
Abstract
Colorectal liver metastasis (CRLM) is challenging in the clinical treatment of colorectal cancer. Limited research has been conducted on how CRLM develops. RNA sequencing data were obtained from the Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA). Four machine learning algorithms were used to screen the hub CRLM-specific genes, including Least Absolute Shrinkage and Selection Operator (Lasso), Random forest, SVM-RFE, and XGboost. The model for identifying CRLM was developed using stepwise logistic regression and was validated using internal and independent datasets. The prognostic value of hub CRLM-specific genes was assessed using the Lasso-Cox method. The in vitro experiments were performed using SW620 cells. The CRLM identification model was developed based on four CRLM-specific genes (SPP1, ZG16, P2RY14, and PRKAR2B), and the model efficacy was validated using GSE41258 and three external cohorts. Five CRLM-specific prognostic hub genes, SPP1, ZG16, P2RY14, CYP2E1, and C5, were identified using the Lasso-Cox algorithm, and a risk score was constructed. The risk score was validated using the GSE39582 cohort. Three genes have both efficacy in identifying CRLM and prognostic value: ZG16, P2RY14, and SPP1. Immune infiltration and enrichment analyses demonstrated that SPP1 was associated with M2 macrophage polarization and extracellular matrix remodeling. In vitro experiments indicated that SPP1 may act as a cancer-promoting factor. The hub CRLM-specific gene SPP1 can help determine the diagnosis, prognosis, and immune infiltration of patients with CRLM.
Similar content being viewed by others


Introduction
Colorectal liver metastasis (CRLM) is the most prevalent form of metastasis in colorectal cancer and is a challenging and key point in the clinical treatment of colorectal cancer1. Tumor metastasis is the discontinuous implantation of a primary tumor that unequivocally marks it as malignant2. However, the process of tumor metastasis and the reasons why tumors tend to metastasize to certain organs are not well known today3. An in-depth understanding of why colon cancer is more likely to metastasize to the liver might be helpful in the treatment of colon cancer liver metastasis. In general, we determine the origin of metastases based on their pathological morphology and immunohistochemical features; for instance, CDX2, an intestine-specific gene, is used as a biomarker for metastatic cancer originating from the intestines4,5. Recently, a study reported the presence of some hepatocyte gene programs and the absence of colon gene programs in tumor cells from liver metastases of colon cancer, which conflicts with our general understanding that tumor metastatic cells should retain the characteristics of the cells of the original tissue6. Therefore, screening for metastasis-specific genes may further elucidate why colon cancer is prone to liver metastasis and provide a research direction for future therapies that may block liver metastasis of colon cancer.
Currently, machine learning based on big data has achieved spectacular results in healthcare7,8. Clinical big data refer to a large volume and variety of data originating from complex sources9,10. Machine learning refers to a discipline of research and algorithms that focuses on finding patterns in data and using those patterns to make predictions11. The combination of clinical big data and machine learning can be highly conductive10,12.
In this study, different algorithms were selected based on machine learning to identify prognostic genes with discriminatory efficacy for colorectal liver metastasis and primary tumors, defined as colorectal liver metastasis-specific genes, exemplified by SPP1. Furthermore, we verified the cancer-promoting properties of SPP1 using cytological analysis. The results of enrichment analysis and immunoinfiltration analysis suggested that the infiltration level of M2 macrophages, which are recognized by scavenger receptors (CD206)13,14, is highly correlated with SPP1 and elicits extracellular matrix (ECM) remodeling15.
Materials and methods
Data collection and screening of colorectal liver metastasis-specific genes
RNA sequencing data and clinical information of colorectal cancer samples were obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/), including four cohorts: GSE41258, GSE49355, GSE68468, and GSE81585. GSE41258 contains 390 samples, including 186 cases of primary colorectal tumors and 47 cases of colorectal liver metastases, and was used as the training set for the present study. Three independent cohorts, GSE49355, GSE68468, GSE81585, and The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/), were used as validation sets.
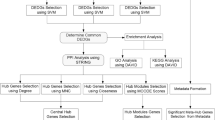
Differential gene expression between primary tumor and liver metastasis was performed by R (4.2.2 version) with ‘limma’ package. The threshold for differentially expressed genes (DEGs) was set at |log2FC|> 1 and p-value < 0.05. We referred to these DEGs as liver-metastasis-specific genes. The process flow diagram for this study is shown in Fig. 1.

Flow diagram of the current study. CRC, Colorectal cancer; CRLM, Colorectal liver metastasis; PPI, Protein–protein interaction; ROC, Receiver operating characteristic curve; DCA, Decision curve analysis; TCGA, The Cancer Genome Atlas; DEGs, Differentially expressed genes; EdU, 5- Ethynyl-2’- deoxyuridine.
Functional enrichment analysis and construction of protein–protein interactions (PPI) network
Gene Ontology (GO) analysis is a way to summarize haphazard liver metastasis-specific genes into a more holistic event. We performed GO analysis with ‘clusterProfiler’ package, ‘org.HS.eg.db’ package16. The statistical significance of pathways is dependent on a normal p-value < 0.05, and FDR q-val < 0.25.
A PPI network was established using the STRING database (http://string-db.org)17. The liver metastasis-specific genes sequences were included in the ‘Multiple proteins’ module of the STRING database for analysis. Besides, we used ‘cytoHubba’ plugin for hub gene analysis, involving ‘Maximal Clique Centrality (MCC)’ and ‘Edge Percolated Component (EPC)’ algorithms.
Machine learning algorithm
Four different machine learning models were applied for the dichotomous classification of liver metastases and primary tumors, including ‘Random Forest (RF),’ ‘Support Vector Machine Recursive Feature Elimination (SVM-RFE)’, ‘Least Absolute Shrinkage and Selection Operator (Lasso)’ and ‘eXtreme Gradient boosting (XGboost).’
The RF model, a supervised machine-learning algorithm based on classification trees proposed by Breiman and Cutler in 2001, improves the prediction accuracy of the model by aggregating a large number of classification trees. We performed the RF algorithm using the ‘randomForest’ R package18 (ntree = 500).
SVM-RFE is an algorithm for the feature extraction of data based on the embedded method. They are widely used in pattern recognition and machine learning. We performed the SVM-RFE algorithm using the ‘e1071, ‘ ‘kernlab’ and ‘caret’ R packages19.
Lasso is a data mining method that simplifies the model to avoid overfitting it. The R package ‘glmnet’ was used for the analysis in this study20.
In data modeling, the XGboost algorithm combines hundreds of tree models with low classification accuracy into a predictive model with high accuracy. In our study, the ‘xgboost,’ ‘tidyverse’ and ‘caret’ R packages were used for the XGboost algorithm21.
We then identified the intersecting genes of the four machine learning models as hub genes for colorectal liver metastasis-specific genes.
The discrimination and generalization performance of liver metastasis-specific genes
We performed univariate and multivariate logistic regression analyses to assess the discrimination performance of hub colorectal liver metastasis-specific genes and develop a model. The regression coefficients of the genes were used to calculate linear predictors for each sample. Furthermore, the predicted probability is based on linear predictors calculated using the following formula:
Model correlation tests were performed in R based on the ‘rms’ and’ ResourceSelection’ R packages. Variance inflation factors (VIF) were calculated to detect multicollinearity in the model. Model fit was assessed using a likelihood ratio test and calibration curve. C-index and receiver operating characteristic (ROC) curves were used to evaluate the predictive power of the model. The decision curve analysis (DCA) curve was used as an indicator of clinical effectiveness. Three independent cohorts, GSE49355, GSE68468, and GSE81585, were used to validate the generalization performance of the model.
Analysis of immune cell infiltration and GSEA enrichment analysis
CIBERSORT and ssGSEA were used to quantify relative immune cell infiltration levels. We performed the CIBERSORT algorithm using the ‘e1071,’ ‘preprocessCore’ and ‘BiocManager’ R packages and the ssGSEA algorithm by ‘GSVA’ R package. The Wilcoxon rank-sum test and Spearman’s correlation analysis were used to clarify the correlation between the expression of colorectal liver metastasis-specific genes and the level of immune cell infiltration. GESA analysis was performed to investigate the hallmarks of SPP1-positive group by ‘clusterProfiler’ R package16,22.
Cell culture and siRNA transfection
The human colon cancer cell line SW620, provided by Zhejiang Meisen Cell Technology Co., LTD, was cultured in DMEM with 10% FBS (Cegrogen) and 1% penicillin–streptomycin (Gibco). All the cells were maintained at 37 °C in a 5% CO2.
For knockdown studies of SPP1, siRNA and GP-transfect-mate were purchased from GenePharma (Shanghai, China). The siRNA sequences used for knockdown are detailed in Supplementary Table 1.
Quantitative real-time PCR (qRT-PCR) was performed to detect the knockdown efficiency using SYBR Green Supermix (TIANGEN Biotech) on an ABI7500 thermocycler. The ΔΔCt method was used to calculate the relative expression levels of the target mRNA. Primers for the internal reference gene and target gene were designed by Sangon Biotech (Shanghai, China) (Supplementary Table 2).
Migration or invasion assays
For transwell assays, cells were plated in the upper chamber of a transwell (Corning Cat). Cells in the upper chamber were placed in serum-free medium and medium containing 30% FBS was added to the lower chamber. To test invasive ability, Matrigel (LYNJUNE) was evenly spread on the upper chamber surface of the small chamber membrane to form a thin film.
For the wound healing assay, cells were plated in a six-well plate, and when the cells grew adherently, they were scratched with a 200μL sterile pipette tip, and the shed cells were washed away with PBS. Serum-free medium was added to continue the culture and the width of the scratch was recorded at 0, 24, and 48 h.
Cell proliferation assays
For the EdU assay, cells were plated in six-well plates and incubated with EdU working solution (APExBIO). After fixation and permeabilization, the EdU reaction complex was added, incubated at room temperature, and protected from light for 30 min. Nuclei were counterstained with Hoechst33342 dye and detected, and photographed using fluorescence microscopy.
Cell counting kit-8(CCK-8) was purchased from MedChemExpress. Cells were diluted into a suspension of 2*10^3 cells/100 μl, and 100 μl of the cell suspension was added to each well in a 96-well plate, with five sub-wells in each group. Add 10ul CCK-8 at 0, 24, 48 and 72 h. Incubate in the incubator for 1.5 h and measure the absorbance (450 nm) using a microplate reader (ThermoFisher).
For the colony formation assay, 500 cells per well were plated in a 6-well plate. The medium was changed every 3–4 days. Fixation and staining were performed after 14 d.
Cell apoptosis assays
For the cell apoptosis assay, flow cytometry (Beckman, FC500) was performed to detect apoptotic cells using an Annexin V-FITC/PI Apoptosis Kit (APExBIO).
Statistical analysis
R was used for all statistical analyses. To analyze the differences between the two datasets, we performed normality tests on the data. For samples that passed the normality test, a paired sample t-test was chosen, and for samples that failed the test, the Wilcoxon rank-sum test was chosen. Statistical significance was set at a two-tailed p-value of 0.05. Spearman’s correlation test was used for the co-expression analysis of genes and immuno-infiltration correlation analysis.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Result
Screening of colorectal liver metastasis-specific genes
To investigate the factors that contribute to the progression of primary colorectal cancer to metastases, we analyzed the differences in expression profiles between primary tumors and liver metastases in the GSE41258 cohort. We found 148 upregulated and 214 downregulated probes with statistical significance in liver metastases (Fig. 2A). A total of 204 differentially expressed genes (DEGs), covering 91 upregulated genes and 113 downregulated genes, were obtained after removing duplicate values and multiple annotated probes (Fig. 2B). We referred to these DEGs as colorectal liver specific genes.

Screening of colorectal liver metastasis-specific genes. (A) Volcano plot of differentially expressed genes (the red dots are 148 up-regulated probes and the blue dots are 214 down-regulated probes). (B) Heat map of 204 differentially expressed genes (DEGs), covering 91 up-regulated genes (upper part) and 113 down-regulated genes (bottom part). GO enrichment analysis on 91 up-regulated (C) and 113 down-regulated liver metastasis-specific genes (D). Protein–protein interaction network was constructed by STRING based on 204 metastasis-specific genes (E), of which hub genes were screened by the ‘cytoHubba’ plugin, involving the ‘MCC’ (F) and ‘EPC’(G) algorithms.
Potential biological function of liver metastasis-specific genes
We performed GO enrichment analysis of 91 upregulated and 113 downregulated liver metastasis-specific genes. The upregulated liver metastasis-specific genes were enriched in the vesicle lumen (GO:0031983), endoplasmic reticulum lumen (GO:0005788), and secretory granule lumen (GO:0034774)’ (Fig. 2C and Supplementary Table 3). While, the down-regulated liver metastasis-specific genes were significantly associated with extracellular matrix-related process, such as ‘Collagen-containing extracellular matrix (GO:0062023)’, ‘Extracellular matrix organization (GO:0030198)’ and ‘Extracellular structure organization (GO:0043062)’ (Fig. 2D and Supplementary Table 4).
Next, we constructed a PPI network for colorectal liver metastasis-specific genes, which was built with 190 nodes and 214 edges, with an average node dimension of 2.25 (Fig. 2E). The screening of hub genes were performed by the ‘cytoHubba’ plugin, involving the ‘MCC’ and ‘EPC’ algorithms (Fig. 2F–G). Based on these two scores, APOH, FGG, AMBP, and FGA were selected as the central genes of the PPI network.
Identification of the hub liver metastasis-specific genes based on machine learning
Four machine-learning algorithms were applied to identify hub liver metastasis-specific genes. The results of the Lasso algorithm suggested that the model fit best when lambda.min was 15 (Fig. 3A). The genes included in the Lasso model were SPP1, ZG16, PIGR and PRKAR2B, etc. For the SVM-RFE algorithm, the root mean square error (RMSE) of the ten-fold cross-validation was minimized when the number of characteristic genes was 19, and the screened genes included CYP2E1, SPP1, ZG16, and PIGR (Fig. 3B). For the RF algorithm, we set the number of iterations of the random forest classifier to 500 so that the out-of-bag (OOB) error was stable and less than 0.05 (Fig. 3C). Twelve genes, including SPP1, PRKAR2B, and ZG16, were identified as liver metastasis-specific genes using the RF algorithm. For the XGboost algorithm, the histogram of the importance of feature genes suggested that C9 ranked first, followed by PRKAR2B, CYP2E1, SPP1, and P2RY14 (Fig. 3D).

Identification of the hub liver metastasis-specific genes based on machine learning. (A) The lasso algorithm determined 15 feature genes based on lambda.min values. (B) The SVM-RFE algorithm determined 19 feature genes based on RMSE. (C) The RF algorithm determined 12 feature genes based on OOB error and MDG. (D) The XGboost algorithm determined 18 feature genes and ranked them by importance. (E) Venn diagram was plotted overlapping the results of the four algorithms described above, obtaining nine genes. Lasso, Least absolute shrinkage and selection operator; SVM-RFE, Support vector machine- recursive feature elimination; RMSE, The root mean square error; RF, Random forest; OOB, Out of bag; MDG, Mean decrease Gini;
Finally, nine intersecting genes were identified: CYP2E1, PRKAR2B, PIGR, C5, TPSAB1, P2RY14, C9, SPP1, and ZG16 (Fig. 3E and Table 1). To obtain the optimal model, we randomly split GSE41258 into a training set and validation set in a ratio of 7:3, and stepwise logistic regression was performed. Four genes (PRKAR2B, P2RY14, SPP1, and ZG16) were identified as hub colorectal liver metastasis-specific genes.
Model development and validation for identifying colorectal liver metastasis
Univariate logistic regression analysis showed good discrimination between liver metastases and primary colorectal tumors by hub colorectal liver metastasis-specific genes. Increased expression of SPP1 (OR = 1.554, 95%CI = 1.187–2.035, P = 0.001) was a risk factor for the development of colorectal liver metastases, while ZG16 (OR = 0.425, 95%CI = 0.292–0.619, P < 0.001), P2RY14 (OR = 0.052, 95%CI = 0.018–0.147, P < 0.001), and PRKAR2B (OR = 0.016, 95%CI = 0.004–0.070, P < 0.001) showed the opposite trend (Table 2).
We further performed multivariate logistic regression analysis (Table 2) and developed a model. The regression coefficients of the genes (Supplementary Table 5) were used to calculate the linear predictors of each sample to identify colorectal liver metastasis. The model is shown in Fig. 4A, and its performance is detailed in Supplementary Table 6. The VIF of all four genes included in the model was less than 3, suggesting that there was no serious multicollinearity (VIFSPP1 = 1.363, VIFZG16 = 1.674, VIFPRKAR2B = 1.358, and VIFP2RY14 = 2.412).

Model development and validation for identifying colorectal liver metastasis (A) Nomogram for identifying colorectal liver metastasis based on four hub liver metastasis-specific genes by stepwise logistic regression. (B–G) The Validation of the efficacy of identifying colorectal liver metastasis by calibration curve, ROC, and DCA curve in GSE41258 (B, Training set, AUCLinear predictors = 0.987; C, Validation set, AUCLinear predictors = 0.968; D, All set, AUCLinear predictors = 0.983), GSE49355 (E, AUCLinear predictors = 0.942), GSE81558 (F, AUCLinear predictors = 0.961), and GSE68468 (G, AUCLinear predictors = 0.873). ROC, Receiver operating characteristic; DCA, Decision curve analysis.
The chi-square value for the likelihood ratio test was 124.8 (P value < 0.001). The Hosmer–Lemeshow goodness-of-fit statistic showed no significant differences (Chi-square = 1.2479, P = 0.9961). The apparent line was similar to the bias-corrected line and close to the ideal line (Fig. 4B). The C-index of the model was 0.987 (0.973–1.000), and the ROC and DCA curves suggested that the efficacy and net benefit of using the model to identify colorectal liver metastasis were better than those of using the hub gene discriminant (AUC Linear predictors = 0.987, AUCPRKAR2B = 0.934, AUCP2RY14 = 0.929, AUCSPP1 = 0.682, AUCZG16 = 0.815). The validation set and full set, as well as the independent cohort validation results of GSE49355, GSE81558, and GSE68468, suggested that the model fit well and the model efficacy was superior (Fig. 4C–G, Validation set: AUCLinear predictors = 0.968, All set: AUCLinear predictors = 0.983, GSE49355: AUCLinear predictors = 0.942, GSE81558: AUCLinear predictors = 0.961, GSE68468: AUCLinear predictors = 0.873).
Prognostic value of the liver metastasis-specific genes
As survival data from the GSE41258 cohort were imperfect, TCGA data were selected for prognostic analysis. Based on the univariate Cox regression analysis (Fig. 5A,B) and the level of differential expression between tumor and normal tissues (Fig. 5C), 204 liver metastasis-specific genes were filtered to obtain nine liver metastasis-specific prognostic genes. Five prognostic hub genes were identified by LASSO regression analysis (Fig. 5D), and risk scores were constructed (Fig. 5E, Supplementary Table 7). This risk score demonstrated a favorable prognostic value in the TCGA cohort (Fig. 5F) and the independent external validation cohort GSE39582 (Fig. 5G).

Prognostic value of the liver metastasis-specific genes. (A) Univariate cox regression analysis was performed to screening prognostic liver metastasis-specific genes. (B) KM curve for predicting OS based on prognostic genes expression grouping. (C) Expression of prognostic liver metastasis-specific genes between normal and tumor in TCGA by unpaired samples. (D) A total of 5 prognostic hub genes were identified by lasso regression analysis and risk scores were constructed. (E) Distributions of risk scores, survival status and expression of 5 prognostic hub genes. KM curves of OS between low-risk and high-risk groups in TCGA (F, Training set) and GSE39582 (G, Testing set). (H) Prognostic genes with discriminatory efficacy for colorectal liver metastasis and primary tumor were defined as colorectal liver metastasis-specific genes, which were SPP1, ZG16 and P2RY14.
Eventually, we identified the prognostic genes with discriminatory efficacy for colorectal liver metastasis and primary tumors, defined as the hub colorectal liver metastasis-specific genes SPP1, ZG16, and P2RY14 (Fig. 5H).
Association of the hub liver metastasis-specific genes with immune infiltration and pathway enrichment analysis
We analyzed the relative infiltration of 22 immune cells in tumor samples from the GSE41258 cohort using the CIBERSORT algorithm. The levels of M0 and M2 macrophage (R = 0.359, P < 0.001) infiltration were higher in SPP1-positive tumors (Fig. 6A). Data from TCGA database similarly showed that the expression of SPP1 was positively correlated with the level of infiltration of macrophages (R = 0.792, P < 0.001, ssGSEA, Fig. 6B).

Association of the hub liver metastasis-specific genes with immune infiltration and pathway enrichment analysis. Correlations between SPP1 and tumor-infiltrating immune cells by CIBERSORT algorithm (A) and ssGSEA algorithm (B). (C) Association between SPP1 and M2 macrophage surface markers. Association between SPP1 and macrophage secretions, including VEGF (D) and TGF-β (E). (F–G) Ridge map of GSEA for DEGs from SPP1-positive tumors compared with SPP1-negative tumors.
Furthermore, we assessed the relationship between SPP1 and M2 macrophage surface markers (Fig. 6C) and found that the expression levels of CD163, CD115(CSF1R), PDL2(PDCD1LG2), and CD206(MRC1) positively correlated with the expression level of SPP1.
Moreover, we evaluated the relationship between SPP1 and M2 macrophage secretion, including VEGF and TGF-β (Fig. 6D,E). The expression levels of most M2 macrophage secretions (including VEGF-B, VEGF-C, VEGF-D, TGF-β1, TGF-β2, and TGF-β3) were higher in SPP1-positive tumors.
In addition, GESA was performed to investigate the potential biological mechanisms by which SPP1 affects tumor progression and metastasis. Immunomodulatory and extracellular matrix remodeling-related pathways were significantly enriched in SPP1-positive tumors (Fig. 6F,G).
Cytologic experimental validation of hub colorectal liver metastasis-specific genes (SW620/SPP1)
From the above bioinformatic analysis, it was found that SPP1 was significantly elevated in patients with colorectal liver metastasis and correlated with poor prognosis. SW620 cells originated from lymph node metastases, which are highly invasive and metastatic, and can rapidly spread to the liver We therefore performed cytological experiments using SW620 cells to explore the role of SPP1 in tumor metastasis and progression.
We knocked down SPP1 by using siRNA with the highest knockdown efficiency (Supplementary Fig. 1). Transwell and wound healing assays showed that the downregulation of SPP1 significantly attenuated the migratory capacity of SW620 cells23,24 (Fig. 7A–C). The results of the EdU (Fig. 7D, Supplementary Fig. 2), CCK-8 (Fig. 7E), and colony formation assays (Fig. 7F) were consistent, suggesting that SPP1 promoted the proliferation of SW620 cells. The flow cytometry results (Fig. 7G) provided evidence that SPP1 knockdown led to increased apoptosis.

Cytologic experimental validation of hub colorectal liver metastasis-specific genes (SW620/SPP1). The cellular migration assay of SW620 cells treated with si-NC and si-SPP1 by transwell assays (A) and wound healing assay (B, C). The cellular proliferation assay of SW620 cells treated with si-NC and si-SPP1 by EdU assay (D), CCK-8 assay (E) and colony formation assay (F). The cellular apoptosis assay of SW620 cells treated with si-NC and si-SPP1 by flow cytometry (G).
Discussion
There remain challenges in the diagnosis and management of colorectal cancer liver metastases25. Clinical diagnosis of colorectal liver metastasis is usually made with the help of postoperative pathologic results, including the pattern of metastasis and the expression of CDX2 and SATB2 detected by immunohistochemistry5,26,27,28. If CDX2 and SATB2 are positive, there is a high confidence that the metastases originate from the colorectum4,29. In other words, liver metastases from colorectal cancer retain specific features of colorectal tumors.
In recent years, numerous studies have suggested reprogramming as a hallmark of tumor progression and metastasis, such as transcriptional and metabolic reprogramming30,31,32. Transcriptional reprogramming is the phenomenon of overall changes in gene expression caused by transcription factors33,34. In colorectal liver metastasis, FOXA2 and HNF1A promote liver metastasis by combining with epigenetic enhancers in liver metastatic cells, thereby inducing liver-specific transcription6. Metabolic reprogramming refers to the mechanism by which cells promote cell proliferation and growth by altering their metabolic patterns in order to satisfy their energy needs, which not only helps the cells to resist external stress but also confers new functions on the cells35,36. The Warburg effect in tumorigenesis is one of the most widely studied cellular metabolic reprogramming modes, whereby tumor cells rapidly proliferate by altering their energy metabolism to adapt to the microenvironment of hypoxia, acidity, and nutrient deficiencies37. In colorectal liver metastasis, MTA1 mediates mitochondrial metabolic reprogramming and drives liver metastasis by increasing oxidative phosphorylation and ATP production38,39. Differences in creatine metabolism between metastatic and primary colorectal tumors mediate SMAD2/3 phosphorylation, which promotes liver metastasis40. Hence, the concept of reprogramming suggests that colorectal liver metastases have a characteristic gene expression pattern different from that of primary tumors.
To clarify this seemingly paradoxical phenomenon, we screened and identified colorectal liver metastasis-specific genes using a machine-learning approach to investigate the mechanism of metastasis. In this study, we screened 204 DEGs between metastatic and primary tumors from the GSE41258 cohort. Nine colorectal liver metastasis-specific genes were identified using four machine learning methods. GSE41258 was divided into training and validation sets in the ratio of 7:3, and based on stepwise logistic regression analysis, four hub liver metastasis-specific genes, SPP1, P2RY14, ZG16, PRKAR2B, were finally identified. We developed a CRLM identification model based on 4-hub colorectal liver metastasis-specific genes, and the model efficacy was validated using the validation set, full set, and external cohorts GSE49355, GSE81558, and GSE68468 using ROC, calibration, and DCA curves. We explored the prognostic value of colorectal liver metastasis-specific genes in TCGA database due to imperfect patient survival information in the GEO database. Five key liver metastasis-specific prognostic genes, SPP1, ZG16, P2RY14, CYP2E1, and C5, were identified using the LASSO-COX algorithm, and a risk score was constructed. The risk score was validated in the GSE39582 cohort.
Three genes have both efficacies in identifying liver metastasis and prognostic value, namely, ZG16, P2RY14, and SPP1. Evidence in the literature suggests that ZG16 is associated with distant and lymphatic metastasis, reduces PD-L1 expression, and may serve as a biomarker of immunotherapy benefit41, consistent with the bioinformatics analysis in this study. Furthermore, ZG16 inhibits stemness in colorectal cancer cells, thereby delaying colorectal cancer progression42. P2RY14 acts as a cancer suppressor in various tumors, such as ovarian cancer43, endometrial cancer44, lung cancer45 and colorectal cancer46,47, but most of these studies were based on bioinformatics analysis. In vitro experiments have demonstrated that activation of the P2Y14 receptor reduces IL-6 production, resulting in the inhibition of glioma progression48. SPP1 has been implicated in the PI3K/AKT pathway49 and β-catenin50 and mediates the epithelial-to-mesenchymal transition process in in vitro experiments of colorectal cancer. However, the cell lines selected for these studies, such as DLD1 and SW480, were non-metastatic. Therefore, the highly invasive and metastatic SW620 cell line was selected for in vitro experiments using SPP1. The results indicated that the knockdown of SPP1 gene expression could repress the proliferation and migration ability of SW620 cells, suggesting that SPP1 might act as a cancer-promoting factor in colorectal cancer.
To investigate the mechanism of SPP1 in the development of colorectal liver metastases, we performed immune infiltration analysis. The results showed a positive correlation between SPP1 and macrophage infiltration, especially M0 and M2 macrophages. Macrophages are a diverse group of leukocytes, with M0 macrophages forming M1 macrophages in response to activation by LPS and Th1 cytokines, such as IFN-γ and TNF-α, and M2 macrophages in response to activation by IL-4, IL-13, IL-10, IL-33, and TGF-β51,52. M1 and M2 macrophages infiltrating solid tumor tissues are referred to as tumor-associated macrophages (TAM), in which M2 macrophages with pro-tumorigenic activity are dominant53,54. Macrophages are extremely plastic, and M2 macrophages can be polarized into M1 macrophages with anti-tumor activity when stimulated by external conditions55. Based on this theoretical background, Egeblad reported an anti-tumor approach by reprogramming TAM56. Surface markers of macrophages are important for distinguishing M1 macrophages from M2 macrophages52. We further investigated the relationship between SPP1 and the surface markers of M2-type macrophages and found that the expression levels of CD163, CD115 (CSF1R), PDL2 (PDCD1LG2), and CD206 (MRC1) were positively correlated with the expression levels of SPP1. These results preliminarily validated the correlation between SPP1 and M2 macrophages. M2 macrophages can be divided into four cell subpopulations, M2a, M2b, M2c, and M2d, depending on the factors activated, in which VEGF secreted by cells of the M2d subpopulation plays a role in promoting angiogenesis and tumor progression51. To investigate whether SPP1 is involved in the pro-carcinogenic process of M2 macrophages, we evaluated the relationship between SPP1 and the VEGF family. The results showed that VEGF-B, VEGF-C, and VEGF-D had higher expression levels in SPP1-positive tumors. Moreover, we found a positive correlation between TGF-β secreted by M2c macrophages involved in apoptosis and the expression of SPP1. The results provide evidence that SPP1 may be involved in the pro-carcinogenic effects of M2 macrophages, which in turn affects tumor progression and metastasis.
Interestingly, the GSEA results demonstrated a correlation between SPP1 and scavenger receptor-associated immunoregulatory hallmarks, which was consistent with the immune infiltration analysis. Significant enrichment of SPP1 in extracellular matrix (ECM) remodeling-related pathways was also demonstrated by GSEA analysis. Tumor-associated ECM degradation is the main cause of normal tissue destruction by tumor cells and infiltrative growth of tumor tissues57. The ECM also plays an extremely important role in tumor immunity, and tumor-associated ECM has the potential to improve tumor immunotherapy58. Whereas not all patients with CRC respond well to immune checkpoint therapy in clinical care59,60, nutritional61,62 and metabolic63 status are important factors that influence the prognosis of these patients. For example, the Royal Marsden Hospital score, a prognostic tool that combines nutritional and inflammatory markers, has shown its potential to predict survival in colorectal cancer and many other cancer types64.
To summarize, we hypothesized that SPP1-positive colorectal cancer promotes M2 macrophage polarization and recruitment to the tumor microenvironment, leading to ECM remodeling, which in turn promotes the development of CRLM (Fig. 8). This study has some limitations. First, we designed this study as a retrospective analysis based on data from TCGA and GEO databases. It may not be possible to homogenize the collection institutions and sequencing methods of the samples. Second, only one hub colorectal liver metastasis-specific gene (SPP1) was validated in in vitro studies. The results of bioinformatics analysis of SPP1-promoted M2 macrophage polarization in the progression of colorectal liver metastasis require further experimental validation.

The model for SPP1 in carcinogenesis of CRC.
Conclusion
In conclusion, machine-learning-based screening of colorectal liver metastasis-specific genes is feasible and accurate, and the models built using this method have good discriminatory efficacy. The hub CRLM-specific gene SPP1 can help determine the diagnosis, prognosis, and immune infiltration of patients with CRLM. However, understanding how SPP1 recruits macrophages in colon cancer and leads to ECM remodeling to mediate tumor metastasis may be a direction for future research.
Data availability
All data presented in this paper are openly available in the TCGA (https://portal.gdc.cancer.gov) and GEO (https://www.ncbi.nlm.nih.gov/geo/) databases. GEO database under accession number GSE41258, GSE49355, GSE68468, and GSE81585.
Abbreviations
- CRLM:
-
Colorectal liver metastasis
- GEO:
-
Gene Expression Omnibus
- TCGA:
-
The Cancer Genome Atlas
- DCA:
-
Decision curve analysis
- ROC:
-
Receiver operating characteristic
- DEGs:
-
Differentially expressed genes
- GO:
-
Gene Ontology
- GSEA:
-
Gene set enrichment analysis
- PPI:
-
Protein–protein interaction
- MCC:
-
Maximal Clique Centrality
- EPC:
-
Edge Percolated Component
- RF:
-
Random Forest
- SVM-RFE:
-
Support Vector Machine Recursive Feature Elimination
- Lasso:
-
Least Absolute Shrinkage and Selection Operator
- XGboost:
-
EXtreme gradient boosting
- VIF:
-
Variance inflation factors
- qRTPCR:
-
Quantitative real-time PCR
- CCK-8:
-
Cell counting kit-8
- RMSE:
-
Root mean square error
- TAM:
-
Tumor-associated macrophages
- ECM:
-
Extracellular matrix
References
Torre, L. A. et al. Global cancer incidence and mortality rates and trends–an update. Cancer Epidemiol. Biomark. Prev. 25(1), 16–27 (2016).
Lambert, A. W., Pattabiraman, D. R. & Weinberg, R. A. Emerging biological principles of metastasis. Cell 168(4), 670–691 (2017).
Lengel, H. B. et al. Genomic mapping of metastatic organotropism in lung adenocarcinoma. Cancer Cell 41(5), 970-985.e3 (2023).
Aasebø, K. et al. CDX2: A prognostic marker in metastatic colorectal cancer defining a better BRAF mutated and a worse KRAS mutated subgroup. Front. Oncol. 10, 8 (2020).
Bellizzi, A. M. An algorithmic immunohistochemical approach to define tumor type and assign site of origin. Adv. Anat. Pathol. 27(3), 114–163 (2020).
Teng, S. et al. Tissue-specific transcription reprogramming promotes liver metastasis of colorectal cancer. Cell Res. 30(1), 34–49 (2020).
Deo, R. C. Machine learning in medicine. Circulation 132(20), 1920–1930 (2015).
Greener, J. G. et al. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 23(1), 40–55 (2022).
Wu, W. T. et al. Data mining in clinical big data: The frequently used databases, steps, and methodological models. Mil. Med. Res. 8(1), 44 (2021).
Yang, J. et al. Brief introduction of medical database and data mining technology in big data era. J. Evid. Based Med. 13(1), 57–69 (2020).
Haug, C. J. & Drazen, J. M. Artificial intelligence and machine learning in clinical medicine, 2023. N. Engl. J. Med. 388(13), 1201–1208 (2023).
Liu, Z. et al. Machine learning-based integration develops an immune-derived lncRNA signature for improving outcomes in colorectal cancer. Nat. Commun. 13(1), 816 (2022).
Shrivastava, R. & Shukla, N. Attributes of alternatively activated (M2) macrophages. Life Sci. 224, 222–231 (2019).
Boutilier, A. J. & Elsawa, S. F. Macrophage polarization states in the tumor microenvironment. Int. J. Mol. Sci. 22(13), 6995 (2021).
Yeung, O. W. et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J. Hepatol. 62(3), 607–616 (2015).
Yu, G. et al. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16(5), 284–287 (2012).
Franceschini, A. et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41(Database issue), D808–D815 (2013).
Breiman, L. Random forests. Mach. Learn. 45(1), 5–32 (2001).
Guyon, I. et al. Gene selection for cancer classification using support vector machines. Mach. Learn. 46(1), 389–422 (2002).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33(1), 1–22 (2010).
Chen, T. & Guestrin, C. XGBoost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD international conference on knowledge discovery and data mining, p. 785–794 (2016).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102(43), 15545–15550 (2005).
Leibovitz, A. et al. Classification of human colorectal adenocarcinoma cell lines. Cancer Res. 36(12), 4562–4569 (1976).
Flatmark, K. et al. Twelve colorectal cancer cell lines exhibit highly variable growth and metastatic capacities in an orthotopic model in nude mice. Eur. J. Cancer 40(10), 1593–1598 (2004).
Brandi, G. et al. Is post-transplant chemotherapy feasible in liver transplantation for colorectal cancer liver metastases?. Cancer Commun. 40(9), 461–464 (2020).
Pinto, R. et al. Precise integration of inducible transcriptional elements (PrIITE) enables absolute control of gene expression. Nucleic Acids Res. 45(13), e123 (2017).
O’Reilly, E. M. & Hechtman, J. F. Tumour response to TRK inhibition in a patient with pancreatic adenocarcinoma harbouring an NTRK gene fusion. Ann. Oncol. 30(Suppl_8), viii36–viii40 (2019).
FitzPatrick, D. R. et al. Identification of SATB2 as the cleft palate gene on 2q32-q33. Hum Mol Genet 12(19), 2491–2501 (2003).
Eberhard, J. et al. A cohort study of the prognostic and treatment predictive value of SATB2 expression in colorectal cancer. Br. J. Cancer 106(5), 931–938 (2012).
Ward, P. S. & Thompson, C. B. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21(3), 297–308 (2012).
Bradner, J. E., Hnisz, D. & Young, R. A. Transcriptional addiction in cancer. Cell 168(4), 629–643 (2017).
Ortmayr, K., Dubuis, S. & Zampieri, M. Metabolic profiling of cancer cells reveals genome-wide crosstalk between transcriptional regulators and metabolism. Nat. Commun. 10(1), 1841 (2019).
Steinparzer, I. et al. Transcriptional responses to IFN-γ require mediator kinase-dependent pause release and mechanistically distinct CDK8 and CDK19 functions. Mol. Cell 76(3), 485-499.e8 (2019).
Kuznetsova, T. et al. Transcriptional and epigenetic regulation of macrophages in atherosclerosis. Nat. Rev. Cardiol. 17(4), 216–228 (2020).
Puchalska, P. & Crawford, P. A. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 25(2), 262–284 (2017).
Habtetsion, T. et al. Alteration of tumor metabolism by CD4+ T cells leads to TNF-α-dependent intensification of oxidative stress and tumor cell death. Cell Metab. 28(2), 228-242.e6 (2018).
Tang, Z. et al. Role of purines in regulation of metabolic reprogramming. Purinergic Signal 15(4), 423–438 (2019).
Ullmann, P. et al. Hypoxia- and MicroRNA-induced metabolic reprogramming of tumor-initiating cells. Cells 8(6), 528 (2019).
Wang, T. et al. MTA1, a novel ATP synthase complex modulator, enhances colon cancer liver metastasis by driving mitochondrial metabolism reprogramming. Adv. Sci. 10(25), e2300756 (2023).
Zhang, L. et al. Creatine promotes cancer metastasis through activation of Smad2/3. Cell Metab. 33(6), 1111-1123.e4 (2021).
Meng, H. et al. ZG16 regulates PD-L1 expression and promotes local immunity in colon cancer. Transl. Oncol. 14(2), 101003 (2021).
Chen, X. et al. Loss of ZG16 is regulated by miR-196a and contributes to stemness and progression of colorectal cancer. Oncotarget 7(52), 86695–86703 (2016).
Shu, C. et al. ncRNA-mediated low expression of P2RY14 correlates with poor prognosis and tumor immune infiltration in ovarian carcinoma. Ann. Transl. Med. 11(1), 10 (2023).
Chen, S. et al. The combined signatures of G protein-coupled receptor family and immune landscape provide a prognostic and therapeutic biomarker in endometrial carcinoma. J. Cancer Res. Clin. Oncol. 149(16), 14701–14719 (2023).
Xu, T. et al. P2RY14 downregulation in lung adenocarcinoma: A potential therapeutic target associated with immune infiltration. J. Thorac. Dis. 14(2), 515–535 (2022).
Wang, Y. R. et al. Insights regarding novel biomarkers and the pathogenesis of primary colorectal carcinoma based on bioinformatic analysis. Comput. Biol. Chem. 85, 107229 (2020).
Makondi, P. T. et al. Development of novel predictive miRNA/target gene pathways for colorectal cancer distance metastasis to the liver using a bioinformatic approach. PLoS One 14(2), e0211968 (2019).
Curet, M. A. & Watters, J. J. P2Y14 receptor activation decreases interleukin-6 production and glioma GL261 cell proliferation in microglial transwell cultures. J. Neurooncol. 137(1), 23–31 (2018).
Wei, R. et al. In vitro and clinical data analysis of Osteopontin as a prognostic indicator in colorectal cancer. J. Cell. Mol. Med. 22(9), 4097–4105 (2018).
Yi, J. et al. Secreted phosphoprotein-1 accelerates the progression of human colorectal cancer through activating β-catenin signaling. Oncol. Lett. 21(5), 372 (2021).
Bosco, M. C. Macrophage polarization: Reaching across the aisle?. J. Allergy Clin. Immunol. 143(4), 1348–1350 (2019).
Chen, Y. & Zhang, X. Pivotal regulators of tissue homeostasis and cancer: macrophages. Exp. Hematol. Oncol. 6, 23 (2017).
Petty, A. J. et al. Targeting tumor-associated macrophages in cancer immunotherapy. Cancers 13(21), 5318 (2021).
Cao, X. et al. Targeting tumor-associated macrophages for cancer immunotherapy. Int. Rev. Cell Mol. Biol. 368, 61–108 (2022).
Dong, Y. et al. Reprogramming immune cells for enhanced cancer immunotherapy: Targets and strategies. Front. Immunol. 12, 609762 (2021).
Sun, L. et al. Activating a collaborative innate-adaptive immune response to control metastasis. Cancer Cell 39(10), 1361-1374.e9 (2021).
Madsen, D. H. & Bugge, T. H. The source of matrix-degrading enzymes in human cancer: Problems of research reproducibility and possible solutions. J. Cell Biol. 209(2), 195–198 (2015).
He, Y. et al. Tumor-associated extracellular matrix: How to be a potential aide to anti-tumor immunotherapy?. Front. Cell Dev. Biol. 9, 739161 (2021).
Rizzo, A. et al. Dose reduction and discontinuation of standard-dose regorafenib associated with adverse drug events in cancer patients: A systematic review and meta-analysis. Ther. Adv. Med. Oncol. 12, 1758835920936932 (2020).
Dall’Olio, F. G. et al. Immortal time bias in the association between toxicity and response for immune checkpoint inhibitors: A meta-analysis. Immunotherapy 13(3), 257–270 (2021).
Guven, D. C. et al. The association between albumin levels and survival in patients treated with immune checkpoint inhibitors: A systematic review and meta-analysis. Front. Mol. Biosci. 9, 1039121 (2022).
Marano, L. et al. Clinical nutrition in surgical oncology: Young AIOM-AIRO-SICO multidisciplinary national survey on behalf of NutriOnc research group. Front. Nutr. 10, 1045022 (2023).
Vitale, E. et al. Associations between “cancer risk”, “inflammation” and “metabolic syndrome”: A scoping review. Biology 13(5), 352 (2024).
Sahin, T. K. et al. Prognostic significance of the Royal Marsden Hospital (RMH) Score in patients with cancer: A systematic review and meta-analysis. Cancers 16(10), 1835 (2024).
Acknowledgements
We thank all of those who participated in this study. We thank Biorender (https://biorender.com/) for assistance with the figures.
Funding
This study was supported by the Medical Innovation Project of Fujian Province [Grant numbers 2020CXB006 and 2021Y9231] owned by C.Y.
Author information
Authors and Affiliations
Contributions
S.Z., H. H., J. Z., and C. Y. were responsible for study design and writing. C.Y., S.G., and C.J. were involved in the study design and were responsible for scientific revision. S.Z., H. H., and J.Z. contributed equally to this paper as co-first authors. C.Y., S.G., and C. J. contributed equally to this work as cocorresponding authors. S.Z., H.H., N.L., Q.W., and E.W. were responsible for the data collection and analysis. S.Z., C.W., S.C., and X.H. contributed to the image painting. S.Z., J.Z., and X.Z. were involved in the cytological experiments. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zheng, S., He, H., Zheng, J. et al. Machine learning-based screening and validation of liver metastasis-specific genes in colorectal cancer. Sci Rep 14, 17679 (2024). https://doi.org/10.1038/s41598-024-68706-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68706-y
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}
{kind=link}