV cluster was supported by a bootstrap value of 100% and shared 81.29–82.52% nucleotide sequence identities with a group of human and porcine MRV genotype 2 strains. Thus, these clades were thought to be sub-genotype clades, 2a and 2b (Fig. 1B, Table 2, Supplementary Table 2).

Phylogenetic analyses based on the nearly complete S1 (A), M2 (B), S2 (C), M1 (D), M3 (E), and L1 (F) gene nucleotide sequences of HH MRVs and MRV strains obtained from the GenBank/EMBL/DDBJ databases. The phylogenetic trees were constructed using the maximum-likelihood method in MEGA7 with best fit models (the GTR + G model for the S1, the TN93 + G + I model for the M2, the T92 + G + I model for the S2, and the GTR + G + I model for the M1, M3, and L1 phylogenetic trees). Bootstrap values above 70 (1000 replicates) are indicated. The bars represent the corrected genetic distances. HH MRVs, bovine MRVs, and Japanese MRVs are indicated in red, purple, and green, respectively.
In the S2, M1, and M3 phylogenetic trees, the HH strains were divided into two groups within the same genotype. The 38 S2 genes of the HH strain formed a cluster sharing 99.77–100% nucleotide sequence identity with each other (named lineage 1), and the 6 S2 genes of the HH strain formed a group with 100% nucleotide identity (named lineage 2). These groups exhibited 93.7–93.86% nucleotide identities to each other and formed a cluster with US bovine MRV 00304/2014 within genotype 1 (Fig. 1C, Supplementary Table 3). M1 gene segments of HH strains formed a cluster of genotype 5 with US bovine MRV 00304/2014. Thirty-six and 12 HH strains grouped with 91.76–91.99% nucleotide sequence identities to lineage 1 and lineage 2, respectively (Fig. 1D, Supplementary Table 3). In the M3 phylogenetic tree, thirty-six and twenty-one HH strains formed lineage 1 and lineage 2, respectively. These two groups showed 91.73–91.77% nucleotide sequence identities and, together with the US bovine MRV 00304/2014, formed a cluster within genotype 1 (Fig. 1E, Supplementary Table 3).
For the L1 phylogenetic tree, HH strains sharing 99.32–100% nucleotide sequence identities with each other branched into one cluster and formed a bovine-only clade with MRV 00304/2014 and MRV GXLZ2301/2023 that fall into genotype 5 (Fig. 1F, Supplementary Table 3).
In the L2, L3, S3, and S4 phylogenetic trees, HH strains shared high nucleotide identities with each other (≥ 98.93%) and branched closely related to MRV 00304 within large clades (genotype 1 of L2, S3, and S4, and genotype 1c of L3) (Table 1, Supplementary Fig. 1A–D, Supplementary Table 3).
Detection of genome constellation patterns
Recently, Diller et al. proposed an MRV genome-based classification system based on sequence differences in each gene segment, in which the genotype of S1 is described first, followed by the genotypes of the remaining segments in order from largest to smallest7. According to this classification system, the 12 HH strains, each having one nucleotide sequence in each gene segment, were classified into three constellation patterns: T3(2)-5-1-1c-5Lin1-2b-1Lin1-1Lin1-1-1 (HH-1, 5, 7, 8ko, 10ko, 13, 14, and 30), T1(1)-5-1-1c-5Lin1-4-1Lin1-1Lin1-1-1 (HH-26 and 28), and T1(1)-5-1-1c-5Lin2-4-1-1Lin2-1Lin2-1-1 (HH-27 and 100ko) for denoting the S1–L1–L2–L3–M1–M2–M3–S2–S3–S4 genes, respectively. The remaining 29 strains contained two different near-complete or partial sequences of the S1, M1, M2, M3, and S2 genes. To obtain strains with a single gene constellation pattern from these strains that contained two nucleotide sequences in one gene segment, plaque cloning was performed (Supplementary Fig. 2). Strains cloned by plaque assay were typed for gene constellation pattern by PCR using primers discriminating S1, M1, M2, M3, and S2 segments (Supplementary Table 4). Four more genome constellations (T1(1)-5-1-1c-5Lin1-4-1Lin2-1Lin1-1-1, T3(2)-5-1-1c-5Lin1-4-1Lin2-1Lin1-1-1, T3(2)-5-1-1c-5Lin1-4-1Lin1-1Lin1-1-1, and T3(2)-5-1-1c-5Lin2-4-1Lin1-1Lin1-1-1) were obtained from 21 clones of HH-2, HH-6, HH-9oya, and HH-11ko (Fig. 2).

Segment-based genome constellation of HH strains including U.S. and Chinese bovine MRV strains.
Discussion
In the present study, MRVs were isolated using MA104 cells from 41 Japanese Black cattle on one farm. We extracted RNA directly from fecal samples and performed deep sequencing using an Illumina MiSeq platform as described previously46; however, only a few MRV reads were obtained (data not shown). Thus, virus isolation using MA104 cells is thought to be a useful tool for MRV detection and characterization. The farm is a fattening and breeding operation that often introduces cattle from other farms. The MRVs isolated in this study might have resulted from two or more strains invading this farm or MRVs that have invaded the farm in the past and are now causing recurrent outbreaks. The detection of multiple virus strains within a single farm has been frequently reported for rotavirus47,48,49, but not for MRV in cattle farms.
Phylogenetic analyses and genomic comparison using HH strain nucleotide sequences obtained in this study and MRV sequences from the GenBank/EMBL/DDBJ databases revealed that the HH strains exhibit the T1(1)/T3-5-1-1c-5-2b/4-1-1-1-1 genome constellation. The HH strains were closely related to the US bovine MRV 00,304/2014 (T1(2)-5-1-1c-5-4-1-1-1-1) in all gene segments except the S1 gene. The HH strains were also closely related to the Chinese bovine MRV GXLZ2301/2023 (T3(2)-5-2-2-2-2b-2-1-1-4) in the S1, L1, and M2 genes. Furthermore, the S1 gene of the Chinese bovine MRV strain YNSZ/V207/2016 (T1(1)-1-1-1c-1-1a-1-1-1-1) was closely related to T1(1) HH strains. None of the gene segments of the HH strain had high similarity to those of Japanese MRV from humans, swine, wild boar, zoo lions, and sewage. These findings suggest that the gene segments of the HH strains might be derived from bovine MRVs in the United States and China. However, imports of live cattle to Japan are conducted under an import quarantine system based on the Livestock Infectious Disease Prevention Act, and are available from Australia and New Zealand, which are currently free of foot-and-mouth disease and have concluded livestock health conditions with Japan. In the past 10 years, approximately 15,000 head of cattle for both breeding and fattening have been imported exclusively from Australia50. Although the status of MRV infection in cattle in Australia is unknown, the gene segments of the HH strains, particularly L1, M1, and M2, which have not been reported from other than cattle MRV, suggest that MRV may have evolved in the cattle population. Since only three complete genome sequences of bovine MRV strains apart from the HH strains have been reported, further investigation of bovine MRV is warranted.
As reoviruses possess segmented genomes, reoviruses, including MRVs, exhibit a high propensity for genetic reassortment, contributing to major evolutionary diversification of circulating viruses6. In this study, there were two gene sequences in the S1, S2, M1, M2, and M3 genes of the HH strains, and seven different genome constellation patterns were identified, suggesting the occurrence of multiple reassortment events. Reassortment of gene segments occurs when two or more reovirus strains co-infect the same cell. In the present case, at least two or more genotypes of MRV were thought to have invaded the farm and repeatedly infected the cattle population. Among the HH strains, the number of strains with near-complete S2 genotype 1 lineage 2 sequences was smaller than that of strains with genotype 1 lineage 1 sequences. This suggests that strains carrying the lineage 2 sequence were less prevalent than strains carrying the lineage 1 sequence, and there was only one pattern of genome constellation. S1 genotypes T1(1) and T3(2) and M2 genotypes 2b and 4 were found in approximately equal numbers in the HH strains (with T3(2) slightly more than T1(1)); however, no T1(1)-2b combinations were detected in strains having one sequence in each gene segment and plaque-cloned strains, suggesting that the T3(2) strain that invaded the farm may have carried the M2-2b gene. It is unclear whether the lack of detected reassortment strains was due to segment incompatibility or other reasons. Pairwise sequence identities between lineage 1 and lineage 2 of the HH strains in the S2, M1, and M3 genes were 93.7–93.86%, 91.76–91.99%, and 91.73–91.77%, respectively, which is high similarity compared to that between genotypes. In the present study, complete sequencing of 41 HH strains revealed that they were divided into two groups and that intra-genotype reassortment can occur frequently. On the other hand, no recombination event was found in any of the segments (data not shown). Since genetic reassortment can occur under artificial conditions51, the reassortant viruses detected in this study may have been generated during cell culture. However, since almost all the gene segments of the HH strains are closely related to bovine-derived MRVs and may have a replication advantage in cattle, it is possible that genetic reassortment could have occurred easily and persisted in cattle. Although HH strains, the US bovine MRV 00304/2014, and Chinese bovine MRV GXLZ2301/2023 formed bovine-only genotypes in the M1, M2, and L1 genes, the Chinese bovine MRV YNSZ/V207/2016 did not branch into a bovine-only genotype and all gene segments were classified into genotype 1, which includes MRVs from many host species. Furthermore, the L2, L3, M1, M3, and S4 gene segments of GXLZ2301/2023 were classified as rare genotypes with few strains. Since only three whole genome sequences of bovine MRV have been sequenced apart from the HH strain, the characteristics of bovine MRV genes and reassortment biases are still unknown; thus, additional complete bovine MRV genome sequencing is needed.
In summary, we identified multiple inter- (S1 and M2) and intra- (M1, M3, and S2) reassortant events in HH strains from Japanese Black cattle on one farm, and 7 genome constellation patterns of MRV strains were found. These bovine MRVs in this study shared sequence similarity to U.S. and Chinese MRVs, not to Japanese MRVs from other animal species, and formed genotypes consisting only of bovine MRVs in the L1, M1, and M2 genes, suggesting that they might have evolved in the cattle population. These results suggest that the bovine MRVs in the present study have evolved mainly through reassortment among bovine MRVs, increasing genetic diversity. These findings shed light on the enhanced genetic diversity and evolution of MRV from not only bovine but also other animal species.
Materials and methods
Sample collection, virus isolation, and plaque assay
The present study was carried out according to the Fundamental Guidelines for Proper Conduct of Animal Experiments and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology of Japan. Forty-one fecal samples were collected from Japanese Black cattle (four diarrheal calves and 37 healthy cattle) kept on a beef farm located on the main island of Japan for enteric virus isolation (Supplementary Table 1). Samples were directly collected from the rectum, transported to the laboratory under refrigeration, and stored at − 80 °C until use for virus isolation. The samples were diluted 1:9 (w/v) with Eagle’s minimal essential medium (EMEM) (Nissui, Tokyo, Japan) and centrifuged at 12,000 × g for 10 min. The supernatant was activated by adding an equal volume of 20 μg/mL trypsin (Sigma-Aldrich, Cat. No. 0303; MO, USA) and incubated for 1 h at 37 °C. The activated samples were inoculated into MA104 cells (RCB0994). The cells were purchased from RIKEN BioResource Research Center (RCB0994). Confluent monolayers of MA104 cells in 48-well plates were washed three times with EMEM and inoculated with 0.1 mL supernatant of the activated fecal sample. After adsorption for 60 min at 37 °C, the cells were washed three times with replacement EMEM containing 1 μg/mL trypsin and incubated for 7 days at 37 °C and 5% CO2. If a CPE was not observed after 7 days of incubation, the cells and supernatant were frozen and thawed three times and harvested; subsequent passages were performed in the same manner. For the plaque assay, MRV HH strain stocks were tenfold serial diluted with EMEM. Confluent MA104 cells were inoculated with the diluted samples and incubated for 60 min at 37 °C. After incubation, the inoculated media were replaced with EMEM containing 3% fetal bovine serum and 3% agarose (SeaPlaque™ Agarose, LONZA, Basel, Switzerland). After 5 days, well-separated plaques were picked through the agar overlay into the plaque area under a microscope. The picked plaque viruses were inoculated into MA104 cells, and when CPE appeared, these were collected and stored at − 80 °C until use for genotype discrimination.
RNA extraction, cDNA libraries construction, and deep sequencing
Total RNA was extracted from the supernatants of cell cultures exhibiting CPE using the MagDEA Dx SV cartridge (Precision System Science Co., Ltd., Chiba, Japan) and magLEAD6gC (Precision System Science Co., Ltd.), followed by DNase I treatment (Takara Bio, Otsu, Japan). cDNA libraries were constructed for deep sequencing using the NEBNext Ultra II RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA, USA), according to the manufacturer’s instructions. After assessing the library quantity on a Qubit® 4.0 Fluorometer (Invitrogen, Carlsbad, CA, USA), deep sequencing using paired-end reads of 151 nucleotides was conducted on a MiSeq benchtop sequencer (Illumina, San Diego, CA, USA) as previously described48.
RT-PCR
Total RNA was extracted from the supernatants of 10% fecal samples using TRIzol LS Reagent (Life Technologies) and subjected to the detection of Group A rotavirus, bovine coronavirus, bovine torovirus, and bovine viral diarrhea virus by qRT-PCR52 using the One Step PrimeScript™ RT-PCR Kit (Perfect Real Time) (Takara Bio). For MRV genotyping RT-PCR, RNA was extracted from the supernatant of cell culture using TRIzol LS Reagent (Life Technologies) and reverse transcription was performed using the PrimeScript™ Reverse Transcriptase (Takara Bio) with random primers. PCR with MRV genotype discrimination primers designed in this study (Supplementary Table 4) was performed using TaKaRa Ex Premier DNA Polymerase Dye plus (TaKaRa bio). The RT-PCR products were resolved via electrophoresis on a 2% agarose gel.
Genome analysis
Deep sequencing data were analyzed using MiSeq Reporter v2.5 (Illumina) to generate FASTQ-formatted sequence data and imported into CLC Genomics Workbench 7.5.5 (CLC bio, Aarhus, Denmark). The resulting sequence data were trimmed, and low-quality sequences were omitted. The processed sequence data were then assembled into contigs using the de novo assembly command in the CLC Genomics Workbench. Near-complete genome sequences of the MRV segments were aligned with MRV sequences obtained from the GenBank/EMBL/DDBJ database using ClustalW53. Phylogenetic analyses were constructed using nucleotide sequences of all segments using the maximum-likelihood method with the best-fit model (the GTR + G model for the S1, the TN93 + G + I model for the M1 and S3, the T92 + G + I model for the S2, the GTR + G + I model for the M1, M3, L1, L2, L3, and the K2 + G + I model for S4 phylogenetic trees) in MEGA754. The phylogenetic trees were evaluated using 1000 replicates in the bootstrap analysis55. Pairwise sequence identity calculations were performed for each gene segment using the CLC Genomics Workbench. Recombination analysis was performed using SimPlot56 and RDP557.
Ethical statement
The present study was carried out according to the Fundamental Guidelines for Proper Conduct of Animal Experiments and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology of Japan. This study is reported in accordance with ARRIVE guidelines (https://arriveguidelines.org).
Data availability
The GenBank/EMBL/DDBJ accession numbers for the sequences of the HH strains determined in this study are LC818341 to LC818808. Other datasets generated or analyzed during the current study are available from the corresponding authors upon reasonable request.
References
Day, J. M. The diversity of the orthoreoviruses: Molecular taxonomy and phylogenetic divides. Infect. Genet. Evol. 9, 390–400. https://doi.org/10.1016/j.meegid.2009.01.011 (2009).
Lee, P. W., Hayes, E. C. & Joklik, W. K. Protein sigma 1 is the reovirus cell attachment protein. Virology 108, 156–163. https://doi.org/10.1016/0042-6822(81)90535-3 (1981).
Weiner, H. L. & Fields, B. N. Neutralization of reovirus: The gene responsible for the neutralization antigen. J. Exp. Med. 146, 1305–1310. https://doi.org/10.1084/jem.146.5.1305 (1977).
Wiener, J. R. & Joklik, W. K. The sequences of the reovirus serotype 1, 2, and 3 L1 genome segments and analysis of the mode of divergence of the reovirus serotypes. Virology 169, 194–203. https://doi.org/10.1016/0042-6822(89)90055-x (1989).
Attoui, H. et al. Sequence characterization of Ndelle virus genome segments 1, 5, 7, 8, and 10: Evidence for reassignment to the genus Orthoreovirus, family Reoviridae. Biochem. Biophys. Res. Commun. 287, 583–588. https://doi.org/10.1006/bbrc.2001.5612 (2001).
McDonald, S. M., Nelson, M. I., Turner, P. E. & Patton, J. T. Reassortment in segmented RNA viruses: Mechanisms and outcomes. Nat. Rev. Microbiol. 14, 448–460. https://doi.org/10.1038/nrmicro.2016.46 (2016).
Diller, J. R., Thoner, T. W. Jr. & Ogden, K. M. Mammalian orthoreoviruses exhibit rare genotype variability in genome constellations. Infect. Genet. Evol. 110, 105421. https://doi.org/10.1016/j.meegid.2023.105421 (2023).
Tai, J. H. et al. Prevalence of reovirus-specific antibodies in young children in Nashville, Tennessee. J. Infect. Dis. 191, 1221–1224. https://doi.org/10.1086/428911 (2005).
Mikuletič, T., Steyer, A., Kotar, T., Zorec, T. M. & Poljak, M. A novel reassortant mammalian orthoreovirus with a divergent S1 genome segment identified in a traveler with diarrhea. Infect. Genet. Evol. 73, 378–383. https://doi.org/10.1016/j.meegid.2019.06.002 (2019).
Cavicchio, L. et al. Unexpected genetic diversity of two novel swine MRVs in Italy. Viruses 12(5), 574. https://doi.org/10.3390/v12050574 (2020).
Fukase, Y. et al. Genetic diversity, reassortment, and recombination of mammalian orthoreoviruses from Japanese porcine fecal samples. Arch. Virol. 167, 2643–2652. https://doi.org/10.1007/s00705-022-05602-8 (2022).
Harima, H. et al. Characterization of mammalian orthoreoviruses isolated from feces of pigs in Zambia. J. Gen. Virol. 101, 1027–1036. https://doi.org/10.1099/jgv.0.001476 (2020).
Hirahara, T. et al. Characteristics of reovirus type 1 from the respiratory tract of pigs in Japan. Nihon. Juigaku. Zasshi 50, 353–361. https://doi.org/10.1292/jvms1939.50.353 (1988).
Lelli, D. et al. First identification of mammalian orthoreovirus type 3 in diarrheic pigs in Europe. Virol. J. 13, 139. https://doi.org/10.1186/s12985-016-0593-4 (2016).
Luo, Y. et al. Prevalence and genomic characteristics of a novel reassortment mammalian orthoreovirus type 2 in diarrhea piglets in Sichuan, China. Infect. Genet. Evol. https://doi.org/10.1016/j.meegid.2020.104420 (2020).
Qin, P. et al. Genetic and pathogenic characterization of a novel reassortant mammalian Orthoreovirus 3 (MRV3) from a diarrheic piglet and seroepidemiological survey of MRV3 in diarrheic pigs from east China. Vet. Microbiol. 208, 126–136. https://doi.org/10.1016/j.vetmic.2017.07.021 (2017).
Thimmasandra Narayanappa, A. et al. A novel pathogenic mammalian orthoreovirus from diarrheic pigs and swine blood meal in the United States. mBio 6, e00593-e1515. https://doi.org/10.1128/mBio.00593-15 (2015).
Ye, D. et al. Molecular characterization of an emerging reassortant mammalian orthoreovirus in China. Arch. Virol. 165, 2367–2372. https://doi.org/10.1007/s00705-020-04712-5 (2020).
Conner, M. et al. Isolation and characteristics of an equine reovirus type 3 and an antibody prevalence survey to reoviruses in horses located in New York State. Vet. Microbiol 9, 5–25. https://doi.org/10.1016/0378-1135(84)90075-0 (1984).
Ahasan, M. S. et al. Molecular characterization of a novel reassortment Mammalian Orthoreovirus type 2 isolated from a Florida White-tailed deer fawn. Virus. Res. https://doi.org/10.1016/j.virusres.2019.197642 (2019).
Besozzi, M. et al. Host range of mammalian orthoreovirus type 3 widening to alpine chamois. Vet. Microbiol. 230, 72–77. https://doi.org/10.1016/j.vetmic.2019.01.012 (2019).
Decaro, N. et al. Virological and molecular characterization of a mammalian Orthoreovirus type 3 strain isolated from a dog in Italy. Vet. Microbiol. 109, 19–27. https://doi.org/10.1016/j.vetmic.2005.05.014 (2005).
Kokubu, T. et al. Isolation of reovirus type 3 from dogs with diarrhea. J. Vet. Med. Sci. 55, 453–454. https://doi.org/10.1292/jvms.55.453 (1993).
Mochizuki, M., Tamazumi, T., Kawanishi, A., Azuma, T. & Shimizu, T. Serotype 2 reoviruses from the feces of cats with and without diarrhea. J. Vet. Med. Sci. 54, 963–968. https://doi.org/10.1292/jvms.54.963 (1992).
Muir, P., Harbour, D. A. & Gruffydd-Jones, T. J. Reovirus type 2 in domestic cats: Isolation and experimental transmission. Vet. Microbiol. 30, 309–316. https://doi.org/10.1016/0378-1135(92)90018-o (1992).
Ichikawa, A. et al. Isolation and genetic characterization of a mammalian orthoreovirus from Vespertilio sinensis in Japan. Arch. Virol. 168, 165. https://doi.org/10.1007/s00705-023-05782-x (2023).
Lelli, D. et al. Detection and characterization of a novel reassortant mammalian orthoreovirus in bats in Europe. Viruses 7, 5844–5854. https://doi.org/10.3390/v7112908 (2015).
Naglič, T. et al. Identification of novel reassortant mammalian orthoreoviruses from bats in Slovenia. BMC Vet. Res. 14, 264. https://doi.org/10.1186/s12917-018-1585-y (2018).
Wang, L. et al. Isolation and identification of a natural reassortant mammalian orthoreovirus from least horseshoe bat in China. PLoS One. 10, e0118598. https://doi.org/10.1371/journal.pone.0118598 (2015).
Yang, X. L. et al. Isolation and identification of bat viruses closely related to human, porcine, and mink orthoreoviruses. J. Gen. Virol. 96, 3525–3531. https://doi.org/10.1099/jgv.0.000314 (2015).
Fehér, E. et al. Isolation and complete genome characterization of novel reassortant Orthoreovirus from common vole (Microtus arvalis). Virus Genes. 53, 307–311. https://doi.org/10.1007/s11262-016-1411-1 (2017).
Gauvin, L. et al. Respiratory infection of mice with mammalian reoviruses causes systemic infection with age- and strain-dependent pneumonia and encephalitis. Virol. J. 10, 67. https://doi.org/10.1186/1743-422X-10-67 (2013).
Lian, H. et al. Novel orthoreovirus from mink, China, 2011. Emerg. Infect. Dis. 19, 985–1988. https://doi.org/10.3201/eid1912.130043 (2013).
Zhang, Y. W. et al. A natural reassortant and mutant serotype 3 reovirus from mink in China. Arch. Virol. 161, 495–498. https://doi.org/10.1007/s00705-015-2670-1 (2016).
Li, X. et al. Isolation and identification of two new strains of mammalian orthoreovirus from Chinese tree shrews. Arch. Virol. 165, 1541–1550. https://doi.org/10.1007/s00705-020-04635-1 (2020).
Li, Z. et al. Isolation and pathogenicity of the mammalian Orthoreovirus MPC/04 from masked civet cats. Infect. Genet. Evol. 36, 5–61. https://doi.org/10.1016/j.meegid.2015.08.037 (2015).
Sedmak, G., Bina, D., Macdonald, J. & Couillard, L. Nine-year study of the occurrence of culturable viruses in source water for two drinking water treatment plants and the influent and effluent of a Wastewater Treatment Plant in Milwaukee, Wisconsin (August 1994 through July 2003). Appl. Environ. Microbiol. 71, 1042–1050. https://doi.org/10.1128/AEM.71.2.1042-1050.2005 (2005).
Kitamura, K. et al. Intertypic reassortment of mammalian orthoreovirus identified in wastewater in Japan. Sci. Rep. 11(1), 12583. https://doi.org/10.1038/s41598-021-92019-z (2021).
Yamamoto, S. P. et al. Novel human reovirus isolated from children and its long-term circulation with reassortments. Sci. Rep. 10, 963. https://doi.org/10.1038/s41598-020-58003-9 (2020).
Hermann, L., Embree, J., Hazelton, P., Wells, B. & Coombs, R. T. Reovirus type 2 isolated from cerebrospinal fluid. Pediatr. Infect. Dis. J. 23, 373–375. https://doi.org/10.1097/00006454-200404000-00026 (2004).
Chua, K. B. et al. A previously unknown reovirus of bat origin is associated with an acute respiratory disease in humans. Proc. Natl. Acad. Sci. USA 104, 11424–11429. https://doi.org/10.1073/pnas.0701372104 (2007).
Cheng, P. et al. A novel reovirus isolated from a patient with acute respiratory disease. J. Clin. Virol. 45, 79–80. https://doi.org/10.1016/j.jcv.2009.03.001 (2009).
Chua, K. B. et al. Investigation of a potential zoonotic transmission of orthoreovirus associated with acute influenza-like illness in an adult patient. PLoS One. 6(10), e25434. https://doi.org/10.1371/journal.pone.0025434 (2011).
Hrdy, D. B., Rosen, L. & Fields, B. N. Polymorphism of the migration of double-stranded RNA segments of reovirus isolates from humans, cattle, and mice. J. Virol. 31, 104–111. https://doi.org/10.1128/JVI.31.1.104-111.1979 (1979).
Anbalagan, S., Spaans, T. & Hause, B. M. Genome sequence of the novel reassortant mammalian Orthoreovirus strain MRV00304/13, isolated from a calf with diarrhea from the United States. Genome Announc. 2(3), e00451-e514. https://doi.org/10.1128/genomeA.00451-14 (2014).
Nagai, M. et al. Full genome analysis of bovine astrovirus from fecal samples of cattle in Japan: Identification of possible interspecies transmission of bovine astrovirus. Arch. Virol. 160, 491–501. https://doi.org/10.1007/s00705-015-2543-7 (2015).
Malik, Y. S. et al. Multispecies reassortant bovine rotavirus strain carries a novel simian G3-like VP7 genotype. Infect. Genet. Evol. 41, 63–72. https://doi.org/10.1016/j.meegid.2016.03.023 (2016).
Papp, H. et al. Review of group A rotavirus strains reported in swine and cattle. Vet. Microbiol. 30, 190–199. https://doi.org/10.1016/j.vetmic.2013.03.020 (2013).
Oki, H. et al. Genomic diversity and intragenic recombination of species C rotaviruses. J. Gen. Virol. https://doi.org/10.1099/jgv.0.001703 (2022).
Morino, S. et al. Analysis of the properties of bovine viral diarrhea virus isolated from the Australian imported cattle herd and the status of possession of neutralizing antibodies in the group. J. Jpn. Vet. Med. Assoc. (in Japanese, In press).
Moody, M. D. & Joklik, W. K. The function of reovirus proteins during the reovirus multiplication cycle: Analysis using monoreassortants. Virology. 173, 437–446. https://doi.org/10.1016/0042-6822(89)90556-4 (1989).
Tsuchiaka, S. et al. Development of a novel detection system for microbes from bovine diarrhea by real-time PCR. J. Vet. Med. Sci. 78, 383–389. https://doi.org/10.1292/jvms.15-0552 (2016).
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F. & Higgins, D. G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic. Acids. Res. 25, 4876–4882. https://doi.org/10.1093/nar/25.24.4876 (1997).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. https://doi.org/10.1093/molbev/msw054 (2016).
Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution. 39, 783–791. https://doi.org/10.1111/j.1558-5646.1985.tb00420.x (1985).
Lole, K. S. et al. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73, 152–160. https://doi.org/10.1128/JVI.73.1.152-160.1999 (1999).
Martin, D. P. et al. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus. Evol. 7(1), veaa087. https://doi.org/10.1093/ve/veaa087 (2020).
Acknowledgements
This work was supported by Livestock Promotional Subsidy from the Japan Racing Association and JSPS KAKENHI (Grant number 21K05947).
Author information
Authors and Affiliations
Contributions
M.O., T.M., and M.N. conceived the experiments, M.O., M.S., H.T., S.M., H.I. and H.M. conducted the experiments, M.O., S.S. and M.N. analyzed the results. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Oba, M., Shimotori, M., Teshima, N. et al. Identification of multiple inter- and intra-genotype reassortment mammalian orthoreoviruses from Japanese black cattle in a beef cattle farm. Sci Rep 14, 19887 (2024). https://doi.org/10.1038/s41598-024-70863-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-70863-z
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
