
Abstract
Recently, a mild elevation of the blood ketone levels was found to exert multifaceted cardioprotective effects. To investigate the effect of angiotensin receptor neprilysin inhibitors (ARNIs) on the blood ketone body levels, 46 stable pre-heart failure (HF)/HF patients were studied, including 23 who switched from angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) to ARNIs (ARNI group) and 23 who continued treatment with ACE inhibitors or ARBs (control group). At baseline, there were no significant differences in the total ketone body (TKB) levels between the two groups. Three months later, the TKB levels in the ARNI group were higher than the baseline values (baseline to 3 months: 71 [51, 122] to 92 [61, 270] μmol/L, P < 0.01). In the control group, no significant change was observed between the baseline and 3 months later. A multiple regression analysis demonstrated that the initiation of ARNI and an increase in the blood non-esterified fatty acid (NEFA) levels at 3 months increased the percentage changes in the TKB levels from baseline to 3 months (%ΔTKB level) (initiation of ARNI: P = 0.017, NEFA level at 3 months: P < 0.001). These results indicate that ARNI administration induces a mild elevation of the blood TKB levels in pre-HF/HF patients.
Similar content being viewed by others


Introduction
In recent years, several studies have focused on the cardioprotective effects of ketone bodies1,2. Sodium glucose cotransporter 2 (SGLT2) inhibitors are now indispensable medications for heart failure, and the elevation of ketone body levels induced by these medicines is thought to contribute to their cardioprotective effects3.
Although fatty acids and glucose are mainly utilized as cardiac energy substrates in normal adult hearts, ketone bodies are an important preferential substrate under pathological conditions, such as heart failure4,5,6. In diabetic patients, the cardiac ketone body uptake increases as an energy source partially replacing glucose7. In addition to being an efficient fuel substrate in heart failure, ketone bodies are involved in multifaceted cardioprotective effects. Cardiac overexpression of beta-hydroxybutyrate dehydrogenase 1 (BDH1), which promotes the oxidation of ketone bodies, promotes increased ketone body utilization, decreases oxidative stress, and protects against heart failure8. Ketone bodies (β-hydroxybutyrate) inhibit NOD-like receptor protein 3 (NLPR3) inflammasome formation and antagonize pro-inflammatory cytokine-triggered mitochondrial dysfunction and fibrosis9. In addition, ketogenesis protects the energy-producing capacity of mitochondria by preventing hyperacetylation of mitochondrial proteins10. Although excessively elevated total ketone body (TKB) levels may cause ketoacidosis, which may have fatal consequences, intentionally induced mild ketonemia is likely to exert cardioprotective effects.
Angiotensin receptor-neprilysin inhibitor (ARNI) reduced cardiovascular deaths and all-cause mortality compared with angiotensin-converting enzyme (ACE) inhibitors in patients with chronic heart failure and a reduced ejection fraction (HFrEF)11. The main reason for this cardioprotective effect of ARNI on HFrEF is considered to be the improvement in the cardiac function via reverse cardiac remodeling12,13. We and others have demonstrated that natriuretic peptides (NPs), which are increased by neprilysin inhibition, can play a crucial role in energy metabolism14,15,16,17, so ARNI may also exert a significant effect on energy metabolism. Previous studies have reported that treatment with ARNI resulted in improved glucose metabolism18,19. We also found that ARNI administration improved insulin resistance in patients with stable chronic heart failure20. However, the mechanism by which ARNI affects ketone body metabolism is not yet fully understood.
In the present study, we investigated the effect of ARNI on blood TKB levels in pre-heart failure/heart failure patients and the possible mechanisms underlying the relationship between ARNI and ketone body metabolism.
Results
A comparison of the clinical characteristics of the two groups at baseline
Table 1 and Supplementary Table S1-S3 show the clinical characteristics of the patients at baseline in this study. The left ventricular ejection fraction (LVEF) of the ARNI group was lower than that of the control group (LVEF, 51.9 ± 2.53 vs. 60.7 ± 1.58%, P = 0.005). The body mass index (BMI) and fasting blood insulin level in the ARNI group were significantly higher than in the control group (BMI, 24.7 [23.5, 27.4] vs. 23.7 [21.0, 25.3] kg/m2, P = 0.027; fasting blood insulin, 9.6 [6.3, 12.7] vs. 5.6 [3.3, 8.0] μU/mL, P = 0.0002). There were no significant differences in TKB levels between the two groups. Furthermore, there were no significant differences in the administration rates of medications for heart failure or antidiabetic agents between the two groups.
Changes in clinical factors from baseline to three months
Figure 1 shows that, at three months, the systolic blood pressure, fasting blood sugar, fasting blood insulin, and uric acid (UA) values had decreased, while the TKB levels had increased in comparison to the baseline levels in the ARNI group (baseline to 3 months: systolic blood pressure, 129 ± 3.48 to 123 ± 2.70 mmHg, P = 0.036; blood sugar, 110 [100, 141] to 102 [99, 128] mg/dL, P = 0.013; insulin, 9.6 [6.3, 12.7] to 7.6 [4.7, 9.3] μU/mL, P < 0.001; UA, 6.44 ± 0.27 to 6.07 ± 0.27 mg/dL, P = 0.017 and TKB level, 71 [51, 122] to 92 [61, 270] μmol/L, P < 0.01). In the control group, there were no significant changes between baseline and 3 months after treatment for any of the clinical factors. In addition, at baseline, there was no marked difference in TKB levels between the two groups; however, after three months, the TKB levels were significantly higher in the ARNI group than in the control group (Fig. 2).

Changes in clinical factors from baseline to 3 months later the initiation of ARNI in patients with pre-heart failure/heart failure. (a) BMI, (b) SBP, (c) DBP, (d) HR, (e) LVEF, (f) FBS, (g) Insulin, (h) HbA1c, (i) TG, (j) HDL, (k) LDL, (l) AST, (m) ALT, (n) LDH, (o) ALP, (p) γ-GTP, (q) TB, (r) Alb, (s) eGFR, (t) UA, (u) BNP, (v) NEFA, (w) TKB. N.S., not significant; ARNI, angiotensin receptor-neprilysin inhibitor; BMI, body mass index; SBP, systolic blood pressure; DBP, diastolic blood pressure; HR, heart rate; LVEF, left ventricular ejection fraction; FBS, fasting blood sugar; Hb, hemoglobin; TG, triglyceride; HDL, high-density lipoprotein cholesterol; LDL, low-density lipoprotein cholesterol; AST, aspartate aminotransferase; ALT, alanine aminotransferase; LDH, lactate dehydrogenase; ALP, alkaline phosphatase; γ-GTP, γ-glutamyltranspeptidase; TB, total bilirubin; Alb, albumin; eGFR, estimated glomerular filtration rate; UA, uric acid; BNP, B-type natriuretic peptide; NEFA, non-esterified fatty acid; TKB, total ketone body.

The comparison of the TKB level between the ARNI group and control group at baseline and three months. TKB, total ketone body; ARNI, angiotensin receptor-neprilysin inhibitor.
Clinical factors at three months influencing the %ΔTKB level
To clarify the clinical factors contributing to the changes in TKB levels, we performed a simple regression analysis among the clinical factors at three months later and the %ΔTKB level (Table 2). The LVEF was significantly negatively correlated with the %ΔTKB (P = 0.007), and the non-esterified fatty acid (NEFA) levels were significantly positively correlated with the %ΔTKB (P < 0.001).
Based on the results of a simple regression analysis, the multiple regression analysis for the %ΔTKB level included the initiation of ARNI, the LVEF, and the NEFA level as independent variables. As shown in Table 3, the multiple regression analysis revealed that the initiation of ARNI and the NEFA level was independently and significantly associated with the %ΔTKB (initiation of ARNI: P = 0.017, NEFA level: P < 0.001), while the LVEF was not significantly correlated with the %ΔTKB.
The relationship among the %ΔB-type natriuretic peptide (BNP) level, %⊿NEFA level and %⊿TKB level
To clarify the relationship among the %ΔBNP level, %⊿NEFA level and %⊿TKB level, we performed a simple regression analysis of the %ΔBNP with the %ΔNEFA, and of the %ΔNEFA with the %ΔTKB. There was no significant relationship between the %ΔBNP and the %ΔNEFA (P = 0.657). However, the %ΔNEFA was significantly related with the %ΔTKB (P < 0.0001) (Supplementary Table S11).
A comparison of blood TKB levels excluding patients who were taking SGLT2 inhibitors
At baseline, there were four patients (two patients in the ARNI group and two patients in the control group) who were taking SGLT2 inhibitors, as shown in Table 1 and Supplementary Table S3. These patients had already been taking SGLT2 inhibitors for more than four weeks prior to baseline. To eliminate the effect of SGLT2 inhibitors, we excluded the four patients who were taking SGLT2 inhibitors, and compared the blood TKB levels in the remaining patients of the group (ARNI group: n = 21, Control group: n = 21). In the control group, there was no marked difference in TKB levels between baseline and 3 months later. In the ARNI group, the TKB levels were significantly higher at 3 months than at baseline (baseline to 3 months: 68 [51, 97] to 92 [67, 225] μmol/L, P < 0.01). In addition, at baseline, there was no marked difference in TKB levels between the 2 groups, but after 3 months, the TKB levels were significantly higher in the ARNI group than in the control group (92 [67, 225] vs. 54 [44, 74] μmol/L, P < 0.01). These results were the same as those found in the whole analysis, which included patients taking SGLT2 inhibitors.
Discussion
The current study is the first to evaluate the effect of ARNI administration on the blood ketone body levels, and the results showed that the administration of ARNI increased the TKB levels in pre-heart failure/heart failure patients. In addition, ARNI administration and an increase in the blood non-esterified fatty acid (NEFA) levels at 3 months were factors that increased the percentage changes in the TKB levels from baseline to 3 months (%ΔTKB level).
In this study, we found that ARNI administration caused an increase in TKB levels in pre-heart failure/heart failure patients. In the liver mitochondria, ketone bodies are produced from acetyl coenzyme A (CoA), which is derived from the β-oxidation of free fatty acids21,22. Although it was reported that blood TKB levels in patients with heart failure were elevated with the deterioration of hemodynamics23, we recently demonstrated that TKB levels closely correlate with NP levels rather than with the severity of their heart failure24. Previous reports demonstrated that, increased NP in heart failure patients activates lipolysis in white adipose tissue (WAT)25,26,27. Another report showed that cold exposure increases the A-type natriuretic peptide (ANP) receptor guanylyl cyclase A (GCA) gene and protein expression in WAT, thus rendering mouse adipocytes responsiveness to ANP-mediated lipolysis22. In addition, we previously demonstrated that exogenous ANP ameliorates high-fat-diet (HFD)-induced insulin resistance by promoting adipose tissue browning as well as by attenuating hepatic steatosis16. It is most likely that the elevation of NPs due to ARNI administration activates lipolysis in adipose tissue, thereby increasing the concentration of fatty acids in the blood, which are taken up by the liver, resulting in ketone body production. In addition, a previous report showed that insulin reduction results in the mobilization of free fatty acids from adipose tissue to the liver28. The present study showed that blood insulin levels were significantly reduced between baseline and three months after the initiation of ARNI. Therefore, insulin reduction was also found to contribute to lipolysis in adipose tissue and consequently it therefore promotes ketone body production.
At baseline, insulin resistance in the ARNI group was higher than in the control group. Insulin resistance is closely related to the pathogenesis of heart failure, and insulin resistance itself is a major factor in the worsening of heart failure29. Patients with increased insulin resistance, which occurs in patients suffering from obesity and diabetes, show the suppression of NPs secretion and an accelerated NPs degradation, and consequently, unexpectedly demonstrate low NPs levels in proportion to the heart failure severity30,31,32. Therefore, ARNI administration to such "low NPs" patients tends to cause a more effective endogenous NPs elevation, thus resulting in an increase in the blood ketone body concentrations, and consequently thereby demonstrates cardioprotective effects.
In the present study, there was no significant increase in the BNP level with the initiation of ARNI. Furthermore, although the %ΔNEFA was significantly related to the %ΔTKB, there was no significant relationship between the %ΔBNP and the %ΔNEFA, and between %ΔBNP and %ΔTKB. BNP shows a lower affinity for neprilysin than ANP, so ARNI increases the concentration of ANP levels rather than the BNP levels33. We lack ANP data in the present study, but we speculate that the initiation of ARNI would significantly increase the ANP level, and there may be significant relationships between %ΔANP and %ΔNEFA and between %ΔANP and %ΔTKB. Furthermore, C-type natriuretic peptide (CNP) and other peptides may also be involved in the elevation of NEFA and TKB, which is an interesting issue to be further investigated in the future.
It is possible that there is another pathway by which ARNI initiation affects TKB levels. In adipocytes, NPs activate peroxisome proliferator-activated receptor γ coactivator (PGC)-1α and uncoupling protein 1 (UCP1) expression, induce mitochondriogenesis, and increase uncoupled and total respiration15. The nuclear receptor peroxisome proliferator activated receptor α (PPARα) is an important element in the metabolic network, where it participates in signaling driven by the main nutrient sensors, such as PGC-1α, and induces hormonal mediators, such as fibroblast growth factor 21 (FGF21), which is produced in the liver34,35,36. Furthermore, in human hepatocytes, PPARα is a strong inducer of 3-hydroxymethylglutaryl-CoA synthase 2 (HMGCS2), a rate-limiting enzyme of endogenous ketogenesis37. Therefore, ARNI may be involved in the induction of HMGCS2 via the PGC-1α-PPARα-FGF21 axis and increased ketone body production.
Several limitations associated with the present study warrant mention. First, it was a retrospective, single-center study with a small sample size, and not only heart failure patients but also pre-heart failure patients with low BNP levels (e.g. Stage B heart failure) were included in this study population. Second, the final decision to switch from ACE inhibitors or ARBs to ARNI was dependent on the physician’s and patient’s will (e.g. some patients refused for economic reasons), which could have influenced their metabolic status per se. Third, at baseline, the type and internal dose of ACE inhibitors or ARBs varied from case to case, as shown in Table S3, and variations in these medications may also influence some clinical factors.
In conclusion, the administration of ARNI increased the TKB levels in pre-heart failure/heart failure patients. In addition, ARNI administration and an increase in blood NEFA levels at 3 months independently affected the %ΔTKB level. These results indicate that ARNI induces a mild elevation of blood TKB levels in pre-heart failure/heart failure patients, which may result in cardioprotective effects similar to those of SGLT2 inhibitors. These results may also shed light on the potential mechanism of ARNI as a treatment for heart failure via the activation of ketone body metabolism.
Methods
Patient population
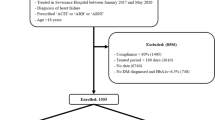
This study is a retrospective observational study, and the study population consisted of 82 patients who regularly visited the outpatient department of Jikei University School of Medicine with pre-heart failure/heart failure between October 2021 and August 2023 and were taking ACE inhibitors or ARBs at any dose (Fig. 3). ACE inhibitors or ARBs were initiated in all patients in the present study for at least four weeks before screening. We excluded 36 patients (as described below) and ultimately analyzed 46 patients in the present study.

Patient flow chart for the present study. We excluded 36 patients, ultimately analyzing 46 patients who considered switching from an ACE inhibitor or ARB to ARNI. ACE, angiotensin-converting enzyme; ARB, angiotensin receptor blocker; ARNI, angiotensin receptor-neprilysin inhibitor.
Patients who switched from ACE inhibitors or ARBs to ARNI were classified into the ARNI group (n = 23), whereas those who continued treatment with ACE inhibitors or ARBs were classified into the control group (n = 23). ARNI treatment was initially started at 100 or 200 mg/day by carefully monitoring the blood pressure and other clinical parameters. For all patients in the present study, no other medications were added or changed from four weeks prior to screening to the end of the observation period, as previously described20.
The exclusion criteria were as follows:
-
i.
Patients hospitalized due to heart failure within 3 months before screening. (n = 5)
-
ii.
Patient with a systolic blood pressure of < 90 mmHg at the time of screening (n = 1)
-
iii.
Patients using insulin injection or injectable glucagon-like peptide-1 (GLP-1) receptor agonists (n = 10)
-
iv.
Patient with other active diseases (e.g. an abdominal aortic aneurysm) at the time of screening (n = 1)
-
v.
Patient undergoing dialysis (n = 1)
-
vi.
Patients who were non-compliant with either the medication or the fasting time before blood tests (n = 6)
-
vii.
Patients < 18 years old or > 85 years old at the time of screening (n = 6)
-
viii.
Patients who added or changed medications (other than ARNI) between 4 weeks prior to screening and the end of the observation period (n = 3)
-
ix.
Patients with missing data on TKB levels (n = 3)
This study was conducted in accordance with the principles expressed in the Declaration of Helsinki and was approved by the medical ethics committee of Jikei University School of Medicine [34-046(11191)]. This was a retrospective study, and the need for informed consent from all subjects and/or their legal guardian(s) was waived by the medical ethics committee. Instead of obtaining informed consent from each patient, we posted a notice about the study design and contact information at a public location in our institution, according to our routine ethical regulations. In this public notification, we ensured that patients had the opportunity to refuse to participate (opt-out) in the study, as previously described20.
Data collection
The clinical characteristics of the patients were measured at baseline and three months later and were retrospectively collected from their medical records. The baseline in this study was the point at which the decision to switch from an ACE inhibitor or ARB to an ARNI was considered. At the baseline, in the control group, the patients continued on ACE inhibitors or ARBs without switching to ARNI either based on the physician's decision or the patient's will. The LVEF was measured using transthoracic echocardiography (TTE). Blood pressure was measured at rest in the outpatient department. Blood samples were collected in the morning after at least 11 h of fasting. We defined the percentage change from baseline to 3 months after the TKB level as %ΔTKB (%ΔTKB [%] = 100 × [TKB at 3 months–TKB at baseline]/TKB at baseline). Similarly, we defined %ΔBNP (%ΔBNP [%] = 100 × [BNP at 3 months–BNP at baseline]/BNP at baseline), %ΔNEFA (%ΔNEFA [%] = 100 × [NEFA at 3 months–NEFA at baseline]/NEFA at baseline), and %ΔLVEF (%ΔLVEF [%] = 100 × [LVEF at 3 months–LVEF at baseline]/LVEF at baseline). Blood TKB levels were measured using an enzyme cycling method with a TKB-L test kit (“Kainos” Laboratories, Inc., Tokyo, Japan).
Statistical analyses
Data are expressed as the mean ± standard error (SE) or as the median (25th, 75th percentile) for significantly skewed variables. For continuous variables, differences between two groups were evaluated using either an unpaired Student’s t-test or the Mann–Whitney rank-sum test, and differences within groups were evaluated using a paired t-test or Wilcoxon’s signed-rank test, as previously described20. For discrete variables, which were expressed as counts and percentages, differences between the two groups were analyzed using the chi-square test, unless the expected value in any cell was < 5, in which case Fisher’s exact test was used. The LVEF measured using TTE were compared among the three time points (baseline, 3 months, and 1 year) using a repeated measure analysis of variance followed by Bonferroni’s test. A multiple linear regression analysis with the forced entry method was performed to determine the contribution of various clinical variables to the %ΔTKB levels. When conducting the multivariate logistic regression analysis, we included predictors with a P value < 0.05 in the single regression analysis.
All statistical analyses were performed using the STATA software program (version 16.0; STATA Corp., College Station, TX, USA). Statistical significance was set at P < 0.05.
Data availability
The data that support the findings of this study are not openly available due to reasons of sensitivity and are available from the corresponding author upon reasonable request.
References
Yurista, S. R. et al. Therapeutic potential of ketone bodies for patients with cardiovascular disease: JACC state-of-the-art review. J. Am. Coll. Cardiol. 77, 1660–1669. https://doi.org/10.1016/j.jacc.2020.12.065 (2021).
Arima, Y. The impact of ketone body metabolism on mitochondrial function and cardiovascular diseases. J. Atheroscler. Thromb. https://doi.org/10.5551/jat.RV22011 (2023).
Ferrannini, E., Mark, M. & Mayoux, E. CV protection in the EMPA-REG OUTCOME trial: A “thrifty substrate” hypothesis. Diabetes Care 39, 1108–1114. https://doi.org/10.2337/dc16-0330 (2016).
Bedi, K. C. Jr. et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation 133, 706–716. https://doi.org/10.1161/circulationaha.115.017545 (2016).
Aubert, G. et al. The failing heart relies on ketone bodies as a fuel. Circulation 133, 698–705. https://doi.org/10.1161/circulationaha.115.017355 (2016).
Ho, K. L. et al. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc. Res. 115, 1606–1616. https://doi.org/10.1093/cvr/cvz045 (2019).
Mizuno, Y. et al. The diabetic heart utilizes ketone bodies as an energy source. Metab. Clin. Exp. 77, 65–72. https://doi.org/10.1016/j.metabol.2017.08.005 (2017).
Uchihashi, M. et al. Cardiac-specific Bdh1 overexpression ameliorates oxidative stress and cardiac remodeling in pressure overload-induced heart failure. Circ. Heart Fail. 10, e004417. https://doi.org/10.1161/circheartfailure.117.004417 (2017).
Deng, Y. et al. Targeting mitochondria-inflammation circuit by β-hydroxybutyrate mitigates HFpEF. Circ. Res. 128, 232–245. https://doi.org/10.1161/circresaha.120.317933 (2021).
Arima, Y. et al. Murine neonatal ketogenesis preserves mitochondrial energetics by preventing protein hyperacetylation. Nat. Metab. 3, 196–210. https://doi.org/10.1038/s42255-021-00342-6 (2021).
McMurray, J. J. et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 371, 993–1004. https://doi.org/10.1056/NEJMoa1409077 (2014).
Januzzi, J. L. Jr. et al. Association of change in N-terminal pro-B-type natriuretic peptide following initiation of sacubitril-valsartan treatment with cardiac structure and function in patients with heart failure with reduced ejection fraction. JAMA 322, 1085–1095. https://doi.org/10.1001/jama.2019.12821 (2019).
Martens, P. et al. Sacubitril/valsartan reduces ventricular arrhythmias in parallel with left ventricular reverse remodeling in heart failure with reduced ejection fraction. Clin. Res. Cardiol. 108, 1074–1082. https://doi.org/10.1007/s00392-019-01440-y (2019).
Miyashita, K. et al. Natriuretic peptides/cGMP/cGMP-dependent protein kinase cascades promote muscle mitochondrial biogenesis and prevent obesity. Diabetes 58, 2880–2892. https://doi.org/10.2337/db09-0393 (2009).
Bordicchia, M. et al. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J. Clin. Investig. 122, 1022–1036. https://doi.org/10.1172/jci59701 (2012).
Kimura, H. et al. Treatment with atrial natriuretic peptide induces adipose tissue browning and exerts thermogenic actions in vivo. Sci. Rep. 11, 17466. https://doi.org/10.1038/s41598-021-96970-9 (2021).
Kimura, H. et al. The thermogenic actions of natriuretic peptide in brown adipocytes: The direct measurement of the intracellular temperature using a fluorescent thermoprobe. Sci. Rep. 7, 12978. https://doi.org/10.1038/s41598-017-13563-1 (2017).
Seferovic, J. P. et al. Effect of sacubitril/valsartan versus enalapril on glycaemic control in patients with heart failure and diabetes: A post-hoc analysis from the PARADIGM-HF trial. Lancet Diabetes Endocrinol. 5, 333–340. https://doi.org/10.1016/s2213-8587(17)30087-6 (2017).
Wijkman, M. O. et al. Effects of sacubitril/valsartan on glycemia in patients with diabetes and heart failure: The PARAGON-HF and PARADIGM-HF trials. Cardiovasc. Diabetol. 21, 110. https://doi.org/10.1186/s12933-022-01545-1 (2022).
Kashiwagi, Y. et al. Effects of angiotensin receptor-neprilysin inhibitor on insulin resistance in patients with heart failure. ESC Heart Fail. 10, 1860–1870. https://doi.org/10.1002/ehf2.14352 (2023).
Puchalska, P. & Crawford, P. A. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 25, 262–284. https://doi.org/10.1016/j.cmet.2016.12.022 (2017).
Carper, D. et al. Atrial natriuretic peptide orchestrates a coordinated physiological response to fuel non-shivering thermogenesis. Cell Rep. 32, 108075. https://doi.org/10.1016/j.celrep.2020.108075 (2020).
Lommi, J. et al. Blood ketone bodies in congestive heart failure. J. Am. Coll. Cardiol. 28, 665–672. https://doi.org/10.1016/0735-1097(96)00214-8 (1996).
Kashiwagi, Y. et al. Close linkage between blood total ketone body levels and B-type natriuretic peptide levels in patients with cardiovascular disorders. Sci. Rep. 11, 6498. https://doi.org/10.1038/s41598-021-86126-0 (2021).
Lafontan, M. et al. Control of lipolysis by natriuretic peptides and cyclic GMP. Trends Endocrinol. Metab. TEM 19, 130–137. https://doi.org/10.1016/j.tem.2007.11.006 (2008).
Szabó, T. et al. Increased catabolic activity in adipose tissue of patients with chronic heart failure. Eur. J. Heart Fail. 15, 1131–1137. https://doi.org/10.1093/eurjhf/hft067 (2013).
Doehner, W., Frenneaux, M. & Anker, S. D. Metabolic impairment in heart failure: The myocardial and systemic perspective. J. Am. Coll. Cardiol. 64, 1388–1400. https://doi.org/10.1016/j.jacc.2014.04.083 (2014).
McGarry, J. D. & Foster, D. W. Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem. 49, 395–420. https://doi.org/10.1146/annurev.bi.49.070180.002143 (1980).
Lopaschuk, G. D., Karwi, Q. G., Tian, R., Wende, A. R. & Abel, E. D. Cardiac energy metabolism in heart failure. Circ. Res. 128, 1487–1513. https://doi.org/10.1161/circresaha.121.318241 (2021).
Wang, T. J. et al. Impact of obesity on plasma natriuretic peptide levels. Circulation 109, 594–600. https://doi.org/10.1161/01.Cir.0000112582.16683.Ea (2004).
Inoue, Y. et al. The impact of an inverse correlation between plasma B-type natriuretic peptide levels and insulin resistance on the diabetic condition in patients with heart failure. Metab. Clin. Exp. 65, 38–47. https://doi.org/10.1016/j.metabol.2015.09.019 (2016).
Oi, Y. et al. Exogenous ANP treatment ameliorates myocardial insulin resistance and protects against ischemia-reperfusion injury in diet-induced obesity. Int. J. Mol. Sci. 23, 8373. https://doi.org/10.3390/ijms23158373 (2022).
Ibrahim, N. E. et al. Effect of neprilysin inhibition on various natriuretic peptide assays. J. Am. Coll. Cardiol. 73, 1273–1284. https://doi.org/10.1016/j.jacc.2018.12.063 (2019).
Lundåsen, T. et al. PPARalpha is a key regulator of hepatic FGF21. Biochem. Biophys. Res. Commun. 360, 437–440. https://doi.org/10.1016/j.bbrc.2007.06.068 (2007).
Badman, M. K. et al. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5, 426–437. https://doi.org/10.1016/j.cmet.2007.05.002 (2007).
Grabacka, M., Pierzchalska, M., Dean, M. & Reiss, K. Regulation of ketone body metabolism and the role of PPARα. Int. J. Mol. Sci. 17, 2093. https://doi.org/10.3390/ijms17122093 (2016).
Raza-Iqbal, S. et al. Transcriptome analysis of K-877 (a novel selective PPARα modulator (SPPARMα))-regulated genes in primary human hepatocytes and the mouse liver. J. Atheroscler. Thromb. 22, 754–772. https://doi.org/10.5551/jat.28720 (2015).
Acknowledgements
Acknowledgement: This work was supported by the JSPS KAKENHI (Grant Number JP24K11300 to Y.K., JP23K07563 to T.N., and JP22K08113 to M.Y.).
Funding
Funding was provided by the JSPS KAKENHI (Grant number: JP24K11300, JP23K07563, JP22K08113).
Author information
Authors and Affiliations
Contributions
Y.K., T.N. and M.Y. were responsible for the conception and design of the study. Y.K. and M.Y. were responsible for the data analysis, writing of the manuscript, and production of the figures and tables. Y.K., T.N., Y.T., Y.O., H.K., K.O. and K.M. contributed to the collection and interpretation of the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kashiwagi, Y., Nagoshi, T., Tanaka, Y. et al. Effects of angiotensin receptor-neprilysin inhibitor on ketone body metabolism in pre-heart failure/heart failure patients. Sci Rep 14, 16493 (2024). https://doi.org/10.1038/s41598-024-67524-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-67524-6
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
