
Abstract
Polygenic risk scores (PRSs) hold promise in their potential translation into clinical settings to improve disease risk prediction. An important consideration in integrating PRSs into clinical settings is to gain an understanding of how to identify which subpopulations of individuals most benefit from PRSs for risk prediction. In this study, using the UK Biobank dataset, we trained logistic regression models to predict the 10 year incident risk of myocardial infarction, breast cancer, and schizophrenia using either just clinical features or clinical features combined with PRSs. For each disease, we identified the top 10% subgroup with the greatest magnitude of improvement in risk prediction accuracy attributed to PRSs in the multi-modal model. Using up to ~ 3.6 k demographic, lifestyle, diagnostic, lab, and physical measurement features from the UK Biobank dataset of ~ 500 k individuals, we characterized these subgroups based on various clinical, lifestyle, and demographic characteristics. The incident cases in the top 10% subgroup for each disease represent distinct phenotypes that differ from other cases and that are strongly correlated with genetic predisposition. Our findings provide insights into disease subtypes and can encourage future studies aimed at classifying these individuals to enhance the targeting of polygenic risk scoring in practice.
Similar content being viewed by others


Introduction
Polygenic risk scores (PRSs) have recently emerged as a tool to use genetic data to predict risk for common diseases, and while they are not yet being routinely used in clinical practice, lately there has been significant interest in implementing these scores in the clinic1,2,3. Translating PRSs into clinical settings will require logistical hurdles to be overcome, such as the need to establish clinical workflows, insurance reimbursement policies, medical provider trainings, and protocols for resource allocation2,4. Thus, a strong justification needs to be in place for the clinical utility of PRSs before they can become a part of routine practice2,5,6,7.
When considered as an independent risk factor, PRSs achieve strong risk discrimination for a growing number of diseases8,22,10. For example, PRSs for heart disease have been shown to identify an ~ 8% swathe of the population at threefold or greater risk of developing the disease compared to the rest of the population, which supports their clinical translation11,12. However, for heart disease prediction, studies that have examined PRSs in the context of non-genetic data that are already routinely available as part of clinical practice have shown minimal improvements in prediction performance attributed to the PRSs, ranging from improvements in area under the curve or C-index (∆AUCPRS or ∆C-indexPRS) of 0.01–0.02 or less13,11,12,13,14,15,16,17,18,19,23.
While there is variability across diseases in the value-add of PRSs for risk prediction over non-genetic data alone (i.e., with the value-add of PRSs for some cancers15 ranging from improvements in C-index of 0.03–0.10), it will be important to identify and characterize subpopulations of individuals for whom calculating PRSs would provide high-yield improvements in risk prediction performance for a given disease. For example, in our study, while the value-add of PRSs for heart disease risk prediction is minimal for the population overall, for the subgroups of individuals that most benefit from PRSs, the improvement in prediction capability is substantial (i.e., ∆AUCPRS of 0.32, Table 1), and characterizing these individuals might enable better targeting of PRSs when implemented in clinical settings.
Few studies to date20,24 have explored this question. Recent studies by Riveros-Mckay et al. and Weale et al., compared the disease prediction value-add of PRSs for heart disease prediction between subgroups of individuals based on age and sex20, and ancestry24, respectively, and found the greatest PRS-attributed prediction improvement in males aged 40–54, with overall consistency across ancestry groups. These studies are limited in that they focused only on differentiating subgroups based on three demographic factors and did not explore more granular phenotypic factors such as lifestyle, physical measurements, clinical lab results, or historical diagnoses in characterizing these subgroups.
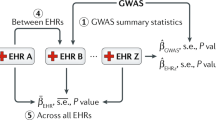
In this study, we expand on previous research through deep phenotyping to characterize the defining clinical, lifestyle, and demographic characteristics of those individuals who most benefit from inclusion of PRSs for risk prediction for three diseases. Using the UK Biobank dataset of ~ 500,000 participants, we systematically trained logistic regression models using clinical features only (non-genetic; NG), and clinical and genetic features together (NG + PRS), respectively, to predict 10 year incident risk of each of the following: first-time myocardial infarction (MI; abbreviated as CAD for coronary artery disease), first-time diagnosis of breast cancer (BC), and first-time diagnosis of schizophrenia (SZ). Then, for each disease, we identify the subgroup comprising the top 10% and bottom 10% of individuals for whom inclusion of PRSs most and least improved their risk prediction accuracy over clinical data alone, respectively (i.e., those with the greatest or least value-add from PRSs). We then characterized each subgroup by leveraging up to 3,648 features comprising data on demographics, lab results, questionnaires, physical measurements, self-reported medications, and historical diagnoses extracted from health records and identifying defining features that distinguish each subgroup. The overview of the approach is detailed in Fig. 1 and Supplementary Fig. S1.

Thresholding approach used to identify patient subgroups. For each random train/validation/test split (ratio of 70/20/10), the best-performing trained NG and NG + PRS models, respectively, were selected and used to obtain the predicted label for each individual in the test set for the disease in question. For each individual, the distance between their true label (0 = control, 1 = incident case) and predicted label (between 0 and 1) was calculated for each of the NG and NG + PRS models. The smaller the distance for each respective model, the better the prediction for a given individual for that model. To evaluate the degree to which adding PRSs improved on or worsened the disease prediction performance for each individual, the difference in distance, \(\Delta\) distance (distanceNG+PRS − distanceNG), was calculated. Any \(\Delta\) distance value greater than 0 indicates an individual-level improvement attributed to adding PRSs to the predictive model, and any value less than 0 indicates that adding PRSs worsened the prediction performance for the individual. The \(\Delta\) distance distribution was then split by percentile, where the top 10% of individuals with the largest \(\Delta\) distance and the bottom 10% with the smallest \(\Delta\) distance were selected for further evaluation. For the initial analysis, this approach was repeated 10× using the test set from each trial and random split of the data. For the subset characterization analysis, this thresholding approach was repeated for the entire population (train, validation, and test sets) after selecting the most “average” pre-trained model out of the 10 trials for each disease (see details in Supplementary Methods).
Results
Disease prediction performance and value-add of polygenic risk scores for different subgroups
We first evaluated the disease prediction value-add of PRSs for the general population prior to splitting the data into subgroups (Table 1, Fig. 2). In support of existing literature, we found that PRSs significantly improved 10 year incident risk prediction performance for first-time myocardial infarction (abbreviated as CAD) and first-time diagnosis of breast cancer (BC). For CAD, there was a minimal but significant ΔAUCPRS of 0.010 ± 0.004 (p-value = 4.05 × 10−3, Cohen’s d = 1.52), but the improvements in F1 score, accuracy, precision, and recall independently were not significant. For BC, risk prediction improved by a greater magnitude, with a ΔAUCPRS of 0.074 ± 0.020 (p-value = 4.92 × 10−9, Cohen’s d = 4.90), and this improvement was significant across all performance metrics assessed. Interestingly, there was no significant improvement in prediction performance for first-time schizophrenia (SZ) despite its high heritability reported in the literature25. Further details on the comparative performance of the single-modality NG and multi-modal NG + PRS models for each disease, including score distributions for cases and controls, and incidence rates and odds ratios at various percentiles of each score, are visualized in Fig. 2a–d and Supplementary Fig. S2.

NG and NG + PRS model performance across diseases. CAD coronary artery disease, BC breast cancer, SZ schizophrenia, NG model with non-genetic feature set, NG + PRS model with combined non-genetic features and polygenic risk scores, AUC area under the curve. (a–c) Plot of first-time incidence rate for each percentile of risk for both NG and NG + PRS risk scores for each disease on the test set across 10 trials. The points in the scatterplot represent the incidence at each percentile for each model at each of the 10 trials (i.e., raw data). The curves visualize the overall trend of the results and were obtained by fitting a fifth order polynomial to the data from each trial and calculating the mean and standard error for each point in the curves. (d) The boxplot illustrates the distribution of the AUCNG and AUCNG+PRS performance metrics for each disease across 10 trials. (e) (Left) Top 10% Subgroup. For the top 10% subgroup there is a substantial improvement in risk discrimination attributed to adding PRSs. For the bottom 30 percentiles of the NG + PRS score, the incidence is 0% and remains < 1% through the 60th percentile. At the top 2 percentiles of the NG + PRS score, the incidence ranges from ~ 47 to ~ 85%, with a higher incidence rate than the NG only model for the top seven percentiles of the score. (Right) Bottom 10% Subgroup. For the bottom 10% subgroup, the NG model achieves strong risk discrimination, with incidence rates ranging from to 0.00% at the lowest percentile up to 23.61% ± 10.92% at the highest percentile of risk, whereas the NG + PRS model achieves worse performance at higher percentiles of the score, such that < 1% of those at the highest percentile of the risk score end up as incident cases.
Once subgroups for each disease were designated using the approach described in Fig. 1 and Supplementary Fig. S1, we calculated the performance of the single-modality NG feature set and the multi-modal NG + PRS feature set for each subgroup, across the ten trials, and then compared the improvements in performance over baseline attributed to the multi-modal feature set. We denote the subgroups as “top 10%” (the 10% of the population that most benefits from PRSs for a given disease) and bottom 10% (the 10% that least benefits from PRSs). As shown in Table 1, there were dramatic improvements in risk prediction performance across all three diseases for the top 10% subgroups. While the ΔAUCPRS for CAD on the entire population was only 0.01, this went up to 0.316 ± 0.042 (p-value = 3.32 × 10−12, Cohen’s d = 7.67) for the top 10% subgroup, with the AUC improving from a low of 0.591 ± 0.053 (NG) up to 0.907 ± 0.024 (NG + PRS). The improvement was more dramatic for BC (ΔAUCPRS = 0.583 ± 0.124; p-value = 4.21 × 10−11, Cohen’s d = 6.59). Even for SZ, the ΔAUCPRS for the top 10% subgroup was 0.270 ± 0.129 (p-value = 2.05 × 10−2, Cohen’s d = 1.197), representing a significant improvement over the nil prediction value-add of PRSs for the overall population. For each disease, the NG + PRS model performance for the bottom 10% subgroup worsened by a similar magnitude compared to the NG model alone. The changes in 10 year incidence at various percentiles for the general population, bottom 10%, and top 10% subgroups for CAD can be further visualized in Fig. 2e. At the top percentile of the NG + PRS score, the incidence rate of first-time myocardial infarction is barely higher than 10% in the general population, however this goes up to nearly ~ 70% on average for the top 10% subgroup. Overall, we found that the subgroups are consistent across trials and effectively stratify individuals based on the degree to which PRSs improve risk prediction performance. Additional details are in Supplementary Fig. S3.
After this initial validation of the subgroups, to obtain a larger sample size for greater power in the subgroup characterization analyses, we selected the most average trial in terms of PRS-attributed prediction performance improvement for each disease, by taking the mean percent of individuals with improved performance attributable to PRSs and then selecting which of the ten trials had a performance improvement closest to the mean value (Supplementary Table S1, Supplementary Fig. S3). We confirmed using a one-way ANOVA that the train, validation, and test sets were not significantly different in the ∆distance distribution values, then combined them into a larger dataset and designated the subgroups from this dataset for the characterization analysis.
Overall trends in characterizing disease subgroups that most benefit from PRSs
As a first step in characterizing each subgroup, the overall counts and percentages of cases and controls were quantified (Supplementary Table S2). While for CAD and SZ, the top 10% subgroups had ~ 1.5× and ~ 2× as high of an incidence rate, respectively, compared to the bottom 10% subgroups (i.e., they were more enriched for incident cases), the incident rates for all subgroups were consistently like those of the overall population (Supplementary Table S3), deviating by less than one percentage point at most. More specifically, the top 10% subgroups had 1,085, 469, and 249 cases, for CAD, BC, and SZ, respectively, and had 30,331, 16,976, and 31,948 controls, respectively.
Given that each subgroup comprised mainly controls and given the likely strong phenotypic differences between cases and controls, we split the analysis of the subgroups by case/control status. We focused primarily on comparing the cases in the top 10% subgroup that most benefitted to those in the remaining 90% of the population (“all others”) with lower value-add from PRSs and then independently repeated the same analysis for the bottom 10% subgroup cases. As a secondary analysis, we repeated the same by comparing the cases in each subgroup, and also explored case/control comparisons within each subgroup. These comparisons are detailed in Supplementary Table S4.
The total number of features explored for each comparison is listed in Supplementary Table S5. The sample sizes and significant features out of the total features assessed for each comparison are in Supplementary Table S6 and are further visualized in the volcano plots in Supplementary Figs. S4 and S5. As expected, due to the lower sample sizes of the true case groups, fewer significant features of larger effect size were identified for the case subgroup comparisons than for the control subgroup comparisons. For the comparison between the top 10% cases and all other cases for CAD, BC, and SZ, we found 17, 8, and 15 distinguishing features, respectively (< 1% of the total features assessed for each disease). This included all four polygenic risk scores for CAD, all three for BC, and one for SZ. Similarly, for the independent comparison between the bottom 10% cases (i.e., those more likely to be considered false negatives when using a multi-modal model that includes PRSs) and all other cases, we found a total of 14, 3, and 13 significant features for CAD, BC, and SZ, respectively, comprising an even smaller percent of total features.
The significant features differentiating the cases in the subgroups that most benefit from PRSs (i.e., top 10%) for each disease are summarized in Tables 2 and 3, including the effect sizes and mean values and population prevalence rates for numerical and binary features, respectively. These results are further visualized in Figs. 3 and 4. In general, across each disease, a trend was that the cases in the subgroup that most benefitted from PRSs also had significantly higher scores for most of the PRSs compared to the rest of the population, as might be expected, except for the schizophrenia PRSs, for which the difference was not significant.

Comparative effect sizes for selected significant features differentiating top 10% or bottom 10% subgroup cases compared to all other cases, respectively, for each disease. CAD coronary artery disease, BC breast cancer, SZ schizophrenia, PRS polygenic risk score, LDL low density lipoprotein, Sibling Hx sibling with a history of heart disease, BMI body mass index, SBP systolic blood pressure, HDL high density lipoprotein, Sibling Hx. of Dep. sibling with a history of depression, Hx history. Note that CAD_6M, CAD_1.7M, CAD_46k, and CAD_202 are polygenic risk scores for heart disease and BC_313, BC_5k, and BC_77 are polygenic risk scores for breast cancer. Note: The x-axis label, Cohen’s value, represents effect size measured by Cohen’s d for numerical features and Cohen’s h for binary features. (a) For CAD, the absolute value effect sizes across all non-genetic significant features for top 10% (Left) and bottom 10% (Right) case comparisons range from ~ 0.15 up to ~ 0.5, representing overall low-to-moderate effect sizes. The top 10% comparison displays the effect sizes for comparisons between the top 10% subgroup cases and all other cases in the population (i.e., the remaining 90% that benefit less from PRSs). The bottom 10% subgroup comparison visualizes the effect sizes for the features that significantly differentiate the bottom 10% subgroup cases from all other cases in the population (i.e., the remaining 90% that benefit more from PRSs, a superset of the top 10% subgroup). (b) For BC, the absolute value effect sizes across all non-genetic significant features are low, ranging from 0.23 to 0.24. The comparisons are shown for the top 10% (Left) and bottom 10% (Right) against the respective rest of the population. (c) For SZ, the absolute value effect sizes across all significant features for cases in the top (Left) and bottom 10% (Right) case comparisons range ~ 0.3 up to ~ 0.8, representing overall moderate-to-high effect sizes.

Comparative prevalence rates and mean values between top 10% or bottom 10% subgroup cases and all other cases, respectively, for selected significant differentiating features for each disease. CAD coronary artery disease (refers to myocardial infarction), BC breast cancer, SZ schizophrenia, BMI body mass index, LDL low density lipoprotein, PRS polygenic risk score. The term, “All Others” represents all other cases in the population that are not in the top 10% subgroup that most benefits from PRSs. (a) For BC three significant features that differentiate the cases in the top 10% subgroup that most benefits from PRSs for risk prediction. One polygenic risk score, BC_5k, is shown to illustrate the difference in distribution of the score between the top 10% cases and all other cases. The top 10% subgroup cases for breast cancer are significantly younger (~ 2 years on average) and slightly leaner, but still overweight on average. (b) For CAD, a selection of numerical features is shown to compare the top 10% subgroup cases that most benefit from PRSs for risk prediction to all other 10-year incident first-time myocardial infarction cases. Apolipoprotein B, LDL, triglycerides, and cholesterol, all of which are biomarkers associated with lipid metabolism, were significantly higher in the top 10% subgroup cases than in other cases, however the mean body mass index was lower, suggesting a slightly leaner phenotype despite signs of poor cardiovascular health. Still, the difference was small (Cohen’s d = − 0.16), with both groups of cases being overweight (BMI > 25 kg/m2) on average. (c) For SZ, some of the binary features with the highest effect size in differentiating the top 10% subgroup cases are shown. Although less 5% of all other 10-year SZ incident cases had a recent marital separation or divorce, this was the case for ~ 17% of the top 10% subgroup cases. Other differentiating factors included having recent financial difficulties (~ 55%) and being male (~ 65%). (d) Some of the most important binary features differentiating the bottom 10% subgroup for SZ are shown.
On the other end of the spectrum, the incident cases in the bottom 10% subgroup (i.e., those that least benefit from PRSs) for each respective disease had lower genetic risk and tended to display opposing characteristics to the cases in the top 10% subgroups, with some important distinctions for SZ and CAD.
Aside from exploring the differentiating characteristics of the true cases that most and least benefit from PRSs for disease prediction, we repeated this comparison for the control groups (i.e., those that do not get a 10 year incident diagnoses of the given disease). For the control comparisons, which were higher powered, we found more significant features differentiating each subgroup. For example, for the comparison between the top 10% subgroup controls for each disease and all other controls, there were 86, 117, and 559 distinguishing features for CAD, BC, and SZ, comprising 2.6%, 3.2%, and 17.2% of the total features, respectively, after the Bonferroni correction for multiple comparisons (α = 0.01). We also compared the cases and controls within each subgroup. The details on our results for these additional analyses are described in Supplementary Tables S4 and S6 and Supplementary Figs. S6–S12.
Heart attack patients that most benefit from PRSs for 10 year incident risk prediction
For CAD, the total features distinguishing the 10 year incident myocardial infarction cases in the top 10% subgroup that most benefit from PRSs from all other true cases were made up of numerical features including physical measurements (systolic blood pressure, hip circumference, body mass index, and waist circumference) and lab results (apolipoprotein B, low density lipoprotein, triglycerides, cholesterol, and direct bilirubin) (Table 2, Figs. 3, 4). The binary features related to smoking status and family history (Table 3, Figs. 3, 4). This group had significantly healthier physical measurements compared to other true incident cases, with lower systolic blood pressure (143.47 ± 18.12 mmHg vs. 148.44 ± 19.35 mmHg for all other cases, Cohen’s d = − 0.27, p-value = 2.09 × 10−16), lower body mass index (albeit nonetheless being overweight on average) (28.07 ± 4.39 kg/m2 vs. 28.80 ± 4.84 kg/m2 for all other cases, Cohen’s d = − 0.16, p-value = 4.87 × 10−7), and lower hip and waist circumference. In contrast, measures of lipid metabolism were significantly worse for the cases in this subgroup, with effect sizes (Cohen’s d) ranging from 0.20 to 0.26 for blood cholesterol, triglycerides, low density lipoprotein, and apolipoprotein B, respectively. This suggests a potentially strong association between genetic risk (as measured by the PRSs) and the lipidemic profile of these individuals within the ~ 10 year window prior to their diagnosis of their first-time heart attack. In contrast, factors such as body mass index or systolic blood pressure do not seem to correlate as strongly with polygenic risk.
The smoking status for the top 10% subgroup was strongly split between current smokers (26% of the top 10% subgroup cases vs. just 16% of all other cases) and never smokers (52% of top 10% subgroup cases vs. 41% of all other cases), with Cohen’s h effect size ranging from 0.20 to 0.23. Meanwhile, past smoking status (i.e., having smoked and then quit) was the most strongly negatively associated with this subgroup (Cohen’s h = − 0.34, p-value = 1.23 × 10−34), suggesting distinct groups of individuals even within this subgroup based on smoking status. It is possible that, for this group, their lack of having been past smokers (one of the non-genetic risk factors used for prediction) ultimately reduced their risk score in the non-genetic risk model only, which was then reversed when PRSs were added to the model. As expected, this group also had a significantly higher sibling history of heart disease than other cases, which correlates well with their genetic predisposition as per the higher polygenic risk scores in this subgroup. Interestingly, while this subgroup comprises more males than females in an approximate 2:1 ratio, there are no significant differences in this ratio between the top 10% group cases and all other cases, so male sex is not a differentiating factor for this subgroup of cases. Details of these characteristics are further visualized in Figs. 3 and 4.
For CAD, as expected, the bottom 10% subgroup incident cases had opposing characteristics to the top 10% subgroup cases in that they had significantly lower genetic risk across all three PRSs, were more likely to have smoked in the past, and were less likely to have a sibling with a history of heart disease. Although males still comprised the majority sex in this subgroup of cases (~ 55%), the ratio of males to females approached 50/50, and thus comprised a significantly greater proportion of females compared to cases in the rest of the population and in the top 10% subgroup specifically.
Breast cancer patients that most benefit from PRSs for 10 year incident risk prediction
For breast cancer, the top 10% subgroup incident cases were differentiated from other cases mostly based on their physical measurements and age. This group of individuals was on average around two years younger than other breast cancer cases (average age ~ 56 vs. ~ 58 for all other breast cancer cases, Cohen’s d = − 0.23, p-value = 9.14 × 10−7) and had slightly lower body mass index (mean of ~ 26.5 kg/m2 vs. ~ 27.7 kg/m2 for all other cases, Cohen’s d = − 0.24, p-value = 1.65 × 10−7), as well as lower hip and waist circumference on average. This group also had significantly lower urate levels, which have been shown to be inversely associated with obesity26. For the true cases in the bottom 10% subgroup that least benefitted from PRSs for breast cancer, the only differentiating features were the three PRSs (BC_313, BC_77, and BC_5k), whereby these individuals had lower polygenic risk for all three scores than the rest of the cases. Although this group had a higher BMI, the effect size was relatively small and did not achieve significance.
Schizophrenia patients that most benefit from PRSs for 10 year incident risk prediction
Finally, for schizophrenia, the key differentiating numerical features for the top 10% subgroup cases that most benefitted from PRSs included testosterone, urate, and age (Table 2). Specifically, top 10% incident cases most likely to be correctly classified in the multi-modal non-genetic risk factors and PRS model than in the non-genetic model alone were significantly younger (average age of 51.78 ± 7.71 years vs. 54.86 ± 8.25 years for all other cases, Cohen’s d = − 0.39, p-value = 1.95 × 10−8), and had higher urate and testosterone levels (Cohen’s d of 0.39 and 0.51, respectively), suggesting that this group is primarily composed of younger males compared to other true 10 year incident schizophrenia cases. This is further supported by the finding that around ~ 2/3 (64%) of the cases in this subgroup consisted of males (Table 3), whereas just ~ 1/3 (35%) of all other cases were male (Cohen’s d = 0.60, p-value = 1.94 × 10−18). In general, one of the most differentiating characteristics of this subgroup was that these individuals had significantly worse financial status, with 56% of these individuals reporting having experienced recent financial difficulties compared to just 19% of all other incident schizophrenia cases (Cohen’s d = 0.79, p-value = 4.95 × 10−35). Related to this, these individuals were significantly more likely to be extremely unhappy with their financial situation. Another distinguishing factor related to their relationships with others, where 11% and 8% of this group reported being moderately unhappy with their friendships and family relationships (Cohen’s d = 0.60 and 0.29, compared to 0.14% and 1.85% for all other cases combined feeling the same way). This group was also more likely than other incident cases to have been a smoker at some point and to have experienced some form of illness, injury, bereavement, or stress in the previous two years before their initial recruitment into the UK Biobank.
The schizophrenia PRSs were not amongst the significant features associated with this subgroup of cases, but this may be due to the low sample sizes and a potentially smaller overall effect size. The SZ_128 and SZ_342 features had no association, but the SZ_1M was higher in this subgroup compared to the other cases (Cohen’s d = 2.66) and could have approached significance (p-value = 1.48 × 10−4) if not for the Bonferroni correction. Overall, our analysis found that those 10 year incident cases who most benefit from inclusion of PRSs for risk prediction comprise a group mostly representative of highly unhappy, stressed younger males with high genetic risk for schizophrenia, lack of relationship satisfaction, and poor financial status. See Figs. 3 and 4 for details.
Cases in the bottom 10% subgroup for schizophrenia were the opposite in some ways to those in the top 10% subgroup (higher levels of happiness and financial or relationship satisfaction), they were significantly more likely to have experienced the death of their spouse or partner in the previous two years (20% of the 125 in this subgroup vs. < 1% in the comparison group; n = 25 vs. n = 9 individuals). Nearly 77% of this subgroup comprised females, compared with ~ 60% on average in the other cases (Cohen’s h = 0.37), but this difference did not achieve significance after correcting for multiple comparisons.
Discussion
In summary, our study showed that the identification and characterization of high-yield subpopulations that most benefit from PRSs is feasible. For CAD, although the improvement in AUC from inclusion of PRSs in the general population was minimal (ΔAUCPRS = 0.01), we identified a 10% swathe of the population for whom inclusion of PRSs in a risk prediction model increased the AUC by 0.32 compared to the counterpart model using non-genetic data alone (AUCNG = 0.59, AUCNG+PRS = 0.91). Similarly, for BC, there was a more substantial improvement in AUC (ΔAUCPRS = 0.58) for the top 10% subgroup compared to the value-add of PRSs in the general population (ΔAUCPRS = 0.07). We were also able to identify a subgroup of individuals for whom PRSs for schizophrenia improved disease prediction performance (AUCNG+PRS = 0.71, ΔAUCPRS = 0.27), despite there being no significant value-add of the PRSs for the general population.
Characterizing the subgroups that derived the most benefit from PRSs (i.e., top 10% subgroups) revealed distinct sub-phenotypes associated with each disease, but these differed based on case/control status. The cases in the top 10% subgroup who most benefitted from PRSs, unsurprisingly, had significantly higher genetic risk as per their polygenic risk scores. The top 10% controls, accordingly, had lower genetic risk (i.e., low polygenic risk scores). The opposite was the case for the bottom 10% cases and controls, respectively. Thus, the identified subgroups were strongly correlated with PRS scores, but split by case/control status. Ultimately, our identified subgroups represented sub-phenotypes for each disease associated with high and low polygenic risk, respectively.
For CAD and SZ, we found that the top 10% subgroup cases displayed nuanced characteristics. For CAD, the top 10% were more likely to be current smokers and had higher lower density lipoprotein, triglycerides, cholesterol, and apolipoprotein B, all of which are significant risk factors for myocardial infarction. For SZ, the top 10% subgroup cases had generally worse life satisfaction and greater levels of financial and life stress, than other cases. They were also more likely to be younger and male, which correlates with known epidemiological risk factors. In contrast, the bottom 10% subgroup was generally happier and more satisfied with life, without financial stressors, and the only major risk factor that differentiated this group from other cases was the recent loss of a spouse. This provides insights into the potential distinct characteristics of 10 year incident schizophrenia cases in association with their genetic risk. For example, it seems that those who are not genetically at-risk might be more likely to get the disease in mid-life following a traumatic experience such as the loss of a loved one. Those more genetically at-risk who get diagnosed might have already had mental health concerns or stressors prior to their diagnosis, potentially resulting from interactions between their genetic predisposition and their environment.
Future work is warranted to explore whether it is possible to leverage the significant features identified in this study in a predictive model to classify individuals likely to be in the top 10% that most benefit from PRSs without a priori knowledge of each individual’s future case/control status. While we attempted to train basic classifiers to use the identified features in this study to find those who most benefit from PRSs for each disease, we did not achieve strong performance when the case/control classes were balanced. This suggests the need for potentially more high-powered analyses in future studies, or the use of more advanced algorithms such as neural networks. Additionally, while our study essentially identified characteristics that correlate with genetic risk, future work is needed to expand on this analysis through Mendelian randomization or genetic correlation analyses to gain deeper insights into the extent to which certain factors, such as poor financial status for schizophrenia, or hyperlipidemia independent of body mass index for heart disease, might be caused by genetic factors captured by these polygenic risk scores.
Another limitation of this study is that we limited the population to European ancestry individuals only. Future studies are needed that not only include individuals of other ancestry groups but that also specifically compare value-add of PRSs when stratifying by ancestry or sex, to replicate the approach used by Riveros-Mckay et al.20 and Weale et al.24 for more diseases. Additionally, our study included a limited selection of non-genetic risk factor features for prediction in the disease risk scores in the NG and multi-modal NG + PRS models. While this was intentional because it was designed to represent a selection of risk factors that would reasonably be associated with the disease and present within the typical range of factors used in other studies, it may be beneficial to expand on this study through the use of additional risk factors for prediction, which might impact the calculated value-add and characteristics of those who most benefit from PRSs. Lastly, the PGS000015 PRS score for BC was generated using a subset of the UK Biobank dataset11, and we do not have access to the unique IDs of UK Biobank participants used to develop this score, so it was not possible to specifically exclude these from our analysis, resulting in potentially inflated performance of the score. This could, in theory, result in those participants that happen to have been used to develop the score having a greater value-add from this PRS, which in turn would influence the characterization of the top 10% group in our study. The magnitude of this effect of this on the characterization subgroups in our study depends on several factors, including: (1) the degree of difference in score performance between train and test sets in the original study (i.e., the train set performance being substantially better than the test set), and (2) the degree of bias in the sample used for the train set in the original study (i.e., a random sample selected for the train set, which was the case in the study, would be better than a train set sample selected non-randomly on specific characteristics). While it is not possible to measure those factors directly without raw data from the original study, the latter issue, which would impact our subgroup characterization analysis, is highly unlikely, as the train/test split is typically done randomly. The former issue alone, if significant, would not conceivably have a major impact on the characterization of our subgroups, assuming the latter factor (i.e., biased train/test split in the original study) is not an issue. Thus, overall, this may reduce the reliability of the study, but we suspect it will not have a major effect on the results for BC.
In conclusion, our study advances our understanding of the subgroups that most and least benefit from inclusion of PRSs in risk prediction models. We found subtypes of cases for each disease with distinct characteristics that can serve as insights for future work aimed at developing better strategies towards targeting the translation of polygenic risk scores in clinical settings.
Methods
Dataset and study population
We utilized the UK Biobank dataset27 containing questionnaire results, physical measurements, diagnoses, and other clinical information for ~ 500,000 individuals. Data in the UK Biobank were collected during assessment visits in which a patient would attend a research site in person, and through online questionnaires or continuously updated national databases (i.e., death or cancer registry, or health records). All participants attended their first assessment visit, which occurred between 2006 and 2010, and all non-genetic features were data collected during each participant’s first assessment visit. Participants with no medical history prior to their first assessment visit, with no available genotype data, or who opted to be removed from the UK Biobank were excluded from the analysis. To avoid potential ancestry-related confounders when analyzing the PRSs, our analysis centered on self-reported White British individuals only.
Cohort definitions
For each disease, individuals were classified based on whether they had a given ICD-10 billing code in their medical record. Five diagnostic datasets available in the UK Biobank were used to identify cases: primary care records, hospital inpatient records, cancer registry data, death registry data, and self-reported questionnaire data (i.e., patients being asked about their medical history). These were encoded using various systems, including ICD-10, ICD-9, a UK Biobank-designed coding system, and read version 2 and 3 codes (Supplementary Fig. S13). We adapted mappings from each coding system to ICD-10 (Supplementary Table S7) and then identified ICD-10 codes that would reasonably identify individuals as being cases for each of the three diseases of interest. These codes are listed in Supplementary Table S8. Any individual diagnosed with at least one code in the list for a given disease at any point in time in their medical history would be considered as a prevalent case. To establish cohorts for incident disease risk prediction, we used the year of each participant’s first UK Biobank assessment visit as the baseline time point for that participant and excluded all participants with a pre-existing diagnosis prior to their assessment visit. The 10 year incident case and control counts for each disease are listed in Supplementary Table S3. Individuals lost to follow-up in the incident prediction time period, as per data field 191 and the death registry, were excluded from the analyses unless they had an incident diagnosis of the disease. Any individuals diagnosed with the disease after the 10 year window were also excluded from the analysis.
Polygenic risk scores (PRSs)
For each disease, we selected PRSs that have previously been published for disease-specific risk prediction. Each PRS consists of a selection of single-nucleotide variants and weights derived from pre-existing large-scale genome-wide association (GWAS) studies. We used the Plink 2.0 software28 to generate a polygenic score for each individual by calculating a weighted sum of allele dosage by GWAS weight for each variant. This was done using the UK Biobank imputed genotypes dataset of 96 million variants in chromosomes 1–22. For each disease, the PRSs used11,23,29,30,31,32,33 are listed in Supplementary Table S9. The rationale for using more than one PRS for each disease, when available, was to maximize the scope of information available in PRSs and avoid limiting the per-disease analysis to one PRS, thus enabling a more robust analysis34. For example, while the selected PRSs for CAD are correlated (Supplementary Fig. S14), collectively they might capture slightly different independent components of genetic risk. The independent performance of each PRS for each disease is described in Supplementary Fig. S15.
Clinical feature selection
To set up the non-genetic feature set for each disease, we identified available UK Biobank data fields with large-enough sample sizes that corresponded to disease-specific risk factor data in the scientific literature and pre-processed the data for CAD13,14,16,22,35, BC15,36, and SZ37. Supplementary Table S10 shows the selected data fields for each risk factor for each disease.
Model training and selection
Each experiment for each disease consisted of a logistic regression model trained using non-genetic (NG) features only or combined non-genetic and polygenic risk score features (NG + PRS). The dataset for each experiment was randomly split into train/validation/test (70/20/10) groups, and random search-based hyperparameter tuning on learning rate and L1 regularization was done using train set after training for up to 50 epochs. For each of 30 combinations of hyperparameters, the best-performing combination in the validation set was selected as the model used to evaluate performance on the test set. This process was repeated 10× in order to get the mean and t-distribution standard deviation of the performance of each model (“experiment”).
Calculation and analysis of ∆AUCPRS for each disease
For each disease, we ran basic statistical analyses on the NG and NG + PRS model performance. We calculated various performance metrics, including area under the curve (AUC), accuracy, precision, recall, true positive rate (TPR), true negative rate (TNR), false positive rate (FPR), false negative rate (FNR), prevalence at each percentile, and odds ratios at each percentile, across each of the 10 trials for each model. Then we compared these metrics between the NG and NG + PRS models using the unpaired Student’s t-test and Cohen’s d to explore the significance and effect size of adding PRSs for each difference in means across the trials.
Identifying subpopulation groups
To identify subgroups for which the disease prediction value-add of PRSs was greater or less than the remainder of the population, the per-individual predictions from each of the trained NG and the NG + PRS models were analyzed.
We used a “thresholding” approach to identify subgroups. This involved directly identifying groups of participants in the test set based on the degree to which their predicted score more closely matched their actual case classification for the disease in the PRS + NG model compared to the NG model. This process is visualized in Fig. 1. For each of the NG and NG + PRS models, we first calculated the distance between the predicted case/control status value and the true value for each participant, where a distance of zero indicates a perfectly accurate prediction for the individual and a distance of one indicates a completely incorrect prediction. Each participant’s distance for the NG + PRS model was subtracted from their distance for the NG model (∆distance = distanceNG − distanceNG+PRS). If the NG + PRS model improved prediction for a given participant, their distance metric would be smaller than for the NG model, indicating a positive result. The opposite would be true if the NG + PRS model worsened performance for that individual. Once the ∆distance values were calculated for each participant, they were ranked and those participants at the tails of the distributions (i.e., with the greatest and least magnitude of improvement) were selected for evaluation. We selected the participants in the top cumulative 10% (90th to 99th percentiles) and in the bottom cumulative 10% (0th to 10th percentiles) of ∆distance for further evaluation. For each subgroup, we independently calculated the ∆AUCPRS for various performance metrics in comparison to rest of the population. This was repeated and compared for each of the 10 different test sets (corresponding to each trial). More details are in Supplementary Fig. S16.
Characterizing subpopulation groups
Once the subpopulation groups were identified, we then compared the differences between the groups. Using the clinical and lifestyle-related data fields in the UK Biobank, we extracted and processed up to ~ 250 non-genetic features including broad information ranging from basic features such as age, sex, and ancestry to more complex features such as family history for various diseases, physical measurements, psychological stress, blood test results, lifestyle factors, and more. In addition to this, we included features corresponding to each individual’s pre-existing diagnoses prior to their first assessment visit, as well as self-reported medications being taken at the time of their assessment visit, for a total of up to 3648 binary and continuous features for BC and 3257 for CAD and SZ. For the latter two, any features specific to biological males or females were excluded. All disease-specific risk factor features used for the prediction models for all diseases were included as part of this dataset, as well as PRSs for all diseases.
We ran several statistical analyses to identify features that differed significantly between the subgroups. To ensure a large sample size for the analyses, we selected the trained logistic regression model from the most “average” trial out of the 10 in terms of PRS value-add (see Supplementary Table S1) and obtained model predictions from the train, validation, and test sets. Using a student’s t-test, we verified that there was no significant difference in mean ∆distance values between the three sample sets, and then we combined them together into a single large dataset for analysis. Sample sizes are described in Supplementary Table S1.
For each of the continuous features, we used an unpaired Student’s t-test to independently compare between (a) the top 10% subgroup of the ∆distance distribution and all others (i.e., the bottom 90%), and (b) bottom 10% subgroup of the ∆distance distribution and all others (i.e., the top 90% of the distribution). For each binary feature, we used a chi-squared test. For each subgroup-specific analysis, we adjusted for multiple comparisons using the Bonferroni correction (α = 0.01) based on the total number of features evaluated for the disease (Supplementary Table S5). Effect sizes of the differences between groups were calculated using Cohen’s d and Cohen’s h for continuous and binary features, respectively. Given that controls for each disease were overrepresented in the comparison analyses, we repeated the analysis independently for just cases and just controls in each subgroup (Supplementary Table S4). Furthermore, to obtain more granular information about how each subgroup (top 10% vs. bottom 10% of ∆distance) compared in terms of differences between the 10 year incident cases and controls in each subgroup, we independently ran t-test and chi-squared analyses across the available features once for each subgroup (top 10%, bottom 10%, and middle 80%) comparing the cases and controls in the subgroup (Supplementary Table S4), and the entire approach is visualized in Supplementary Fig. S1.
Data availability
The data that support the findings of this study were not collected by the authors but are available from the UK Biobank. There are restrictions to the availability of these data, which were used under license 17984 for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of UK Biobank which limits sharing of the data beyond the current analysis for which it was licensed. Access to the data can be requested via application through the UK Biobank website: https://www.ukbiobank.ac.uk/.
References
Wand, H. et al. Improving reporting standards for polygenic scores in risk prediction studies. Nature 591, 211–219 (2021).
Torkamani, A., Wineinger, N. E. & Topol, E. J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 9, 581 (2018).
Lewis, A. C. F. & Green, R. C. Polygenic risk scores in the clinic: New perspectives needed on familiar ethical issues. Genome Med. 13, 14 (2021).
Hao, L. et al. Development of a clinical polygenic risk score assay and reporting workflow. Nat. Med. 28, 1006–1013 (2022).
Lewis, C. M. & Vassos, E. Polygenic risk scores: From research tools to clinical instruments. Genome Med. 12, 44 (2020).
Kumuthini, J. et al. The clinical utility of polygenic risk scores in genomic medicine practices: A systematic review. Hum. Genet. https://doi.org/10.1007/s00439-022-02452-x (2022).
Adeyemo, A. et al. Responsible use of polygenic risk scores in the clinic: Potential benefits, risks and gaps. Nat. Med. 27, 1876–1884 (2021).
Khan, A. et al. Genome-wide polygenic score to predict chronic kidney disease across ancestries. Nat. Med. 28, 1412–1420 (2022).
Paul, K. C., Schulz, J., Bronstein, J. M., Lill, C. M. & Ritz, B. R. Association of polygenic risk score with cognitive decline and motor progression in Parkinson disease. JAMA Neurol. 75, 360–366 (2018).
Shams, H. et al. Polygenic risk score association with multiple sclerosis susceptibility and phenotype in Europeans. Brain. https://doi.org/10.1093/brain/awac092 (2022).
Khera, A. V. et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018).
Schork, A. J., Schork, M. A. & Schork, N. J. Genetic risks and clinical rewards. Nat. Genet. 50, 1210–1211 (2018).
Mars, N. et al. Polygenic and clinical risk scores and their impact on age at onset and prediction of cardiometabolic diseases and common cancers. Nat. Med. 26, 549–557 (2020).
Elliott, J. et al. Predictive accuracy of a polygenic risk score-enhanced prediction model vs a clinical risk score for coronary artery disease. JAMA 323, 636–645 (2020).
Kachuri, L. et al. Pan-cancer analysis demonstrates that integrating polygenic risk scores with modifiable risk factors improves risk prediction. Nat. Commun. 11, 6084 (2020).
Isgut, M., Sun, J., Quyyumi, A. A. & Gibson, G. Highly elevated polygenic risk scores are better predictors of myocardial infarction risk early in life than later. Genome Med. 13, 13 (2021).
Khan, S. S., Cooper, R. & Greenland, P. Do polygenic risk scores improve patient selection for prevention of coronary artery disease? JAMA 323, 614–615 (2020).
Sun, L. et al. Polygenic risk scores in cardiovascular risk prediction: A cohort study and modelling analyses. PLoS Med. 18, e1003498 (2021).
Abraham, G. et al. Genomic prediction of coronary heart disease. Eur. Heart J. 37, 3267–3278 (2016).
Riveros-Mckay, F. et al. Integrated polygenic tool substantially enhances coronary artery disease prediction. Circ. Genom. Precis. Med. 14, e003304 (2021).
Hindy, G. et al. Genome-wide polygenic score, clinical risk factors, and long-term trajectories of coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 40, 2738–2746 (2020).
Mosley, J. D. et al. Predictive accuracy of a polygenic risk score compared with a clinical risk score for incident coronary heart disease. JAMA 323, 627–635 (2020).
Inouye, M. et al. Genomic risk prediction of coronary artery disease in 480,000 adults: Implications for primary prevention. J. Am. Coll. Cardiol. 72, 1883–1893 (2018).
Weale, M. E. et al. Validation of an integrated risk tool, including polygenic risk score, for atherosclerotic cardiovascular disease in multiple ethnicities and ancestries. Am. J. Cardiol. 148, 157–164 (2021).
Hilker, R. et al. Heritability of schizophrenia and schizophrenia spectrum based on the nationwide Danish twin register. Biol. Psychiatry 83, 492–498 (2018).
Gong, M. et al. Converging relationships of obesity and hyperuricemia with special reference to metabolic disorders and plausible therapeutic implications. Diabetes Metab. Syndr. Obes. 13, 943–962 (2020).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Mavaddat, N. et al. Prediction of breast cancer risk based on profiling with common genetic variants. J. Natl. Cancer Inst. 107, 036 (2015).
Mavaddat, N. et al. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am. J. Hum. Genet. 104, 21–34 (2019).
Ripke, S. et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Trubetskoy, V. et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604, 502–508 (2022).
Zheutlin, A. B. et al. Penetrance and pleiotropy of polygenic risk scores for schizophrenia in 106,160 patients across four health care systems. AJP 176, 846–855 (2019).
Clifton, L., Collister, J. A., Liu, X., Littlejohns, T. J. & Hunter, D. J. Assessing agreement between different polygenic risk scores in the UK Biobank. Sci. Rep. 12, 12812 (2022).
Lloyd-Jones, D. M. et al. Framingham risk score and prediction of lifetime risk for coronary heart disease. Am. J. Cardiol. 94, 20–24 (2004).
Kelsey, J. L., Gammon, M. D. & John, E. M. Reproductive factors and breast cancer. Epidemiol. Rev. 15, 36–47 (1993).
McDonald, C. & Murray, R. M. Early and late environmental risk factors for schizophrenia. Brain Res. Rev. 31, 130–137 (2000).
Author information
Authors and Affiliations
Contributions
M.I., as the primary author, conceptualized the ideas in this work, designed the experimental framework, processed the data, analyzed the results, and wrote the paper. F.G. provided insights on modifications to the experimental methodology. L.G., A.S., and K.C. helped with pre-processing the UK Biobank phenotype, diagnostic, and polygenic risk score data. A.H. and S.D. provided helpful insights on how to structure the writing for the paper. M.D.W. provided support for the completion of the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Isgut, M., Giuste, F., Gloster, L. et al. Identifying and characterizing disease subpopulations that most benefit from polygenic risk scores. Sci Rep 14, 22124 (2024). https://doi.org/10.1038/s41598-024-63705-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-63705-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
