
Abstract
Sphaeropsidins are iso-pimarane diterpenes produced by phytopathogenic fungi that display promising anticancer activities. Sphaeropsidin A, in particular, has been shown to counteract regulatory volume increase, a process used by cancer cells to avoid apoptosis. This study reports the hemi-synthesis of new lipophilic derivatives obtained by modifications of the C15,C16-alkene moiety. Several of these compounds triggered severe ER swelling associated with strong proteasomal inhibition and consequently cell death, a feature that was not observed with respect to mode of action of the natural product. Significantly, an analysis from the National Cancer Institute sixty cell line testing did not reveal any correlations between the most potent derivative and any other compound in the database, except at high concentrations (LC50). This study led to the discovery of a new set of sphaeropsidin derivatives that may be exploited as potential anti-cancer agents, notably due to their maintained activity towards multidrug resistant models.
Similar content being viewed by others

Introduction
Changes in expression of ion channels and transporters play an integral part in cancer and represent promising targets for therapeutic intervention1,2. Indeed, 90% of human tumor samples are found to contain ion channel or transporter mutations3. These are often important mechanisms responsible for the development of cancer cell resistance to apoptosis4. One such mechanism of resistance involves the impairment of the reduction in cell volume, an important hallmark of apoptosis5. It precedes the cytochrome C release, caspase-3 activation, DNA laddering or any other detectable characteristic of apoptosis6,7. The reduction in cell volume is brought about by the decrease in intracellular ionic strength via the loss of K+ and Cl−8. Cells resist the apoptotic volume decrease by activating compensatory mechanisms such as regulatory volume increase (RVI) that aims to restore the volume of shrunken cells9.
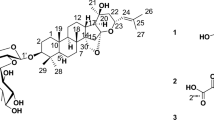
We previously discovered that sphaeropsidin A (1, Fig. 1A), which is the main phytotoxin produced by Diplodia cupressi10 showing several interesting biological activities11, overcomes apoptosis resistance by inducing a marked, rapid (within 6 h) and sustained cellular shrinkage12. This phenotypic change leads to apoptotic cell death 24–48 h later according to TUNEL staining12. The lack of close correlation of the differential cellular sensitivities with the 60 cancer cell line NCI panel containing > 763,000 compounds suggested a possibly novel mechanism of action. Sphaeropsidin A inhibits NKCC activity at low micromolar concentration, similar to its GI50 ranging from 1.6 to 6.3 µM (except for leukemia cell lines), but other cellular systems including anion exchangers have also been suggested as targets12.

Synthetic sequences involved in the project. (A) Two possible hydrolysis pathways for acetate 2. (B) Esterification of 1 at C-6 hydroxyl. (C) Cross-metathesis of 1 at C15,C16-alkene. (D) Preparation of pyrene alkene 14. (E) Structure of sphaeropsidin B (15). (F) Synthesis of pyrene-conjugated sphaeropsidin B 16. (G) Preparation of pyrene alkene with a longer linker (18) and a shorter linker (19). (H) Structures of additional alkenes utilized in cross-metathesis reactions with 1. (I) Structures of additional cross-metathesis products synthesized in this work.
To identify compounds more potent than sphaeropsidin A itself we set out to prepare derivatives containing structural changes at sites that were previously shown to retain activity. For example, work in our laboratories indicated that the C7-ketone, C9-OH and C8,C14-alkene functionalities are critical for activity, whereas the C6-OH and C15,C16-alkene can tolerate modifications (Fig. 1A)13,14. It should also be noted that previous research has demonstrated that the C6-OH of sphaeropsidin A is readily modified, be it through simple acetylation13,15,16,17 or esterification14 whilst structural extension of sphaeropsidin A’s terminal alkene via cross-metathesis has been a fairly recent disclosure14 (for a recent review including the structural extension and modification of the sphaeropsidin A skeleton see18).
The present report describes the preparation of seventeen new derivatives of sphaeropsidin A containing modifications in the tolerant sites and evaluation of these compounds as potential anti-cancer agents. The results show that we not only were able to improve the antiproliferative potency compared to the natural product itself, but also that several modifications actually led to interesting, and potentiality novel cellular effects related to severe endoplasmic reticulum swelling.
Results
Hemi-synthesis of sphaeropsidin A derivatives and their in vitro IC50 determination
We first intended to investigate whether the C6-hydroxyl functional group could be esterified with retention of activity, in light of our previous finding that the potency of C6-acetate 2 (Fig. 1A) was equal to that of sphaeropsidin A13. It could be hypothesized that acetate 2 readily undergoes intracellular hydrolysis to regenerate the C6-OH of the natural product. If this hydrolysis occurred through the attack of a water molecule on the carbonyl group of the acetate (path a), then introducing bulkier esters at C6 should prevent hydrolysis with a likely abolishment of the activity. In contrast, the attack at the C20 lactone carbonyl (path b) would result in regeneration of activity due to the departure of the C6-ester. To this end we prepared esters 3–5 (Fig. 1B) using optimized conditions employing a 10-fold excess of a selected acid, 15-fold excess of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) and dimethylaminopyridine (DMAP). Ester 3 was aliphatic, ester 4 aromatic, while ester 5 incorporated a polar carboxyl group. As can be seen in Table 1, all compounds 3–5 displayed low micromolar activities that were not very different from those of the natural product itself. The delay in the cellular shrinkage of a minimum of 24 h by esters 3 and 4 versus SphA 1, as determined by videomicroscopy in SKMEL-28 cells (Fig. S1), prompted us to suggest that these compounds indeed undergo intracellular hydrolysis, probably through path b (Fig. 1A). We deduced this because for path a we would have expected the bulkier groups to slow down the direct hydrolytic reversion back to SphA. Confirmation by stability studies in these physiological conditions should be conducted in the future. Altogether, these results suggest that derivatizing the C6-hydroxyl as an ester can be utilized for a prodrug design, for example to enable tumor targeting.
Next, the derivatization of the C15,C16-alkene was performed using an olefin cross-metathesis (CM) strategy. Figure 1C shows that the reaction was quite tolerant of diverse functionality on the alkene component. We found that the reaction went to completion (based on disappearance of sphaeropsidin A) after 10 equivalents of the alkene were added in three portions during 2 h intervals.
Surprisingly, evaluation of compounds 6, 7, 9, 10, 11, 12, containing non-polar functional groups, gave activities comparable to those of sphaeropsidin A. These results suggested that this part of the molecule could be modified for drug optimization during pre-clinical development. Of interest, compound 8 containing a polar carboxyl group was found to be inactive. To test the limit of the size of a non-polar substituent obtained through the cross-metathesis process, we appended a pyrene moiety to obtain conjugate 13 (additionally, it was considered that the fluorescent properties of this fragment might be useful for biodistribution studies as presently done in Fig. 4). The pyrene cross-metathesis precursor 14 was prepared from commercially available 1-pyrenebutanol as illustrated in Fig. 1D. Interestingly, evaluation of compound 13 in our cancer cell panel revealed a significant increase in potency with an IC50 5–10 times lower than compound 1 (Table 1; p = 0.0039 by Mann Whitney comparison). Compound 14 was determined to be inactive when utilized alone and its efficacy in combination with 1 (as a 1:1 mixture) was equivalent to 1 alone (not significant (NS: p > 0.05 by Mann Whitney comparison)). This suggested that 13 does not engage two separate targets in cancer cells, i.e. one target due to the sphaeropsidin A component (1) and another due to the pyrene portion (14). Furthermore, the activity of sphaeropsidin B (15, Fig. 1E), which is markedly lower than that of sphaeropsidin A, was also significantly improved upon its conjugation with the pyrene moiety (compound 16, Fig. 1F; p = 0.0039 by Mann Whitney comparison), while again, the combined compound treatment with 14 and 15 was not significantly better than 15 alone (Table 1; NS). Clearly, pyrene conjugates 13 and 16 therefore engage new targets in cancer cells that are different from those affected by the natural products or pyrene element separately.
The pyrene-substituted sphaeropsidin A derivative 13 induces cellular anti-cancer effects that differ from those of sphaeropsidin A and are related to severe ER swelling
Morphological comparison of cancer cells treated with 13 and 1 using phase contrast microscopy revealed that compound 13 induces severe vacuolization (black arrows) in cancer cells after 15–24 h depending on the cell model (SKMEL-28 melanoma and U373 glioblastoma cell models respectively), while 1 induces rapid cellular shrinkage (white arrows) within 6 h of treatment in both cell lines (Fig. 2A). Investigation of the vacuole origin in cancers cells treated with 13 was carried out using fluorescent probes for mitochondria, lysosome and endoplasmic reticulum (ER) cell compartments. While the vacuoles were negative to ER tracker®, MitoTracker® or LysoTracker® (data not shown), vacuoles accumulated a fusion protein made of GFP fused to the ER signal sequence of calreticulin and the ER retention signal (KDEL) (Fig. 2B). This fusion protein has been designed to be expressed and retained in the ER; specifically, calreticulin is a multifunctional protein that acts as a major Ca2+-binding protein of the lumen of the endoplasmic reticulum. The fusion with the KDEL sequence allows its retention in the ER. Compound 13 appeared thus to induce severe swelling of the ER leading to progressively enlarging vacuoles (Fig. 2B). Upon treatment with 13, dysfunction of the ER membrane and/or sulfonylurea receptors targeted by the conventional ER tracker® dye could explain the absence of staining with that method. The origin of the vacuoles induced by compound 13 was further confirmed by transmission electron microscopy in both cell models (ER-derived vacuoles (ER V) marked by a red star in Fig. 3).

Microscopic study of the effects of 1 and derivative 13 on SK-MEL-28 human melanoma and U373 glioblastoma cells. (A) Representative pictures of brightfield morphological microscopic study of the effects. Black arrows: cytoplasmic vacuoles; white arrows: cell shrinkage. (B) Derivative 13 induces severe ER swelling in SK-MEL-28 human melanoma and U373 glioblastoma cells. Representative pictures of the staining of cells with the Cell Light® system. Upper pictures of each panel represent merged brightfield and fluorescent images while lower pictures of each panel are fluorescent pictures alone. Green fluorescence results from the transfection of fusion protein (GFP-KDEL-calreticulin based expression assay).

Ultrastructural changes induced by compounds 1 and 13 in SK-MEL-28 human melanoma and U373 glioblastoma cells. ER: endoplasmic reticulum; M: mitochondria; G: golgi apparatus; N: nucleus; MN: nuclear membrane; ER V: ER-derived vacuoles; CC: condensed chromatin; red stars: ER vacuoles; red arrows: Christmas trees, red arrow heads: lipid droplets; bullet-ended arrow: mitophagy.
Although the appearance of the ER membrane is altered, the continuity of the ER V membrane with the nuclear envelope is clearly demonstrated in Fig. 3K. In addition to strong ER structural alterations, we also observed an increase in lipid droplets (red arrow heads) in the cytoplasm and changes in nucleolar ultrastructure characterized by “Christmas trees” formation (red arrows; Fig. 3E,K). The latter are composed of pre-rRNA assembled with chromatin19,20,21. A rapid swelling of the ER is associated with an increased demand for membrane and ribosomes that could lead to the features observed, i.e. lipid droplets and nucleolar “Christmas trees”. Importantly, these features were not observed with respect to cells treated with compound 1 (Fig. 3C,D,I,J). These cells displayed more affected mitochondrial structures including mitophagy and few ER-derived vacuoles (see red bullet-ended arrows in Fig. 3D,J).
Compound 13 penetrates into the cell, reaching notably the ER and resulting in strong proteasomal inhibition leading to cell death
Using the fluorescent properties of 13, microscopic observation suggests ER and nucleoli localization for compound 13 (Fig. 4A), the sites where the ultrastructural modifications were noticed in TEM (Fig. 3). Accordingly, high magnification TEM captions of the ER-vacuoles (Fig. 4Bb,Bc) showed that they contain ultrastructure materials ascribed to compound 13 itself after comparison in terms of size and morphology with TEM images of a dried solution of 13 prepared in DMSO (Fig. 4Ba). Quantitative data revealed that 13 is getting inside the cell in a time- and concentration-dependent manner (Fig. 4C). This compound is incorporated by SKMEL-28 cells quicker than U373 glioma cells, a feature that could explain at least partly why 13-induced effects are observed earlier and more markedly in those cells (Fig. 2A,B; note that time points for TEM analyses were chosen consistently for each cell line). According to the rapid cell penetration of 13, effects are irreversible in both cell lines after 6 h of treatment as the compound had already penetrated the cells.

Compound 13 cellular internalization study and its consequences on proteasomal activity and cell death. (A) 13 fluorescence observed by fluorescent microscopy in SK-MEL-28 cells over time. Cells were treated with 1 µM of 13 or left untreated (CT). White arrows point nucleoli. (B) (Ba) TEM image of 13 obtained from dried 13 DMSO stock solution. (Bb) TEM illustration of an ER-V-containing materials zoomed in Bc where measurements of the size of the structure were similar (from 8.7 to 19.5 nm) to the one made on picture Ba (15.7 nm). (C) quantification of the fluorescence levels of cells treated with 13 at 1 µM over a 24 h period (Ca) or with various concentrations of 13 for 6 h (Cb). Data are expressed as mean ± SEM of the three replicates of the experiment. (D) Proteasomal inhibition induced by 13 treatment. Cells were treated with 13 or 1 for 6 h while treatment with the positive control MG132 lasted for 24 h. Data are expressed as mean ± SEM of three experiments. (E) Evaluation of cell death induced by 13 treatment. (Ea): Percentage of cells stained by annexin-V alone (A+/PI−: light color bars) and stained by both annexin-V and propidium iodide (A+/PI+; dark color bars) assessed by flow cytometry after treatment with 1 or 13. Data are presented as mean ± SEM of three independent experiments conducted each in simplicate. 10,000 events per sample were analyzed. (Eb): Caspase 3 activity measurement by fluorescence. For this experiment, A549 non-small cell lung carcinoma cells were used and treated with etoposide (50 µM for 24 h). Data are expressed as mean ± SEM of two independent experiments conducted each in duplicates and triplicates respectively.
Again, the effects were more pronounced with respect to SKMEL-28 cells (Fig. S2). Noteworthily, we were unable to observe the presence of compound 1 within the cells by TEM, but this is not excluding its intracellular penetration. HPLC dosages are currently envisaged to compare the intracellular penetration of compounds 1 and 13.
Proteasomal perturbations have been previously shown to lead to severe ER-swelling22. To evaluate this hypothesis, we studied the cellular proteasomal trypsin-like, chymotrypsin- like and caspase-like catalytic activities in both cell lines in the presence of 1 and 13. Figure 4D shows that 13 inhibits chymotrypsin-like and caspase-like activities respectively by more than 60% after only 6 h of treatment in both models, similarly to the positive control MG-13223,24. By contrast, compound 1 appeared to stimulate caspase-like activity (Fig. 4D). However, whether proteasomal inhibition is a causal or consequent event to the ER swelling observed with 13 remains to be determined. Compound 13-induced effects lead to cell death as assessed by annexin-V/PI staining (Fig. 4Ea). Nonetheless, the cell death pathway triggered, e.g. apoptotic versus paraptotic-like seems to be cell-type dependent when considering caspase-8, -9 and -3 activities data (Fig. 4Eb; Fig. S3).
Indeed, caspase -3 (Fig. 4Eb) and -8 (Fig. S3) activities were markedly increased after 15 h of treatment with 13 in SKMEL-28 cells according to high levels of annexin-V positive stained cells (Fig. 4Ea). By contrast, in U373 cells, no caspase activation could be detected (Fig. 4Eb; Fig. S3), at least until 24 h of treatment, although more than 60% of the cells were already positive to annexin-V (Fig. 4Ea). Notably, phosphatidyl-serine externalization could be observed in paraptosis-like cell death too25. On the other hand, compound 1 did not seem to induce strong caspase activation in any of these models, a feature that is compatible with its effects described before on the regulatory volume increase allowing the bypass of conventional apoptotic pathways. Finally, various drug resistant cell models, including the p53 null RKO model, highly ABCB1 overexpressing KB-C-1 cells as well as cell lines with acquired cisplatin and oxaliplatin resistance appeared equally sensitive to 13-induced in vitro anti-cancer effects according to MTT data (Table S1). These encouraging results and the low vulnerability towards multiple important drug resistance mechanisms prompted us to further consider compound 13 as a new promising anti-cancer agent. However, the role of the pyrene moiety had first to be considered and deciphered.
The DNA targeting properties of the pyrene moiety are not responsible for the ER swelling induced by compound 13 and analogues
Due to the mechanistic differences observed between compounds 1 and 13, as well as the increase in potency conferred by the pyrene substitution, compound 13 was accepted for testing by the NCI. The mean GI50 of 0.19 µM against the 60 cancer cell line panel was consistent with our results, which showed a mean IC50 0.3 µM with few variations between the cell lines (Table 1; Fig. S4). By contrast, the leukemia cell lines displayed a mean LC50 40 times higher than the global mean LC50 of 1.5 µM. Compound 1 was found to be less active against leukemia cell lines12. Nevertheless, of significance was that a comparison of the response profile of compound 13 to > 763,000 compounds of the database with the COMPARE algorithm26 did not show any correlation between compound 13 and 1 at both GI50 and LC50 levels. Moreover, at the GI50 level, no other compound from the database correlated with 13 (the highest correlation coefficient was only 0.39) suggesting that 13 displays different anti-proliferative mechanisms when compared to the other database compounds, including 1. In contrast, the COMPARE analysis revealed seven compounds correlating with 13 with respect to LC50 profile with CC > 0.7, namely teroxirone, caracemide, tamoxifen, acodazole, amsacrine, pyrazolo acridine and cytembena (Table S2). Three compounds out of the seven are known for their capability to intercalate into DNA and be associated with topoisomerase inhibition27,28,29,30,31.
These correlations with DNA interacting agents could thus be conferred by the pyrene moiety. Although this correlation appeared only at the LC50 concentration, we investigated such activity in vitro for both compounds 13 and 16 in comparison to their respective parental natural products 1 and 15. All four compounds inhibited the catalytic activity of human TopIIa in cell-free decatenation assays, albeit they displayed considerable differences in the strength of the effect (Fig. 5). 1 and 15 reduced decatenated DNA levels only at the highest applied concentration of 500 µM (250 and ~ 8 times more than their respective IC50 values), at which they led to ~ 50% TopoIIa inhibition (Fig. 5b). Derivatives 13 and 16 both exhibited a concentration-dependent trend of TopIIa inhibition that was observed starting from 1 µM (Fig. 5c). Compound 13 exerted slightly stronger inhibitory properties, being significantly different from the solvent control at concentrations ≥ 1 µM, whereas 15 significantly inhibited the enzyme at 50 µM, although fluorescence images might also indicate a chemical-induced fragmentation of the incubated kDNA (Fig. 5a). These results were obtained in cell-free assays; the intracellular concentrations are probably less than those at which we observed 50% TopoIIa inhibition.

Decatenation Assay of compounds 1, 15, 13 and 16. (A) Representative gels show the inhibition of TopIIa by the tested compounds, resulting in the gradual disappearance of decatenated DNA bands as compared to the solvent control (SC). The original uncropped images of the representative gels are supplied in Figs. S5 and Figs. S6. (B, C) show the relative quantification of decatenated DNA as T/C [%] values, which are expressed as means + SD of at least 3 independent experiments. Significant differences to the solvent control were calculated by one-way ANOVA, followed by Fisher’s LSD post-hoc testing, and are indicated by “*” (p < 0.05), “**” (p < 0.01), or “***” (p < 0.001).
To get more insight into the contribution of the pyrene moiety to the in vitro anti-cancer effects of 13, we prepared a second small set of derivatized compounds focused on differences involving the pyrene unit. Using our olefin CM process, we synthesized compounds incorporating a longer (23, Fig. 1I) and shorter (24, Fig. 1I) linkers, oxygen in the linker (25, Fig. 1I), a smaller aromatic naphthalene group (26, Fig. 1I), and a linear long-chain aliphatic substituent (27, Fig. 1I). Figure 1G illustrates the preparation of the requisite alkenes 18 and 19, while Fig. 1H shows the structures of 20 and 21 prepared using literature methods. In addition, alkene 22 was commercially available.
Although compounds 25, 26 and 27 did not display increased in vitro anti-proliferative effects in comparison to 1 (Table 2), morphological evaluation of their effects highlighted in all cases the induction of ER-derived vacuolization (Fig. 6). Cell shrinkage was also observed with respect to 25 and 26, while evidence of altered plasma membrane was observed after treatment with 27 (Fig. 6). This feature may be attributed to the long alkene chain that may disturb the plasma membrane. These three compounds led to cell detachment from their culture support (more efficiently than 9 and in a similar manner to natural product 1). Considering these results, the ER swelling induced by several sphaeropsidin derivatives likely depends on the presence of a lipophilic substituent linked to C-16. This small set of pyrene-modified compounds nevertheless provides evidence that the pyrene substituent is not required to induce the severe ER swelling and therefore opens up new avenues for sphaeropsidin-based chemical optimization for anti-cancer purposes. Finally, the relevance of the length of the carbon chain linking the pyrene and the sphaeropsidin scaffolds was evaluated with compounds 23 (longer chain) and 24 (shorter chain). According to the results of Table 2, it appears that the chain linker has a minimal length requirement to increase the anti-cancer activity of the parental compound.

Compound 9, 25, 26 and 27 activity –induced vacuolization and other morphological effects in SK-Mel-28 human melanoma cells and U373 human glioblastoma cells. Representative pictures of brightfield images of cells treated with each compound as indicated on the picture itself. Right panel illustrates the staining of U373 cells stained with the Cell Light® system (merged brightfield and fluorescent pictures shown).
Discussion
Several reports, including our work, highlighted the interesting anti-cancer effects in vitro of sphaeropsidin A (1)12,13,14. In particular, 1 has been shown to display both anti-migratory and cytotoxic effects against melanoma cells12. These have been associated partly with ion homeostasis disruption via NKCC1 inhibition and/or the targeting of anion exchangers.12 Ion channels and transporters represent targets of growing interest since they appear to be involved in cancer development, chemoresistance and migration4,31,32,33. Accordingly, several inhibitors of ion channels, transporters or pumps are being studied in clinical trials either alone (for example the TRPV6 calcium channel inhibitor SOR-C13)34 or in combination with approved chemotherapeutic agents (for example digoxine in combination with trametinib, vemurafenib, cisplatin or decitabine; clinical trials.gov)35. Despite the deregulation of their targets’ expression in cancer cells32, specific delivery to the tumor may be needed to avoid toxicity36. This may be particularly the case with respect to natural product 1, knowing the high expression levels of NKCC and ion exchangers in the kidneys37,38. In addition, we recently reported on 1’s instability in physiological medium that also needs to be overcome for in vivo administration39.
In this study we thus investigated derivatization on two sites on the sphaeropsidin A (1) skeleton, namely the C6-OH and C15,C16-alkene previously shown to be tolerant of modifications without losing in vitro anti-cancer activity13,14. In terms of a brief overview of the structure activity relationships (SAR) extractable from the small library of compounds synthesized, the following aspects could be determined. As mentioned previously, the sphaeropsidin A-derived esters (2–6) all had comparable mean IC50 values similar to their parent sphaeropsidin A 1 (IC50 = 2.07 ± 0.15 μM), indicating probable hydrolysis of these compounds in situ. When sphaeropsidin A’s C15=C16 alkene was metathetically extended to a long aliphatic chain (27, mean IC50 = 2.06 ± 0.45 μM) the activity was retained, while a much shorter butyl extension to give 9 gave the best aliphatic derivative (mean IC50 = 0.88 ± 0.16 μM). In terms of further comparison of this set of compounds, terminally substituted aliphatic chains featuring bromo atoms (10 and 11) or methyl ester groups (6 and 7), all had similar mean activities compared to the parent sphaeropsidin A 1. Of interest, was that the carboxylic acid derivative of 6, namely compound 8, lost its activity completely and had a mean IC50 of > 94 μM. Concerning the sphaeropsidin A 1 derivatives generated by metathesis extension with alkenes bearing aromatic functional groups, the styrene-derived compound 12 was only slightly less active than its parent 1. On the other hand, the synthesis of a sphaeropsidin A 1 derivative with a three methylene alkyl chain tethered to the 1-position on the pyrene scaffold gave the most active compound 13 (mean IC50 = 0.37 ± 0.04 μM). Extending this chain by one methylene gave compound 23 with similar activity (mean IC50 = 0.49 ± 0.07 μM), whilst decreasing the aliphatic tether by one CH2 reversed the activity back to that of the parent compound (24, mean IC50 = 1.40 ± 0.34 μM). Finally, the introduction of an oxygen atom into the 3 carbon-tethered pyrene (to give 25) or to a 1-naphthyl group (to afford 26) resulted in sphaeropsidin A 1 derivatives with activities indistinguishable from the parent compound (mean IC50’s of 3.34 ± 0.16 and 2.14 ± 0.33 μM respectively).
The most potent derivative of this study, i.e. compound 13, induces rapid and severe ER-swelling as assessed through fluorescent GFP-KDEL-calrecticulin expression assay and electron microscopy (Figs. 2B and 3). Whether this swelling results from a proteasomal inhibition leading to an ER stress40,41 or that the swelling of the ER is the causal event resulting in an inhibition of the proteasome still remains to be investigated. We indeed observed rapid and strong inhibition of chymotrypsin-like and caspase-like proteasomal activities as early as 6 h after treatment initiation with 13 (Fig. 4D). On the other hand, 1 may target NKCC transporters and induces rapid cell shrinkage associated with ion homeostasis disruption12. Hypertonic stress has been previously shown to trigger accumulation of unfolded and oxidized protein in the ER that stimulate degradation mechanisms42. Under such conditions, increased autophagy and proteasomal activities were previously described, similarly to what we observed herein with respect to 1 (including mitophagy and capsase-like increased activity shown in Figs. 3 and 4D, respectively)43,44. Interestingly, NKCC transporters targeted by 1 have been found in abundance within the ER and the Golgi41. Although they might be non-functional at those sites45, their presence in the ER may facilitate the accumulation of 13 in that organelle after its rapid cellular penetration as observed in Fig. 4A–C. We hypothesize that the addition of a hydrophobic substituent on C-16 may increase the penetration of the compound into the cell, allowing new intracellular distribution. The addition of a hydrocarbon chain is well known to allow an increased cellular uptake in a length dependent manner46. To further compare the cellular bioavailability of 1 versus 13, but also to other derivatives including for example 25, 26 and 27, we needed to develop analytical methods and proposed to use them according to Teuscher et al.47.
Additional effects linked to ER stress and/or swelling have been observed in 13-treated cancer cells; these include an increase in lipid droplet content and the appearance of nucleolar “Christmas trees” (Fig. 3). Indeed, the ER stress activates lipogenesis through the three main actors of the unfolded protein response (UPR), namely IRE-1 α, Perk and Atf-648,49. The IRE-1 α/XBP-1 pathway activates phosphatidylcholine synthesis. PERK/eIF2 α pathway stimulates Chop and PPARɣ expression promoting lipogenesis, while Atf-6 stimulates phospholipid synthesis. On the other hand, “Christmas trees”, formed of pre-transcripted rRNA assembled to chromatin, reflect over-activation of ribosomal biosynthesis27,50. The lipid droplets and “Christmas trees” may thus be induced by the ER-swelling to face the need for ER-membrane.
The role of the pyrene substituent in the anti-cancer effects observed was further considered. Unsurprisingly, it could contribute to the intercalating properties of 13 at high concentrations only, according to the NCI COMPARE data and in vitro DNA decatenation assay (Fig. 5). Even derivatives bearing only the alkyl tail, such as 27, also induced strong ER-derived vacuolization, suggesting that the pyrene moiety is not required to induce the ER-swelling observed. By contrast, the pyrene skeleton seems to result in increased potency of the in vitro anti-cancer effects when comparing 13’s IC50 values to those of 1, 9, 25 and 26 (Table 2). This hypothesis is also supported by the relevance of the length of the carbon chain separating the pyrene unit from the sphaeropsidin A skeleton and its effect on in vitro potency (comparison between 13, 23 and 24; Table 2); when the chain is too short, we observe lower potency. Finally, the chemical linkage of the two scaffolds appeared also mandatory to get increased anti-cancer effects and ER-swelling as co-treatment with 1 and 14 did not show any additive effects (Table 1).
Importantly, 13-induced effects were shown to trigger cell death in both SK-MEL-28 and U373 cell models with resistance to various pro-apoptotic stimuli51. In general, the activation of the unfolded protein response (UPR) triggered by ER-stress induces cell cycle arrest and apoptotic cell death30. This may be the case with respect to SK-MEL-28 cells that displayed strong capsase-3 activation upon 13-treatment (Fig. 4E). By contrast, we observed phosphatidylserine externalization and sub-G1 increase (data not shown) in U373 without caspase activation (Fig. 4E, Fig. S3). In these cells, 13-induced effects may lead to paraptosis-like cell death according to the ER swelling, the proteasomal inhibition, the externalization of the phosphatidylserines and the absence of caspase activation52,53. Cell death pathway induction may actually depend on the cell type according to their biochemical characteristics, including for example the expression levels of proteins involved in the UPR. Grp78 is indeed overexpressed by U373 cells in comparison to melanoma cells54. Upon ER-stress, Grp78 chaperone dissociates from IRE-1α to be activated and then contributes to unfolded proteasomal protein decay55.
Considering our recent findings that the instability of 1 in physiological media is enhanced in the presence of amino-acids39, we also worked on sphaeropsidin B skeleton characterized by the C7-alcohol (instead of 1’s ketone functionality). Importantly, the analogue of 13 prepared using the sphaeropsidin B skeleton (derivative 16) turned out to be an order of magnitude more potent than sphaeropsidin B itself, displaying an IC50 in the low micromolar range and it induced severe ER-swelling at relevant concentrations. Future efforts include evaluation of sphaeropsidin B derivatization series at C15–C16 alkene, development of formulations based on C6-derivatization involving esterase-sensitive prodrug design as well as developing formulation methods to address solubility and stability issues for future in vivo evaluation.
Conclusions
We recently discovered that sphaeropsidin A, a fungal phytotoxin isolated from Diplodia cupressi, overcomes apoptosis resistance in cancer cells by inducing a marked and rapid cellular shrinkage through the impairment of regulatory volume increase12. Cell shrinkage was determined to be the cause, not the consequence of apoptosis, and was related to the rapid loss of intracellular chloride12. Our results suggest that the prolonged effect of SphA on chloride concentrations, leading to sustained cellular shrinkage, triggers apoptosis directly, bypassing the classical signaling pathways and overcoming apoptosis resistance12.
Here, we describe very unusual chemistry of sphaeropsidin A and hemi-synthesis of seventeen new derivatives involving lipophilic substitutions at the C15,C16-alkene. These compounds were obtained in one-step by an efficient cross-metathesis reaction of the natural product with terminal alkene-containing compounds. Our lead derivative containing a pyrene moiety at this position led to potency enhancement of 5–10 times over that of the natural product itself and altered the biological mode of action of the natural product. These analogues now triggered severe ER swelling associated with strong proteasomal inhibition and consequently cell death, a feature that was not observed with respect to the mode of action of the natural product. Importantly, the profile of responses of the 60 cancer cell lines of the NCI to the lead compound did not match with any profile of the NCI compound collection. This study led to the discovery of a new set of sphaeropsidin A derivatives that may be exploited as potential anti-cancer agents, notably due to their novel cell killing mechanism allowing to overcome cancer cell resistance to chemotherapy as assessed by its effects on multidrug resistant models.
Materials and methods
Chemistry
General
All reagents, solvents and catalysts were purchased from commercial sources (Acros Organics and Sigma-Aldrich) and used without purification. All reactions were performed in oven-dried flasks open to the atmosphere or under nitrogen as described and monitored by thin layer chromatography (TLC) on TLC precoated (250 µm) silica gel 60 F254 glass-backed plates (EMD Chemicals Inc.). Visualization was accomplished with UV light. Flash column chromatography was performed on silica gel (32–63 µm, 60 Å pore size). 1H and 13C NMR spectra were recorded on Bruker 300, 400 and 500 MHz spectrometers. Chemical shifts are reported in ppm (d) relative to the TMS internal standard. Abbreviations are as follows: s (singlet), d (doublet), dd (double doublet), t (triplet), dt (double triplet), ddt (doublet of double triplets), q (quartet), m (multiplet). HRMS analyses were performed using Waters Synapt G2 LCMS. Compounds 1456, 2057, 2158 were prepared according to literature methods. Sphaeropsidin A (1) was obtained by purification of the organic extract of Diplodia cupressi culture filtrates as reported previously10.
General procedure for Steglich esterification
The desired carboxylic acid (0.28 mmol), DMAP (7.1 mg, 0.058 mmol) and EDCI (67.3 mg, 0.43 mmol) were added to a round-bottom flask, under N2 and dissolved in 2.5 mL of CH2Cl2. The reaction was stirred for 1.5 h and monitored by TLC to observe the activation of the acid. Sphaeropsidin A (1) (10 mg, 0.028 mmol) was added in one portion and reaction was stirred overnight to produce the desired analogue (as witnessed by TLC). The solvent was evaporated under reduced pressure after which the crude material was dissolved in EtOAc, washed 3 times with deionized water and brine, and dried over MgSO4. The desired analogue was purified by flash chromatography (n-hexane/CH2Cl2, 40:1) or by preparatory TLC (CHCl3) to obtain the compounds as described below:
3 (45%): 1H NMR (400 MHz, CDCl3) δ 6.53 (d, J = 1.8 Hz, 1H), 5.79 (dd, J = 17.5, 10.7 Hz, 1H), 5.08 (dd, J = 17.7, 0.5 Hz, 1H), 5.05 (dd, J = 10.0, 0.7 Hz, 1H), 2.86 (s, 1H), 2.76–2.57 (m, 2H), 2.34 (td, J = 6.9, 2.6 Hz, 2H), 2.29–2.16 (m, 2H), 1.99 (t, J = 2.6 Hz, 1H), 1.96–1.89 (m, 2H), 1.87–1.54 (m, 6H), 1.39–1.30 (m, 1H), 1.27–1.20 (m, 2H), 1.19 (s, 3H), 1.10 (s, 3H), 1.08 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 189.6, 174.1, 169.3, 148.5, 144.7, 135.9, 113.1, 105.4, 83.2, 71.7, 69.5, 56.2, 51.0, 40.6, 38.9, 33.1, 32.8, 32.7, 29.7, 26.7, 24.5, 23.5, 22.7, 22.5, 17.9, 17.8. HRMS m/z (ESI+) calcd for C26H32NaO6 [M + Na]+ 463.2091, found 463.2075.
4 (75%): 1H NMR (400 MHz, CDCl3) δ 8.13–8.05 (m, 2H), 7.68–7.58 (m, 1H), 7.54–7.44 (m, 2H), 6.61 (br. s, 1H), 5.80 (dd, J = 17.5, 10.6 Hz, 1H), 5.07 (dd, J = 17.5, 0.7 Hz, 1H), 5.04 (dd, J = 10.6, 0.7 Hz, 1H), 2.63 (s, 1H), 2.25 (d, J = 13.5 Hz, 1H), 1.97–1.56 (m, 7H), 1.41–1.28 (m, 3H), 1.25 (s, 3H), 1.17 (s, 3H), 1.11 (s, 3H). HRMS m/z (ESI+) calcd for C27H30NaO6 [M + Na]+ 473.1935, found 473.1920.
5 (55%): 1H NMR (400 MHz, CDCl3) δ 6.54 (d, J = 1.7 Hz, 1H), 5.80 (dd, J = 17.5, 10.6 Hz, 1H), 5.08 (dd, J = 17.2, 0.5 Hz, 1H), 5.06 (dd, J = 10.9, 0.5 Hz, 1H), 2.86 (s, 1H), 2.65–2.47 (m, 2H), 2.45–2.39 (m, 2H), 2.22 (br. d, J = 13.3 Hz, 1H), 1.94–1.87 (m, 1H), 1.85–1.82 (m, 1H), 1.81–1.75 (m, 5H), 1.75–1.54 (m, 5H), 1.40–1.32 (m, 1H), 1.27–1.21 (m, 1H), 1.19 (s, 3H), 1.10 (s, 3H), 1.09 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 189.8, 178.1, 174.1, 169.4, 148.5, 144.7, 135.9, 113.1, 105.4, 71.8, 56.2, 53.5, 40.6, 38.9, 34.1, 33.5, 32.8, 32.7, 29.7, 26.7, 24.5, 24.0, 24.0, 22.7, 22.5, 17.9. HRMS (ESI) m/z calcd for C26H34NaO8 [M + Na]+ 497.2146, found 497.2154.
General procedure for cross-metathesis
Sphaeropsidin A (1) (10 mg, 0.028 mmol) and Grubbs II catalyst (7.4 mg, 0.008 mmol) were dissolved in CH2Cl2 in a round-bottom flask, a condenser applied, placed under N2 and brought to reflux. The desired alkene (0.28 mmol) was added in three portions over 2 h intervals. The solvent was evaporated under reduced pressure and purified by flash column chromatography (n-hexane/Et2O, 9:1) or by preparatory TLC (n-hexane/EtOAc, 7:3).
6 (38%): 1H NMR (400 MHz, CDCl3) δ 6.82 (d, J = 1.8 Hz, 1H), 5.52–5.36 (m, 2H), 3.67 (s, 3H), 2.72 (s, 1H), 2.30 (t, J = 7.4 Hz, 2H), 2.26–2.16 (m, 1H), 2.13–2.04 (m, 3H), 1.90–1.79 (m, 3H), 1.72 (t, J = 7.4 Hz, 2H), 1.68–1.60 (m, 5H), 1.37–1.32 (m, 2H), 1.20 (s, 3H), 1.19 (s, 3H), 1.06 (s, 3H). HRMS (ESI) m/z calcd for C25H34NaO7 [M + Na]+ 469.2197, found 469.2185.
7 (52%): 1H NMR (400 MHz, CDCl3) δ 6.80 (d, J = 1.7 Hz, 1H), 5.55–5.42 (m, 2H), 5.19 (s, 1H), 3.67 (s, 3H), 2.71 (s, 1H), 2.44–2.31 (m, 4H), 2.22 (br. d, J = 9.2 Hz, 1H), 1.92–1.75 (m, 5H), 1.67–1.54 (m, 3H), 1.42–1.31 (m, 2H), 1.19 (s, 3H), 1.19 (s, 3H), 1.06 (s, 3H). HRMS (ESI) m/z calcd for C24H33O7 [M + H]+ 433.2221, found 433.2223.
8 (25%): 1H NMR (400 MHz, CDCl3) δ 6.79 (d, J = 1.7 Hz, 1H), 5.53–5.36 (m, 2H), 5.14 (s, 1H), 2.72 (s, 1H), 2.32 (t, J = 6.7 Hz, 2H), 2.25–2.21 (m, 1H), 2.15–2.08 (m, 1H), 1.96–1.74 (m, 4H), 1.67–1.54 (m, 5H), 1.40–1.20 (m, 4H), 1.20 (s, 3H), 1.19 (s, 3H), 1.07 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 192.6, 174.7, 153.4, 137.2, 132.5, 130.4, 128.7, 103.7, 71.2, 57.2, 51.5, 40.5, 38.7, 33.2, 32.7, 32.4, 31.9, 29.9, 27.0, 25.2, 24.1, 23.0, 22.5, 18.1. HRMS (ESI) m/z calcd for C24H33O7 [M + H]+ 433.2221, found 433.2223.
9 (48%): 1H NMR (400 MHz, CDCl3) δ 6.84 (d, J = 1.8 Hz, 1H), 5.54–5.35 (m, 2H), 5.20 (s, 1H), 2.72 (s, 1H), 2.22 (br. d, J = 8.0 Hz, 1H), 2.02 (q, J = 6.4 Hz, 2H), 1.91–1.77 (m, 3H), 1.69–1.54 (m, 5H), 1.40–1.28 (m, 6H), 1.19 (s, 3H), 1.19 (s, 3H), 1.06 (s, 3H), 0.89 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 191.8, 174.7, 153.9, 136.2, 132.5, 129.8, 103.7, 71.2, 57.2, 51.4, 40.5, 38.7, 32.7, 32.5, 32.3, 31.6, 30.3, 27.1, 24.9, 23.0, 22.5, 22.3, 18.1, 14.1. HRMS (ESI) m/z calcd for C24H34NaO5 [M + Na]+ 425.2298, found 425.2283.
10 (52%): 1H NMR (400 MHz, CDCl3) δ 6.83 (d, J = 1.7 Hz, 1H), 5.52–5.40 (m, 2H), 5.19 (s, 1H), 3.41 (t, J = 6.7 Hz, 2H), 2.71 (s, 1H), 2.26–2.21 (m, 1H), 2.10–2.03 (m, 2H), 1.92–1.79 (m, 4H), 1.68–1.49 (m, 9H), 1.39–1.34 (m, 1H), 1.20 (s, 3H), 1.19 (s, 3H), 1.07 (s, 3H). HRMS (ESI) m/z calcd for C24H33BrNaO5 [M + Na]+ 503.1404, found 503.1561.
11 (65%): 1H NMR (400 MHz, CDCl3) δ 6.83 (d, J = 1.8 Hz, 1H), 5.56 (dt, J = 15.8, 1.0 Hz, 1H), 5.46 (dt, J = 15.8, 6.5 Hz, 1H), 5.19 (s, 1H), 3.65 (s, 1H), 3.39 (t, J = 6.9 Hz, 2H), 2.72 (s, 1H), 2.64–2.54 (m, 2H), 2.23 (br. d, J = 6.3 Hz, 1H), 1.94–1.78 (m, 4H), 1.71–1.56 (m, 3H), 1.40–1.29 (m, 2H), 1.20 (s, 3H), 1.19 (s, 3H), 1.09 (s, 3H).HRMS (ESI) m/z calcd for C22H29BrNaO5 [M + Na]+ 475.1091, found 475.1091.
12 (51%): 1H NMR (400 MHz, CDCl3) δ 7.41–7.28 (m, 4H), 7.25–7.21 (m, 1H), 6.94 (d, J = 1.7 Hz, 1H), 6.41 (d, J = 16.3 Hz, 1H), 6.18 (d, J = 16.3 Hz, 1H), 5.20 (s, 1H), 3.65 (s, 1H), 2.74 (s, 1H), 2.25 (br. d, J = 8.1 Hz, 1H), 2.02–1.87 (m, 4H), 1.86–1.73 (m, 1H), 1.66–1.57 (m, 2H), 1.41–1.32 (m, 2H), 1.21 (s, 3H), 1.20 (s, 3H), 1.20 (s, 3H). HRMS (ESI) m/z calcd for C26H30NaO5 [M + Na]+ 445.1985, found 445.1981.
13 (75%): 1H NMR (300 MHz, CDCl3) δ 8.25 (d, J = 9.3 Hz, 1H), 8.20–8.07 (m, 4H), 8.06–7.95 (m, 3H), 7.85 (d, J = 7.8 Hz, 1H), 6.82 (d, J = 1.6 Hz, 1H), 5.57–5.40 (m, 2H), 5.20 (s, 1H), 3.34 (t, J = 7.6 Hz, 2H), 2.70 (s, 1H), 2.24–2.14 (m, 2H), 2.01–1.89 (m, 3H), 1.87–1.70 (m, 3H), 1.65–1.51 (m, 4H), 1.41–1.28 (m, 3H), 1.19 (s, 6H), 1.04 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 191.7, 174.7, 153.6, 136.9, 136.7, 132.5, 131.6, 131.0, 129.9, 129.2, 128.7, 127.6, 127.4, 127.4, 126.7, 126.0, 125.2, 125.1, 125.0, 124.9, 124.8, 123.5, 103.7, 71.1, 57.1, 51.3, 40.4, 38.7, 33.1, 32.7, 32.6, 32.3, 31.5, 30.2, 27.0, 24.9, 22.9, 22.5, 18.1. HRMS (ESI) m/z calcd for C39H39O5 [M − H]− 587.2803, found 587.2816.
16 (86%): 1H NMR (300 MHz, CDCl3) δ 8.25 (d, J = 9.3 Hz, 1H), 8.19–8.06 (m, 4H), 8.03–7.94 (m, 3H), 7.85 (d, J = 7.8 Hz, 1H), 5.83 (s, 1H), 5.46 (br. s, 2H), 4.24 (s, 1H), 3.33 (t, J = 7.6 Hz, 2H), 2.55 (s, 1H), 2.23–2.06 (m, 3H), 2.00–1.88 (m, 3H), 1.67–1.44 (m, 5H), 1.34–1.16 (m, 4H), 1.19 (s, 3H), 1.10 (s, 3H), 0.96 (s, 3H), 0.96–0.87 (m, 2H). HRMS (ESI) m/z calcd for C39H43O5 [M + H]+ 591.3105, found 591.3111.
23 (61%): 1H NMR (500 MHz, CDCl3) δ 8.26 (d, J = 9.2 Hz, 1H), 8.17–8.09 (m, 4H), 8.05–7.97 (m, 3H), 7.86 (d, J = 7.8 Hz, 1H), 6.81 (d, J = 1.8 Hz, 1H), 5.48–5.36 (m, 2H), 5.19 (s, 1H), 3.35 (t, 2H), 2.69 (s, 1H), 2.19 (d, J = 9.1 Hz, 1H), 2.10 (q, J = 6.9 Hz, 2H), 1.90–1.80 (m, 4H), 1.77–1.69 (m, 2H), 1.61–1.55 (m, 8H), 1.19 (3, 3H), 1.19 (3, 3H), 1.02 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 191.8, 174.7, 153.7, 137.1, 136.7, 132.6, 131.6, 131.1, 129.9, 129.4, 128.8, 127.7, 127.5, 127.3, 126.7, 126.0, 125.3, 125.2, 125.0, 125.0, 124.8, 123.6, 103.7, 71.1, 57.1, 51.4, 40.5, 38.7, 33.5, 32.7 (2C), 32.3, 31.3, 30.3, 29.4, 27.1, 24.9, 23.0, 22.5, 18.1. HRMS (ESI) m/z calcd for C40H42NaO5 [M + Na]+ 625.2924; found 625.2938.
24 (55%): 1H NMR (CDCl3, 500 MHz) δ 8.25 (d, J = 9.3 Hz, 1H), 8.20–8.08 (m, 4H), 8.06–7.96 (m, 3H), 7.83 (d, J = 7.7 Hz, 1H), 6.74 (s, 1H), 5.61–5.57 (m, 1H), 5.35 (d, J = 16.4 Hz, 1H), 5.16 (s, 1H), 3.43 (t, J = 7.6 Hz, 2H), 2.66 (s, 1H), 2.59 (d, J = 7.5 Hz, 2H), 2.19 (d, J = 8.3 Hz, 1H), 1.86–1.63 (m, 6H), 1.57 (s, 4H), 1.21 (s, 3H), 1.18 (s, 3H), 0.98 (s, 3H). HRMS (ESI) m/z calcd for C38H38NaO5 [M + Na]+ 597.2611; found 597.2613.
25 (18%): H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 9.2 Hz, 1H), 8.26–8.12 (m, 4H), 8.10–7.96 (m, 4H), 6.81 (d, J = 1.7 Hz, 1H), 5.76–5.59 (m, 2H), 5.27–5.02 (m, 3H), 4.14 (d, J = 4.6 Hz, 2H), 2.67 (s, 1H), 2.19 (br. d, J = 10.3 Hz, 1H), 1.89–1.79 (m, 3H), 1.71–1.55 (m, 5H), 1.33–1.27 (m, 2H), 1.19 (s, 3H), 1.18 (s, 3H), 1.06 (s, 3H). HRMS (ESI) m/z calcd for C38H37O6 [M − H]− 589.2596, found 589.2599.
26 (41%): 1H NMR (400 MHz, CDCl3) δ 8.29–8.19 (m, 1H), 7.86–7.73 (m, 1H), 7.53–7.33 (m, 4H), 6.86 (d, J = 1.7 Hz, 1H), 6.81 (d, J = 7.4 Hz, 1H), 5.67–5.64 (m, 2H), 5.20 (s, 1H), 4.18 (t, J = 6.4 Hz, 2H), 2.70 (s, 1H), 2.69–2.61 (m, 2H), 2.22 (d, J = 5.9 Hz, 1H), 1.97–1.78 (m, 3H), 1.67–1.55 (m, 5H), 1.39–1.27 (m, 2H), 1.19 (s, 6H), 1.09 (s, 3H). HRMS (ESI) m/z calcd for C32H35O6 [M − H]− 515.2439, found 515.2441.
27 (57%): 1H NMR (500 MHz, CDCl3) δ 6.84 (d, J = 1.8 Hz, 1H), 5.50–5.37 (m, 2H), 5.20 (s, 1H), 2.72 (s, 1H), 2.22 (d, J = 9.1 Hz, 1H), 2.05–1.96 (m, 2H), 1.96–1.77 (m, 3H), 1.75–1.52 (m, 6H), 1.26 (s, 25H), 1.19 (s, 3H), 1.19 (s, 3H), 1.06 (s, 3H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 191.8, 174.7, 153.9, 136.2, 132.5, 129.9, 103.7, 71.2, 57.2, 51.4, 40.5, 38.7, 32.9, 32.7, 32.3, 32.1, 30.3, 29.9, 29.8, 29.8, 29.8, 29.8, 29.7, 29.6, 29.5, 29.5, 29.3, 27.1, 24.9, 23.0, 22.8, 22.5, 18.1, 14.3. HRMS (ESI) m/z calcd for C34H54NaO5 [M + Na]+ 565.3863; found 565.3867.
Synthesis of CM precursor 18
To a solution of 1-pyrenebutanol (200 mg, 0.728 mmol) and CBr4 (266 mg, 0.801 mmol) in CH2Cl2, in a round-bottom flask, PPh3 (210 mg, 0.801 mmol) was added at 0 °C. The resulting mixture was allowed to warm to rt, and the reaction progress were monitored by TLC. After the complete consumption of starting alcohol (~ 3 h), the solvent was evaporated under reduced pressure. To the resulting brown residue, n-hexane was added while stirring, resulting in a white precipitate which was removed by filtration, and after which the filtrate was concentrated under reduced pressure. Column purification of the corresponding residue on silica gel (EtOAc/hexane (5/95)) afforded bromide 17 (205 mg, 0.607 mmol, 83%). Compound 17 (100 mg, 0.297 mmol) was then dissolved in THF in a round-bottom flask. To this solution copper iodide (62 mg, 0.326 mmol) was added at rt and the reaction was stirred for 30 min. At the same temperature, vinylmagnesium bromide (4.24 mL, 2.97 mmol) was added dropwise over 20 min and then reaction mixture was heated at 40 °C. The reaction progress was monitored by TLC. After complete consumption of the starting bromo compound (6 h), the reaction was quenched with aqueous NH4Cl. The reaction mixture was extracted with Et2O (3 × 5 mL), the combined organic layers were washed with brine (5 mL), dried with MgSO4, and concentrated under reduced pressure. The crude product was initially purified by chromatographic column using EtOAc/n-hexane (2/98) yielding a 1:0.7 mixture of compounds 18 and 19 (59 mg, 0.207 mmol, 70%). This mixture of alkenes was again subjected to a gravity column using silica gel and pentane as an eluent. This column purification was found to be successful in separating the two alkenes.
18 (34%): 1H NMR (CDCl3, 500 MHz) δ 8.28 (d, J = 9.3 Hz, 1H), 8.19–8.08 (m, 4H), 8.06–7.94 (m, 3H), 7.87 (d, J = 7.8 Hz, 1H), 5.83 (ddt, J = 16.9, 10.2, 6.7, 6.7 Hz, 1H), 5.06–4.94 (m, 2H), 3.36 (d, J = 7.9 Hz, 2H), 2.19–2.13 (m, 2H), 1.89 (d, J = 146.4 Hz, 2H), 1.71–1.54 (m, 2H). 13C NMR (CDCl3, 126 MHz) δ 139.0, 137.2, 131.6, 131.1, 129.9, 128.8, 127.7, 127.4, 127.3, 126.7, 125.9, 125.3, 125.2, 125.0, 124.9, 124.8, 123.6, 114.7, 33.9, 33.6, 31.5 29.2. HRMS (ESI) m/z calcd for C22H21 [M + H]+ 285.1638; found 285.1641.
Synthesis of cross-metathesis precursor 19
Compound 17 (100 mg, 0.297 mmol) was dissolved in 1:1 ratio of dimethylformamide and toluene in a round-bottom flask. To this solution, DBU (89 µL, 0.59 mmol), tetrabutylammonium iodide (11 mg, 0.030 mmol), were added and the resulting solution was heated at 110 °C for 10 h. The reaction mixture was then quenched with the addition of water and extracted with ether (3 × 5 mL). Combined organic layers were washed with brine (5 mL), dried with MgSO4, and concentrated in vacuo. The crude product was purified by chromatographic column using EtOAc/n-hexane (3/97) as an eluent. The corresponding alkene product 19 (20 mg, 0.078 mmol, 26%) was isolated as a colorless liquid.
19 (26%): 1H NMR (CDCl3, 500 MHz) δ 8.28 (d, J = 9.3 Hz, 1H), 8.18–8.10 (m, 4H), 8.05–7.97 (m, 3H), 7.88 (d, J = 7.8 Hz, 1H), 6.19–5.85 (m, 1H), 5.23–4.90 (m, 2H), 3.45 (t, J = 7.9 Hz, 2H), 2.66–2.53 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 138.3, 136.3, 131.6, 131.1, 130.0, 128.8, 127.7, 127.4, 127.4, 126.8, 126.0, 125.3, 125.2, 125.0, 125.0, 124.9, 123.5, 115.3, 36.0, 33.2. HRMS (ESI) m/z calcd for C20H16Na [M + Na]+ 279.1144; found 279.1143.
Cell biology assays
Cell culture
Cancer cell lines were obtained from either the European Collection of Cell Cultures (ECACC; Salisbury, UK) or the American Type Culture Collection (ATCC; Manassas, VA). The two glioma cell lines were the U373 (ECACC code 08061901) and Hs683 (ATCC HTB-138) cell lines. The carcinoma cell lines were the A549 non-small cell carcinoma cell line (ATCC code CCL 185) and the MCF-7 mammary breast carcinoma cell line (ATCC code HTB-22). The melanoma cell lines were the mouse B16F10 cells (ATCC code CRL-6475) and the human SKMEL-28 cells (ATCC code HTB-72). The ovarian cancer cell line A2780 and its cisplatin-resistant subline A2780cis were obtained from Sigma-Aldrich (St. Louis, MO, USA). Colon cancer HCT116 cells were obtained from ATCC (CCL-247) and the oxaliplatin-resistant subline HCT116/OxR established in the W. Berger lab59. The HeLa cell derivative KB-3-1 and the colchicine-selected, highly ABCB1-overexpressing subline KB-C-1 were generously donated by Dr. D. W. Shen, Bethesda, MD. RKOp53wt colon cancer cells together with the RKOp53KO (homozygous knock-out) subline60 were generously provided by Dr. Vogelstein from John Hopkins University, Baltimore. The correct origin of all cell models was confirmed by STR during performance of the experiments. All cell lines were cultured in RPMI1640 culture medium supplemented with 10% heat-inactivated fetal bovine serum, 2% l-glutamine (0.6 mg/mL), 2% penicillin/streptomycin (200 IU/mL and 200 μg/mL) and 0.1 mg/mL gentamicin.
Colorimetric MTT assay
The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) colorimetric assay has been conducted as described previously61. Briefly, cells were seeded 24 h before their treatment with each compound for a period of 72 h. The concentrations tested ranged from 10 nM to 100 µM. Each assay was conducted in sextuplicates. The number of independent experiments is mentioned in the legend of the tables.
Reversibility assay
The reversibility of the effects induced by compounds 13 was evaluated through qualitative microscopic observation of the cell cultures (morphology and confluence of the cell culture) over time after having been exposed or not to the compound for 1, 3 or 6 h. Negative control corresponds to untreated cells while positive control corresponds to cells treated with 13 continuously over the whole experiment. Briefly, 6 × 104 cells per well were seeded in 6 well plates and incubated for 24 h before their use. Culture medium was then replaced by fresh medium with or without compound 13. After 1, 3 or 6 h of treatment, cells were washed and the culture medium was renewed. Pictures were taken at different time points starting at t = 0 when the treatment was applied to the cells, 6 h, 24 h and 72 h thereafter.
Phase contrast microscopy
For the morphological evaluation of the effects induced by compounds 1 and 13, 6 × 104 cells were seeded on glass coverslips placed in 6 well plates a minimum 24 h before their treatment. At the end of the incubation period with the treatment, coverslips were rinsed once in PBS buffer and directly mounted on slides to allow visualization of the cell with an Axio.Imager.M2 microscope (G X40 and G X100).
Fluorescent microscopy
To decipher the origin of vacuoles, fluorescent probes staining different cellular compartments were used: the lysosomal Red DND-99 LysoTracker®, the glibenclamide Red ER tracker®, the green FM MitoTracker® that were all purchased from Lifes Technologies (Invitrogen, Belgium) and used according to manufacturer’s instructions. The CellLight ER-GFP (Thermofisher, Belgium) staining of the ER is based on the transfection of a plasmid coding for a fusion protein made of GFP fused to the ER signal sequence of calreticulin and the KDEL ER retention signal. Briefly, 6 × 104 cells were seeded on glass coverslips in 6 well plates. After a minimum of 24 h, cells were treated or not with the compounds of interest. Note that concentration and duration of the treatment were determined separately for each compound prior to the experiment on the basis of the apparition of marked morphological changes in phase contrast microscopy. Sixteen hours before the end of the experiment, 4 µL/well of the reagent were added to allow transfection and expression of the GFP-fused protein by the cells. Coverslips were then washed twice in PBS and mounted on slides without fixation step to take captions with the Axio.Imager.M2 microscope (Carl Zeiss, Belgium). To evaluate the cellular distribution of 13, the Axio.Imager.M2 was specifically equipped with an M340L4 excitation source (Thorlabs) and the pseudoconfocal Zeiss ZEN deconvolution system kindly provided for demo by Carl Zeiss (Belgium) to allow direct visualization of the compound 13 itself thanks to its intrinsic fluorescent properties.
Transmission electron microscopy
For TEM, cells were fixed with glutaraldehyde 2.5% (EM grade, Electron Microscopy Sciences, EMS) at 4 °C overnight, washed tree times in cacodylate buffer (pH 7.4) and postfixed 1 h at room temperature in 1% OsO4, (EMS), 1.5% potassium ferrocyanide (Sigma-Aldrich) in 0.15 M cacodylate buffer. This was immediately followed by a second incubation in 1% OsO4 for 1 h at room temperature. After washing in distilled water, samples were stained with 1% uranyl acetate (EMS) and serially dehydrated in increasing ethanol concentrations. Samples were then embedded in epoxy resin (Agar 100 resin, Agar Scientific Ltd) and left to polymerize for 2 days at 60 °C. Ultrathin sections (50–70 nm thick) were collected with a Leica UC6 ultra-microtome on formvar-carbon-coated copper grids and further stained with uranyl acetate and lead citrate by standard procedures. Observations were made on a Tecnai 10 transmission electron microscope (FEI) and images were captured with a Veleta camera and processed with SIS iTEM software (Olympus).
Fluorimetric quantification of 13 cellular content
The fluorescent properties of 13 (maximal excitation at 340 nm and emission at 380 nm in PBS buffer with 5% FBS) allowed us to quantify the cellular content of 13 over time after exposure to the compound at various concentrations. Briefly, 5 × 103 cells/well were seeded in 24 well plates 24 h before their treatment with 13 for 2–24 h. At the end of the treatment, cells where washed three times with PBS and further detached with trypsin/EDTA (100 µL per well). 150 µL of PBS with 5% FBS were added to each well to inactivate trypsin/EDTA. 100 µL of the cell suspension were then transferred to black 96 well plates for fluorescent measurement (Ex: 340 nm; Em: 380 nm; SynergyMX Biotek, USA). The fluorescence levels were finally adjusted according to the cell concentration and normalized to 104 cells.
Catalytic proteasomal activities measurement in cells
The chemotrypsin-like, trypsin-like and caspase-like activities of the proteasome were evaluated in cells with the G8660, G8861 and G8761 kits respectively according to the manufacturer’s instructions (Promega, USA). Briefly, the assays are based on the use of short amino-acid sequences coupled with luciferin and that can be specifically cleaved by each catalytic unit of the proteasomal complex. For this purpose, cells were seeded in 96 well plates 24 h before their treatment with 1 (5 µM), 13 (1 µM) for 6 h or with the positive control MG132 (1 µM) for 24 h. At the end of the treatments, cells were washed three times with PBS and permeabilized with digitonin (0.5 mg/mL; 50 µL per well) under shaking for 10 min. Then, 50 µL of a mixture of the substrate of interest and luciferase were added for 15 min under slow shaking. Luminescence levels quantified with the Active-Glo LR-100 luminometer (DSLabs, Suisse) were normalized according the number of viable cells evaluated in parallel in the same conditions by means of MTT assay.
Cell death evaluation
Cell death induced by compounds 1 and 13 was evaluated by means of the double annexin-V/propidium iodide staining by flow cytometry with the FITC Annexin V apoptosis detection kit from BD Pharmingen (USA) according to the manufacturer’s instructions. Briefly, cells were cultured and treated in 25 cm2 flasks. At the end of the treatment, all cells (adherent and floating cells) were collected, washed and resuspended at 106 cells/mL. 100 µL of the cell suspension were stained with 5 µL of FITC-annexin-V and 5 µL of propidium iodide for 15 min at rt in the dark. 400 µL of buffer were then added before data acquisition of 10,000 events per sample (Gallios, Beckman Coulter, Belgium). Caspase activation is usually evaluated to confirm or not the apoptotic feature of the cell death induced. For this purpose, we used the caspase-3, -8 and 9 multiplex assay (Ab219915, Abcam, UK) following manufacters’ instructions. In this assay, cells were exposed to short peptidic sequences linked to different fluorochromes and that could be specifically cleaved by each of these caspases. This assay thereby allowed simultaneous evaluation of the three caspase activities thanks to the quantification of the three fluorophores released in a SynergyMX plate fluorimeter (Biotek, USA).
Topoisomerase activity assay
The topoisomerase (Top) activity assay kit, including kinetoplast DNA (kDNA and recombinant human TopIIa enzyme, was obtained from TopoGen (Port Orange, FL, USA), agarose from PeqLab (Erlangen, Germany), 50 × TAE buffer from Roth (Karlsruhe, Germany) and ethidium bromide solution (10 mg/mL) from Sigma-Aldrich (Taufkirchen, Germany). Decatenation assays were performed as described previously62 with slight modifications according to the manufacturer’s protocol. 100 µM of the recently described dual topoisomerase poison P8-D6 served as a positive control63, and 5% DMSO as a solvent control. Briefly, the compounds were dissolved in DMSO and diluted to 20× the concentration to be tested. 1 µL of the resulting stock solutions were mixed with 18 µL of a buffer containing 200 ng kDNA and 1 mM ATP. Subsequently, 1 µL of a 1u/µL dilution of TopIIa enzyme was added and incubated at 37 °C for 30 min. Afterwards, the reaction was stopped by addition of Stop buffer, and electrophoresis was carried out on a 1% agarose gel in TAE buffer. The gel was stained with 0.5 µg/mL ethidium bromide, washed with water, and fluorescence images were captured with the LAS4000 system (Fujifilm, Duesseldorf, Germany). All steps were carried out on ice.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Bell, D. C., Leanza, L., Gentile, S. & Sauter, D. R. News and views on ion channels in cancer, is cancer a channelopathy?. Front. Pharmacol. https://doi.org/10.3389/fphar.2023.1258933 (2023).
Capatina, A. L., Lagos, D. & Brackenbury, W. J. Targeting ion channels for cancer treatment, current progress and future challenges. Rev. Phys. Biochem. Pharmacol. 183, 1–43. https://doi.org/10.1007/112_2020_46 (2022).
Molenaar, R. J. Ion channels in glioblastoma. ISRN Neurol. 590249(2011), 590249. https://doi.org/10.5402/2011/590249 (2011).
Zhao, J. Y., Li, M., Xu, J. & Cheng, W. The modulation of ion channels in cancer chemo-resistance. Front. Oncol. 12, 945896. https://doi.org/10.3389/fonc.2022.945896 (2022).
Pasantes-Morales, H. Channels and volume changes in the life and death of the cell. Mol. Pharmacol. 90, 358–370. https://doi.org/10.1124/mol.116.104158 (2016).
Bortner, C. D. & Cidlowski, J. A. Ions, the movement of water and the apoptotic volume decrease. Front. Cell Dev. Biol. 8, 611211. https://doi.org/10.3389/fcell.2020.611211 (2020).
Maeno, E., Takahashi, N. & Okada, Y. Dysfunction of regulatory volume increase is a key component of apoptosis. FEBS Lett. 580, 6513–6517. https://doi.org/10.1016/j.febslet.2006.10.074 (2006).
Masselli, M. et al. Deregulation of ion channel and transporter encoding genes in pediatric gliomas. Front. Oncol. 2, 53. https://doi.org/10.3389/fonc.2012.00053 (2012).
Yurinskaya, V. E. et al. Dual response of human leukemia U937 cells to hypertonic shrinkage, initial regulatory volume increase (RVI) and delayed apoptotic volume decrease (AVD). Cell Physiol. Biochem. 30, 964–973. https://doi.org/10.1159/000341473 (2012).
Evidente, A. et al. A phytotoxic pimarane diterpene of Sphaeropsis sapinea f. sp. cupressi, the pathogen of a canker disease of cypress. Phytochemistry 42, 1541–1546. https://doi.org/10.1016/0031-9422(96)00206-3 (1996).
Masi, M. et al. Bioactive metabolites from pathogenic and endophytic fungi of forest trees. Curr. Med. Chem. 25, 208–252. https://doi.org/10.2174/0929867324666170314145159 (2018).
Mathieu, V. et al. Sphaeropsidin A shows promising activity against drug-resistant cancer cells by targeting regulatory volume increase. Cell Mol. Life Sci. 72, 3731–3746. https://doi.org/10.1007/s00018-015-1902-6 (2015).
Lallemand, B. et al. Evaluation of in vitro anticancer activity of sphaeropsidins A-C, fungal rearranged pimarane diterpenes, and semisynthetic derivatives. Phytochem. Lett. 5, 770–775. https://doi.org/10.1016/j.phytol.2012.08.011 (2012).
Li, Y. et al. Aspergillus candidis is a newly recognized source of sphaeropsidin A, Isolation, semisynthetic derivatization and anticancer evaluation. Bioorg. Med. Chem. Lett. 27, 5436–5440. https://doi.org/10.1016/j.bmcl.2017.11.001 (2017).
Sparapano, L., Bruno, G., Fierro, O. & Evidente, A. Studies on structure–activity relationship of sphaeropsidins A-F, phytotoxins produced by Sphaeropsis sapinea f. sp. cupressi. Phytochem. 65, 189–198. https://doi.org/10.1016/j.phytochem.2003.11.006 (2004).
Evidente, A. et al. In vitro antibacterial activity of sphaeropsidins and chemical derivatives toward Xanthomonas oryzae pv. oryzae, the causal agent of rice bacterial blight. J. Nat. Prod. 74, 2520–2525. https://doi.org/10.1021/np200625m (2011).
Wang, X.-N. et al. Smardaesidins A-G, Isopimarane and 20-nor-Isopimarane Diterpenoids from Smardaea sp., a Fungal Endophyte of the moss Ceratodon purpureus. J. Nat. Prod. 74, 2052–2061. https://doi.org/10.1021/np2000864 (2011).
Masi, M. & Evidente, A. Sphaeropsidin A: A pimarane diterpene with interesting biological activities and promising practical applications. Chem. Bio. Chem. 22, 3263–3269. https://doi.org/10.1002/cbic.202100283 (2021).
Raška, I., Koberna, K., Malinsky, J., Fidlerova, H. & Masata, M. The nucleolus and transcription of ribosomal genes. Biol. Cell 96, 579–594. https://doi.org/10.1016/j.biolcel.2004.04.015 (2004).
Montanaro, L., Govoni, M., Orrico, C., Treré, D. & Derenzini, M. Location of rRNA transcription to the nucleolar components, disappearance of the fibrillar centers in nucleoli of regenerating rat hepatocytes. Cell Struct. Funct. 36, 49–56. https://doi.org/10.1247/csf.10017 (2011).
Peculis, B. A. Ribosome biogenesis, ribosomal RNA synthesis as a package deal. Curr. Biol. 12, R623–R624. https://doi.org/10.1016/S0960-9822(02)01135-1 (2002).
Nunes, A. T. & Annunziata, C. M. Proteasome inhibitors, structure and function. Semin. Oncol. 44, 377–380. https://doi.org/10.1053/j.seminoncol.2018.01.004 (2017).
Chen, S. F. et al. Apoptotic effect of MG-132 on human tongue squamous cell carcinoma. Biomed. Pharmacother. 65, 322–327. https://doi.org/10.1016/j.biopha.2011.04.009 (2011).
Chen, H. Y. et al. Upregulated ROS production induced by the proteasome inhibitor MG-132 on XBP1 gene expression and cell apoptosis in Tca-8113 cells. Biomed. Pharmacother. 68, 709–713. https://doi.org/10.1016/j.biopha.2014.07.011 (2014).
Wang, L., Ding, P., Zhang, Y., Han, W. & Ma, D. An alternative form of paraptosis-like cell death, triggered by TAJ/TROY and enhanced by PDCD5 overexpression. J. Cell Sci. 117, 1525–1532. https://doi.org/10.1242/jcs.00994 (2004).
Paull, K. D. et al. Display and analysis of patterns of differential activity of drugs against human tumor cell lines, Development of mean graph and COMPARE algorithm. J. Natl. Can. Inst. 81, 1088–1092 (1989).
Cassileth, P. A. & Gale, R. P. Amsacrine, a review. Leukem. Res. 10, 1257–1265. https://doi.org/10.1016/0145-2126(86)90331-0 (1986).
Trump, D. L. et al. Phase I clinical trial and pharmacokinetic devaluation of acodazole (NSC 305884), an imidazoquinoline derivative with electrophysiological effects on the heart. Cancer Res. 47, 3895–3900 (1987).
Ramaswamy, B., Mrozek, E., Kuebler, J. P., Bekaii-Saab, T. & Kraut, E. H. Phase II trial of pyrazoloacridine (NSC#366140) in patients with, metastatic breast cancer. Invest. New Drugs 29, 347–351. https://doi.org/10.1007/s10637-009-9338-1 (2011).
Fu, W. et al. A novel acridine derivative, LS-1–10 inhibits autophagic degradation and triggers apoptosis in colon cancer cells. Cell Death Dis. 8, 3086–3099. https://doi.org/10.1038/cddis.2017.498 (2017).
Schwab, A., Fabian, A., Hanley, P. J. & Stock, C. Role of ion channels and transporters in cell migration. Physiol. Rev. 92, 1865–1913. https://doi.org/10.1152/physrev.00018.2011 (2012).
Lastraioli, E., Iorio, J. & Arcangeli, A. Ion channel expression as promising cancer biomarker. Biochim. Biophys. Acta Biomem. 1848, 2685–2702. https://doi.org/10.1016/j.bbamem.2014.12.016 (2015).
Arcangeli, A. & Becchetti, A. Novel perspectives in cancer therapy, targeting ion channels. Drug Resist. Updat. 21, 11–19. https://doi.org/10.1016/j.drup.2015.06.002 (2015).
Fu, S. et al. First-in-human phase I study of SOR-C13, a TRPV6 calcium channel inhibitor, in patients with advanced solid tumors. Invest. New Drugs 35, 324–333. https://doi.org/10.1007/s10637-017-0438-z (2017).
Frankel, A. E. et al. Digoxin plus trametinib therapy achieves disease control in BRAF wild-type metastatic melanoma patients. Neoplasia 19, 255–260. https://doi.org/10.1016/j.neo.2017.01.010 (2017).
Kaushik, V., Yakisich, J. S., Kumar, A., Azad, N. & Lyer, A. K. V. Ionophores, potential use as anticancer drugs and chemosensitizers. Cancers 10, 1–21. https://doi.org/10.3390/cancers10100360 (2018).
Castrop, H. & Schießl, I. M. Physiology and pathophysiology of the renal Na+-K+-2Cl- cotransporter (NKCC2). Am. J. Physiol. Renal. Physiol. 307, 991–1002. https://doi.org/10.1152/ajprenal.00432.2014 (2014).
Singh, R. et al. Plasma membrane targeting of endogenous NKCC2 in COS7 cells bypasses functional golgi cisternae and complex N-glycosylation. Front. Cell Develop. Biol. 4, 1–17. https://doi.org/10.3389/fcell.2016.00150 (2017).
van der Westhuyzen, A. E. et al. Deciphering the chemical instability of sphaeropsidin A under physiological conditions—Degradation studies and structural elucidation of the major metabolite. Org. Biomol. Chem. 18, 8147–8160. https://doi.org/10.1039/D0OB01586E (2020).
Aki, T., Nara, A. & Uemura, K. Cytoplasmic vacuolization during exposure to drugs and other substances. Cell Biol. Toxicol. 28, 125–131. https://doi.org/10.1007/s10565-012-9212-3 (2012).
Shubin, A.V., Demidyuk, I.V., Komissaarov, A.A., Rafieva, L.M. & Kostrov, S.V. (2016) Cytoplasmic vacuolization in cell death and survival. Oncotarget. 7, 55863–55889. https://doi.org/10.18632/oncotarget.10150.
Peña-Oyarzun, D. et al. (2017) Hyperosmotic stress stimulates autophagy via polycystin-2. Oncotarget 8, 55984–55997. https://doi.org/10.18632/oncotarget.18995
Tanaka, K. & Matsuda, N. Proteostasis and neurodegeneration, the roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta Mol. Cell Res. 1843, 197204. https://doi.org/10.1016/j.bbamcr.2013.03.012 (2014).
Kaur, J. & Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 16, 461–472. https://doi.org/10.1038/nrm4024 (2015).
Singh, R., Almutairi, M. M., Pacheco-Andrade, R., Almiahuob, M. Y. M. & Di Fulvio, M. Impact of hybrid and complex N-glycans on cell surface targeting of the endogenous chloride cotransporter Slc12a2. Int. J. Cell Biol. 505294, 1–20. https://doi.org/10.1155/2015/505294 (2015).
Durand, E. et al. The nonlinear effect of alkyl chain length in the membrane interactions of phenolipids, Evidence by X-ray diffraction analysis. Eur. J. Lipid. Sci. Technol. 119, 1–7. https://doi.org/10.1002/ejlt.201600397 (2017).
Teuscher, K. B., Zhang, M. & Ji, H. A versatile method to determine the cellular bioavailability of small-molecule inhibitors. J. Med. Chem. 60, 157–169. https://doi.org/10.1021/acs.jmedchem.6b00923 (2017).
Zheng, Z., Zhang, C. & Zhang, K. Role of unfolded protein response in lipogenesis. World J. Hepatol. 2, 203–207. https://doi.org/10.4254/wjh.v2.i6.203 (2010).
Bravo, R. et al. Endoplasmic reticulum and the unfolded protein response, dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 301, 210–290. https://doi.org/10.1016/B978-0-12-407704-1.00005-1 (2013).
Derenzini, M., Montanaro, L. & Trerè, D. Ribosome biogenesis and cancer. Acta Histochem. 119, 190–197. https://doi.org/10.1016/j.acthis.2017.01.009 (2017).
Lefranc, F. et al. In vitro pharmacological and toxicological effects of norterpene peroxides isolated from the Red Sea sponge Diacarnus erythraeanus on normal and cancer cells. J. Nat. Prod. 76, 1541–1547. https://doi.org/10.1021/np400107t (2013).
Yoon, M. J., Kim, E. H., Lim, J. H., Kwon, T. K. & Choi, K. S. Superoxide anion and proteasomal dysfunction contribute to curcumin-induced paraptosis of malignant breast cancer cells. Free Rad. Biol. Med. 48, 713–726. https://doi.org/10.1016/j.freeradbiomed.2009.12.016 (2010).
Yoon, M. J. et al. Stronger proteasomal inhibition and higher CHOP induction are responsible for more effective induction of paraptosis by dimethoxycurcumin than curcumin. Cell Death Dis. 5, 1–14. https://doi.org/10.1038/cddis.2014.85 (2014).
Kang, B. R., Yang, S.-H., Chung, B.-R., Kim, W. & Kim, Y. S. Cell surface GRP78 as a biomarker and target for suppressing glioma cells. Sci. Rep. 6(3), 4922. https://doi.org/10.1038/srep34922 (2016).
Xu, W. & Neckers, L. Gr(i)p the ER to stress out melanoma. Cancer Cell 29, 769–771. https://doi.org/10.1016/j.ccell.2016.05.006 (2016).
Safronov, A. V. et al. Direct observation of bis(dicarbollyl)nickel conformers in solution by fluorescence spectroscopy, an approach to redox-controlled metallacarborane molecular motors. Inorg. Chem. 53, 10045–10053. https://doi.org/10.1021/ic402372c (2014).
Busch, B. B., Staiger, C. L., Stoddard, J. M. & Shea, K. J. Living polymerization of sulfur ylides. synthesis of terminally functionalized and telechelic polymethylene. Macromolecules 35, 8330–8337. https://doi.org/10.1021/ma020729n (2002).
Wu, N.-Y., Xu, X.-H. & Qing, F.-L. Copper-catalyzed regioselective borylfluoromethylation of alkenes. ACS Catal. 9, 5726–5731. https://doi.org/10.1021/acscatal.9b01530 (2019).
Jungwirth, U. et al. Anticancer activity of methyl-substituted oxaliplatin analogs. Mol. Pharmacol. 81, 719–728. https://doi.org/10.1124/mol.111.077321 (2012).
Dinhof, C. et al. p53 loss mediates hypersesnsitivity to ETS transcription factor inhibition based on PARylation-mediated cell-death induction. Cancers 12, 3205. https://doi.org/10.3390/cancers12113205 (2020).
Mathieu, V. et al. Amaryllidaceae alkaloids decrease the proliferation, invasion, and secretion of clinically relevant cytokines by cultured human colon cancer cells. Biomolecules. 12, 1267. https://doi.org/10.3390/biom12091267 (2022).
Baechler, S. A., Schroeter, A., Walker, J., Aichinger, G. & Marko, D. Oxidative metabolism enhances the cytotoxic and genotoxic properties of the soy isoflavone daidzein. Mol. Nutr. Food Res. 58, 1269–1281. https://doi.org/10.1002/mnfr.201300531 (2014).
Aichinger, G. et al. The aza-analogous benzo[c]phenanthridine P8–D6 acts as a dual Topoisomerase I and II poison, thus exhibiting potent genotoxic properties. Molecules 25, 1524. https://doi.org/10.3390/molecules25071524 (2020).
Acknowledgements
A.K. is grateful to the Denise M. Trauth Endowed Presidential Research Professorship. We thank the “Fondation Universitaire” for their contribution to the publication fees.
Funding
This project was supported by the Grants from the National Institutes of Health (GM131717-01) and CA227680-01A1), and the Belgian Tumor Support Group (BBTS; Belgium). The CMMI is supported by the European Regional Development Fund and the Walloon Region. The National Research Foundation (NRF, Pretoria, South Africa) is thanked for support (Grant UID 113322). Stellenbosch University is acknowledged for research support.
Author information
Authors and Affiliations
Contributions
Conceptualization, A.K., V.M.; methodology, A.E., W.A.L.v.O., A.K., V.M.; investigation, A.I., R.S., A.R.H., A.E.v.d.W., S.B.W., J.d.M., L.M., G.A., M.V., D.P-M., V.M.; writing—original draft preparation, A.K., V.M.; writing—review and editing, A.E., W.A.L.v.O., A.K., V.M.; supervision, D.M., W.B., A.E., W.A.L.v.O., A.K., V.M.; project administration, A.E., W.A.L.v.O., A.K., V.M.; funding acquisition, A.E., W.A.L.v.O., A.K., V.M. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
A.K., V.M., W.A.L.v.O, A.E., A.I. and S.B.W. are inventors of the patent application PCT/US23/35648 (date of application 21 October 2023). The novel chemical entities and their mode of action described in the paper are part of the patent application. Remaining Authors have no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ingels, A., Scott, R., Hooper, A.R. et al. New hemisynthetic derivatives of sphaeropsidin phytotoxins triggering severe endoplasmic reticulum swelling in cancer cells. Sci Rep 14, 14674 (2024). https://doi.org/10.1038/s41598-024-65335-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-65335-3
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
