
Abstract
The primary aim of this study was to estimate the incidence of posterior fossa anomalies (PFA) and assess the associated outcomes in King Abdulaziz Medical City (KAMC), Riyadh. All fetuses diagnosed by prenatal ultrasound with PFA from 2017 to 2021 in KAMC were analyzed retrospectively. PFA included Dandy–Walker malformation (DWM), mega cisterna magna (MCM), Blake's pouch cyst (BPC), and isolated vermian hypoplasia (VH). The 65 cases of PFA were 41.5% DWM, 46.2% MCM, 10.8% VH, and 1.5% BPC. The annual incidence rates were 2.48, 2.64, 4.41, 8.75, and 1.71 per 1000 anatomy scans for 2017, 2018, 2019, 2020, and 2021, respectively. Infants with DWM appeared to have a higher proportion of associated central nervous system (CNS) abnormalities (70.4% vs. 39.5%; p-value = 0.014) and seizures than others (45% vs. 17.9%; p-value = 0.041). Ten patients with abnormal genetic testing showed a single gene mutation causing CNS abnormalities, including a pathogenic variant in MPL, C5orf42, ISPD, PDHA1, PNPLA8, JAM3, COL18A1, and a variant of uncertain significance in the PNPLA8 gene. Our result showed that the most common PFA is DWM and MCM. The autosomal recessive pathogenic mutation is the major cause of genetic disease in Saudi patients diagnosed with PFA.
Similar content being viewed by others
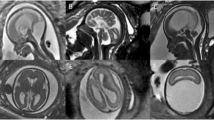


Introduction
Central nervous system (CNS) malformations are the second most common fetal anomalies, following cardiac anomalies. Therefore, prenatal diagnosis is very important because of the high rates of perinatal morbidity and mortality1; besides, it would help with counseling and further follow-up. One of the CNS anomalies is posterior fossa anomalies (PFA), which can be associated with hypotonia, developmental delay, microcephaly, or hydrocephalus2.
Posterior fossa abnormalities include Dandy–Walker malformation (DWM), mega cisterna magna (MCM), Blake’s pouch cyst (BPC), and isolated vermian hypoplasia (VH)3. The spectrum of PFA is found in about 1 in 5000 live births4. The variability of the prognosis and neurodevelopmental outcome depends on different factors, including the type of PFA and whether it is isolated PFA or associated with another CNS or extra CNS abnormalities, also if it is associated with chromosomal abnormalities or not5,6. Associated CNS anomalies include ventriculomegaly, hydrocephalus, and agenesis of the corpus callosum. Extra CNS anomalies may involve the kidney, heart, extremity, facial abnormalities, single umbilical artery, liver, and orthopedic malformations7.
Based on previous studies, PFA is usually associated with other structural anomalies, either CNS or extra-CNS. In general, isolated PFA of any type has better neurodevelopmental outcomes than cases with additional intracranial or extracranial abnormalities. MCM would have better neurodevelopmental outcomes than DWM, especially if isolated8.
In Saudi Arabia, we know little about the fetal and neonatal outcomes of PFA, although there is a high rate of genetic disease and consanguineous marriage. Ohaegbulam et al. found that the incidence of DMW in Tabuk city, Saudi Arabia was 1.24 for males and 0.78 for females, and it was associated with 3.5% of infantile hydrocephalus9.
This study aims to estimate the incidence and identify the different types of posterior fossa abnormalities in the King Abdulaziz Medical City (KAMC) population and assess the outcomes associated with different types of PFA, such as neurologic development, from 2017 to 2021.
Results
Sixty-five posterior fossa anomalies cases were identified in the KAMC—Riyadh, Saudi Arabia, between 2017 and 2021. The annual incidence rates were 2.48, 2.64, 4.41, 8.75, and 1.71 per 1000 anatomy scans for 2017, 2018, 2019, 2020, and 2021, respectively (Fig. 1). DWM and MCM were the most frequent PFA in this study. Between 2017 and 2019, the majority of the PFA were DWM, whereas MCM took over afterward during 2020 and 2021 (Fig. 1).

Frequency and Incidence* of Posterior Fossa Abnormalities in the KAMC—Riyadh, Saudi Arabia between 2017 and 2021. *Incidence was calculated as number of the posterior fossa abnormalities per total number of anatomy scans. DWM Dandy–Walker malformation, MCM mega cisterna magna, BPC Blake's pouch cyst, VH isolated vermian hypoplasia.
Over the 5 years, the cases of PFA were distributed as follows: 41.5% DWM, 46.2% MCM, 10.8% VH, and 1.5% BPC. The mean and standard deviation (SD) of maternal age was 31.6 ± 6.5 years. Approximately 14% of the mothers were primigravid. The prevalence of miscarriage was 38.5% among the mothers in this study. Approximately one-third of mothers in the study reported they were married consanguineously. Further, associated CNS abnormalities and extra CNS abnormalities were found in 52.3% and 41.5%, respectively, of the cases. A full description of the maternal and infant characteristics is included in Table 1.
Table 2 includes the maternal and infant characteristics of the two major posterior fossa anomalies diagnosed in the KAMC. Infants with DWM appear to have a higher proportion of associated CNS abnormalities than those without DWM (70.4% vs. 39.5%; p-value = 0.014). On the other hand, infants with MCM appear to have a significantly lower proportion of associated CNS abnormalities than those without MCM (36.7% vs. 65.7%; p-value = 0.019). Further, infants with DWM appear to have a higher proportion of dysmorphology than those without DWM (60.9% vs. 30%; p-value = 0.025). Further, compared to those without DWM, those with DWM had an abnormal neurological exam (70% vs. 37.9%; p-value = 0.027) and abnormal head and neck examination (63.2% vs. 29.2%; p-value = 0.026). Also, Infants with DWM appear to have a higher proportion of seizures than those without DWM (45% vs. 17.9%; p-value = 0.041). Finally, those with DWM were more likely to undergo neurosurgical intervention than those without DWM (30% vs. 6.9%; p-value = 0.049). On the other hand, those with MCM were not significantly different in the distribution of several maternal and infant characteristics from those without MCM (Table 2).
A diagnosis of Dandy-Walker malformation was found in 27 pregnancies in total; CNS abnormalities were diagnosed in 20 cases, including absent cavum septum pellucidum in six fetuses and bilateral dilated lateral ventricles with absent cavum septum pellucidum in four, two fetuses were diagnosed with agenesis of the corpus callosum and eight with dilated lateral ventricles. Regarding the extra-CNS, a total of 10 pregnancies were identified, seven with cardiac anomalies and three with renal anomalies. Eleven fetuses diagnosed with MCM had associated CNS abnormalities: four with absent cavum septum pellucidum, four dilated lateral ventricles, two with agenesis of corpus callosum, and one with small cerebellum. Extra-CNS abnormalities in MCM were found in 11 cases, four with renal anomalies, including absent single kidney, polycystic kidney, and dilated renal pelvis. Three of MCM had hydrops fetalis. One was diagnosed with Down syndrome, the second one ended with stillbirth, and there was missing data for the third case. Cardiac anomalies were identified in three pregnancies with MCM and echogenic bowel in one case. VH was diagnosed in seven cases; three of them had associated CNS abnormalities, two with abnormal absent cavum septum pellucidum, one with dilated posterior fossa, and one diagnosed postnatally with Joubert syndrome. Only one of the VH cases had extra-CNS abnormalities, including atrial septal defect and dilated large bowel. Black's pouch case had dilated 4th ventricle and echogenic bowel. Dysmorphology was identified in 23 infants; (Table 3) shows the details of this dysmorphology and the associated PFA.
Fetal karyotype was performed in 21 cases, providing five abnormal karyotypes, two fetuses with MCM associated with CNS anomalies and three fetuses with DWM associated with CNS and non-CNS abnormalities. The abnormal karyotyping results were 4 trisomy 18 and 1 trisomy 13.
Genetic testing was done for 17 infants; 12 had abnormal results. Among the 12 patients, two infants had abnormal karyotypes, one had trisomy 18, and the other had a heterozygous likely pathogenic deletion within the cytogenic band 2q.23.1(147940954_148024399). The rest have a single gene mutation causing CNS abnormalities; four patients have pathogenic variants in different genes known to have CNS abnormalities, one patient with a pathogenic variant in the MPL gene, two affected infants with different variants in C5orf42 gene, the fourth patient has a pathogenic variant in ISPD gene (Table 4).
We also identified four patients with likely pathogenic variants in the following genes PDHA1, PNPLA8, JAM3and in COL18A1 genes causing CNS manifestations. Also, one patient has a different variant of uncertain significance in the PNPLA8 gene that suggests its rule for pathogenicity. Finally, we found two variants in two different genes in an infant with ventriculomegaly osteopetrosis (Table 4).
Discussion
Our study’s findings showed a single-tertiary center experience and indicated that DWM and MCM were the most common PFA, consistent with other research3. Incidence rates per 1000 anatomy scans were 2.48, 2.64, 4.41, 8.75, and 1.71 for 2017, 2018, 2019, 2020, and 2021, respectively. DWM is a rare disease with an estimated incidence of 1 per 30,000 births10,11. A common CNS finding in DWM cases is ventriculomegaly3, which might need a ventriculoperitoneal (VP) shunt in severe cases. A previous study found that 62.7% of DWM cases required VP shunts; however, our findings showed fewer patients needed VP shunts, 30%12. The majority of DWM cases in our study have been associated with CNS and extra CNS anomalies. DWM infants, therefore, have a high rate of seizure occurrence. Previous neurosurgical series indicate that the combination of DWM and hydrocephalus is associated with abnormal neurologic development in 40–70% of survivors13,14. Our findings showed a slightly higher percentage, with 78.6% of DWM cases diagnosed postnatally as developmental delay; Hydrocephaly was developed in only six cases. However, hydrocephalus was reported as the most frequent CNS complication in DWM, and Venkatesan et al. results showed there is a high risk of requiring a ventriculoperitoneal shunt and developing epilepsy during early childhood15,16,17. In addition, the results of a previous systematic review demonstrate an increased risk of abnormal neurodevelopmental outcomes in children with a prenatal diagnosis of isolated DWM18.
In general, the reported neurodevelopmental outcome of isolated MCM is favorable18, similar to our study of seven cases of isolated MCM with a normal neurodevelopmental outcome. However, concurrent CNS abnormalities were related to a worse prognosis in children with MCM8; in our study, all cases of MCM associated with CNS abnormalities were diagnosed later as a developmental delay, except one patient was diagnosed with cytomegalovirus (CMV) infection.
The precise diagnosis of isolated BPC is not always simple and is not confirmed prenatally in some cases3,19. Our result showed one case of BPC was diagnosed prenatally but was not confirmed postnatally as the diagnosis was DWM with no ventriculomegaly by postnatal MRI. The existence of extra CNS anomalies was correlated with a worse prognosis compared to the absence of such abnormalities5. CNS-associated abnormalities were 52% in cases of DWM and MCM, which is consistent with a recent study's finding that CNS was the most frequent ultrasound abnormality20. The most frequently observed extra CNS anomalies were renal and congenital heart disease in 27.7% and 23.1% of all PFA cases, respectively, which is similar to the previous study's result20. Our result is consistent with the previous report, which showed the most frequent extra CNS anomalies were infantile polycystic kidneys in DWM infants21. In addition, eight cases with congenital heart disease were found to have developmental delays. According to Seker et al., cardiac anomalies play a crucial impact in infant prognosis22. There have been reports of concomitant dysmorphology similar to our study's findings, including deformities of the face and a cleft palate and lip20,23,24.
Genetic and chromosomal abnormalities are important factors in DMW etiology25. Although a normal karyotype can coexist with DWM, the 3rd, 9th, 13th, and 18th chromosomes are most frequently linked to concurrent chromosomal abnormalities26. In our study, genetic testing was performed on 17 patients, of which 12 had abnormal results. Total (fetal and postnatal) karyotype was performed in 23 cases, of which six had abnormal results. We have found 12 infants with abnormal results, two with abnormal karyotypes, and the remaining ten with gene mutations. Among the patients with abnormal karyotypes, we found four cases with trisomy 18 and one with trisomy 13, known in previous studies to be associated with posterior fossa anomalies25. The other case has microdeletion in the cytogenetic band 2q23.1, which was not known to be associated with posterior fossa abnormalities27. From this study, we cannot conclude if the PFA are a coincidence or an expanding phenotype for this microdeletion. Further study is needed to clarify this association. The remaining ten infants have a single gene mutation, including the MPL gene, which causes rare inherited bone marrow failure syndrome, which was reported in one case report to be associated with hypoplastic cerebellar vermis with a communication between the fourth ventricle and the cisterna magna28. Two affected infants with different variants in the C5orf42 gene caused Joubert syndrome type 17, which was not specifically associated with posterior fossa anomalies. However, Joubert syndrome was reported in some cases with PFA, including DWM29. The fourth patient has a pathogenic variant in the ISPD gene causing Muscular dystrophy-dystroglycanopathy, which is known to be associated with DWM30. We also identified four patients with likely pathogenic variants, including PDHA1 causing Pyruvate dehydrogenase E1-alpha deficiency, JAM3 causing hemorrhagic destruction of the brain, subependymal calcification, and congenital cataracts (HDBSCC) with the neonatal onset and COL18A1 gene causing Knobloch syndrome, type 1. All genes are known to be associated with CNS manifestations; however, none of them is reported to be associated with PFA31,32,33. In addition, two patients were found to have a PNPLA8 gene mutation, which causes mitochondrial myopathy with lactic acidosis, and it was not reported previously with PFA34. Finally, we have identified one infant with two variants in two different genes; one gene is MTMR2, causing Charcot-Marie-Tooth disease, type 4B1, and the other is aTCIRG1, causing osteopetrosis, autosomal recessive 135,36. The latter has been reported in one case of severe autosomal recessive osteopetrosis associated with Dandy–Walker syndrome and agenesis of the corpus callosum37.
Non-genetic contributor to PFA is infection38. According to certain reports, PFA may be related to CMV infection39,40. Intracranial calcification is the most frequent abnormality observed in infants with congenital CMV infection on neuroradiologic imaging, in addition to other intracranial abnormalities that include ventriculomegaly, white matter abnormalities, neuronal migration abnormalities, and extensive destructive encephalopathy40. We reported one case of congenital CMV in which prenatal ultrasound showed mega cisterna magna associated with right ventriculomegaly.
The consanguineous marriage rate is 51% in Saudi Arabia41. According to our findings, one-third of the parents of fetuses with PFA were consanguineous, and 61% of these fetuses have abnormal genetic testing. Our results show that half (7 out of 13) of the genetic testing is an autosomal recessive disease which is supported by a previous study that demonstrates autosomal recessive pathogenic mutation is the major cause of genetic disease in Saudi patients42. In addition, previous data showed a high proportion of DWM in consanguineous marriages, reaching 44%21.
To our knowledge, this is the only study of fetal posterior fossa anomalies in Saudi Arabia. Although it is limited because it is retrospective, with missing data and a small sample size, it provides information on posterior fossa anomalies in our population and the association of genetic disease in consanguineous marriage. MRI criteria for diagnosing DWM were classic since it is a retrospective study from 2017 to 2021, and this is one of the limitations. The criteria that most accurately characterize the modern DWM phenotype are an enlarged tegmentovermian angle, an obtuse fastigial recess, an unpaired caudal lobule (tail sign), inferior, predominant VH, and inferolateral displacement of the tela choroidea/choroid plexus43.
Conclusion
The most common PFA in the KAMC population is DWM and MCM. Developmental delay is more likely when there are associated CNS anomalies. The autosomal recessive pathogenic mutation is the major cause of genetic disease in Saudi patients diagnosed with PFA, and the possibility of identifying new phenotypic associations is high. Therefore, preconception and genetic counseling programs should be implemented for the consanguineous parents.
Methods
This was a retrospective, single-center cohort study conducted between January 2017 and December 2021 in King Abdulaziz Medical City (KAMC), Riyadh, Saudi Arabia. KAMC is one of the largest tertiary centers in the region, with 1973 beds. The Department of Obstetrics and Gynecology performs about 8000 deliveries each year, with mother and fetal outcomes similar to international standards44. Initially, cases were identified from the perinatal review meeting list held weekly to discuss the new fetal cases in the clinic. All fetuses diagnosed with PFA were included. The diagnosis was made according to transabdominal ultrasound mid-trimester examination images using the standard axial planes for screening exams, including trans ventricular, trans thalamic, and trans cerebellar planes45, which were reviewed and confirmed by a fetal-medicine consultant. A fetal MRI was ordered when diagnostics certainty was required. The MRI criteria for diagnosing DWM include lack of the lower part of the vermis, anterior rotation, upward displacement, and hypoplasia of the remaining vermis. The fastigium's angle may be fattened or absent. There is a large bossing posterior fossa with torcular elevation, and normal or hypoplastic cerebellar hemispheres are displaced anterolaterally46. The data were obtained from the medical records; all maternal and infant information was collected from BESTcare, the hospital’s electronic health system. The extracted maternal data included maternal age, gravidity, parity, gestational age at diagnosis, gestational age at delivery, family history of genetic disease, fetal death, stillbirth, and consanguineous marriage. Fetal records were reviewed for birth weight, neonatal intensive care unit (NICU) admission, neonatal death, neuroimaging, dysmorphology, extra-CNS organ involvement, seizure, developmental and cognitive outcome, neurosurgery intervention, and genetic testing, including whole genome sequencing WGS, whole exome sequencing WES and chromosomal analysis.
Posterior fossa abnormalities were defined based on a proposed morphological approach, including:
-
1.
Dandy–Walker malformation (DWM) was defined by the classic triad of enlargement of the posterior fossa with an elevated cerebellar tentorium, complete or partial agenesis of the cerebellar vermis, and dilatation of the fourth ventricle.
-
2.
Mega cisterna magna (MCM) was defined as enlargement of the cisterna magna > 10 mm with normal cerebellar vermis.
-
3.
Blake’s pouch cyst (BPC) was defined as the presence of an upwardly displaced normal cerebellar vermis, normal appearance of the fastigium, tentorium, and size of the cisterna magna.
-
4.
Isolated vermian hypoplasia (VH) was defined as a normally formed vermis of smaller size, and the posterior fossa is otherwise of normal size and anatomy47.
Marriage between two blood-related individuals with a common ancestor, like a second cousin or closer relative, is considered consanguineous marriage48. No formal evaluation of neurodevelopment was conducted. However, the outcome was deemed normal if the child had normal routine developmental screenings.
Research ethics approval
The study was approved by the institutional review board of King Abdullah International Medical Research Center (KAIMARC) (IRB/1833/21). The research was conducted in compliance with the relevant regulations and rules. In accordance with the hospital policy, informed consent was obtained from all individuals and/or their legal guardian(s) for all genetic testing.
Data analysis
Data were analyzed using SPSS version 21.0 software (SPSS Inc., Chicago, IL, USA). Descriptive statistics were reported as frequencies (n) and percentages (%) for categorical/nominal variables and mean and standard deviation (SD) for continuous variables. In addition, the annual incidence of posterior fossa anomalies was calculated as the annual number of cases divided by the total number of anatomy scans in the corresponding year.
Since the two major posterior fossa anomalies diagnosed in the KAMC were DWM and MCM, we aimed to assess their relationships and several maternal and infant characteristics. Thus, we used the chi-square test, Fisher exact test when required, to explore the relationship between each type of the anomalies and several maternal and infant characteristics measured on a categorical scale. We also used independent samples t-test, Mann–Whitney where applicable, to compare the means of other maternal and infant characteristics measured in interval scale across DWM and MCM anomalies.
Data availability
The data supporting this study's findings are available from the corresponding author upon reasonable request.
References
Milani, H. J. F. et al. Ultrasonographic evaluation of the fetal central nervous system: Review of guidelines. Radiol. Bras. 52, 176–181 (2019).
Niesen, C. E. Malformations of the posterior fossa: Current perspectives. Proc. Semin. Pediatr. Neurol. 9(4), 320–334 (2002).
D’Antonio, F. et al. Systematic review and metaanalysis of isolated posterior fossa malformations on prenatal ultrasound imaging (part 1): Nomenclature, diagnostic accuracy and associated anomalies. Ultrasound Obstet. Gynecol. 47, 690–697 (2016).
Parisi, M. A. & Dobyns, W. B. Human malformations of the midbrain and hindbrain: Review and proposed classification scheme. Mol. Genet. Metab. 80, 36–53 (2003).
Patek, K. J. et al. Posterior fossa anomalies diagnosed with fetal MRI: Associated anomalies and neurodevelopmental outcomes. Prenat. Diagn. 32(1), 75–82 (2012).
Ecker, J. L. et al. The sonographic diagnosis of Dandy–Walker and Dandy–Walker variant: Associated findings and outcomes. Prenat. Diagn. 20(4), 328–332 (2000).
Long, A., Moran, P. & Robson, S. Outcome of fetal cerebral posterior fossa anomalies. Prenat. Diagn. 26(8), 707–710. https://doi.org/10.1002/pd.1485 (2006).
Bolduc, M. E. & Limperopoulos, C. Neurodevelopmental outcomes in children with cerebellar malformations: A systematic review. Dev. Med. Child Neurol. 51(4), 256–267 (2009) (Epub 2009 Feb 3).
Ohaegbulam, S. C. & Afifi, H. Dandy–Walker syndrome: Incidence in a defined population of Tabuk, Saudi Arabia. Neuroepidemiology 20, 150–152 (2001).
Pilu, G. et al. Ultrasound of the fetal central nervous system. Curr. Opin. Obstet. Gynecol. 12, 93–103 (2000).
Altman, N. R., Naidich, T. P. & Braffman, B. H. Posterior fossa malformations. Am. J. Neuroradiol. 13(2), 691 (1992).
D’Antonio, F. et al. Systematic review and meta-analysis of isolated posterior fossa malformations on prenatal imaging (part 2): Neurodevelopmental outcome. Ultrasound. Obstet. Gynecol. 48(1), 28–37 (2016).
Hirsch, J. F. et al. The Dandy–Walker malformation: A review of 40 cases. J. Neurosurg. 61(3), 515–522. https://doi.org/10.3171/jns.1984.61.3.0515 (1984).
Sawaya, R. & McLaurin, R. L. Dandy–Walker syndrome. Clinical analysis of 23 cases. J. Neurosurg. 55(1), 89–98. https://doi.org/10.3171/jns.1981.55.1.0089 (1981).
Ecker, J. L., Shipp, T. D., Bromley, B. & Benacerraf, B. The sonographic diagnosis of Dandy–Walker and Dandy–Walker variant: Associated findings and outcomes. Prenat. Diagn. 20(4), 328–332 (2000).
Di Nora, A. et al. Dandy–Walker malformation and variants: Clinical features and associated anomalies in 28 affected children-a single retrospective study and a review of the literature. Acta Neurol. Belg. 123(3), 903–909. https://doi.org/10.1007/s13760-022-02059-z (2023).
Venkatesan, C. et al. Short- and long-term outcomes of prenatally diagnosed Dandy–Walker malformation, vermian hypoplasia, and blake pouch cyst. J. Child Neurol. 36(12), 1111–1119. https://doi.org/10.1177/08830738211049115 (2021) (Epub 2021 Nov 10).
D’Antonio, F. et al. Systematic review and meta-analysis of isolated posterior fossa malformations on prenatal imaging (part 2): Neurodevelopmental outcome. Ultrasound Obstet. Gynecol. 48(1), 28–37 (2016).
Kau, T. et al. Blakeʼs Pouch Cysts and differential diagnoses in prenatal and postnatal MRI. Clin. Neuroradiol. 30, 435–445 (2020).
Sun, Y., Wang, T., Zhang, N., Zhang, P. & Li, Y. Clinical features and genetic analysis of Dandy–Walker syndrome. BMC Pregnancy Childbirth. 23(1), 40. https://doi.org/10.1186/s12884-023-05367-1 (2023).
Has, R. et al. Dandy–Walker malformation: A review of 78 cases diagnosed by prenatal sonography. Fetal Diagn. Ther. 19(4), 342–347 (2004).
Şeker, E. et al. Long term outcomes of fetal posterior fossa abnormalities diagnosed with fetal MRI. J. Turk. Ger. Gynecol. Assoc. https://doi.org/10.4274/jtgga.galenos.2022.2022-7-13 (2022).
Guibaud, L. et al. Prenatal diagnosis of “isolated” Dandy–Walker malformation: Imaging findings and prenatal counselling. Prenat. Diagn. 32(2), 185–193. https://doi.org/10.1002/pd.3828 (2012).
Aydın, E., Turgal, M., Can, S. & Özyüncü, Ö. Posterior fossa anomalies: Related anomalies and the methods of pregnancy termination. Perinat. J. 24(2), 88–95. https://doi.org/10.2399/prn.160242006 (2016).
Murray, J. C., Johnson, J. A. & Bird, T. D. Dandy–Walker malformation: Etiologic heterogeneity and empiric recurrence risks. Clin. Genet. 28(4), 272–283. https://doi.org/10.1111/j.1399-0004.1985.tb00401.x (1985).
Imataka, G., Yamanouchi, H. & Arisaka, O. Dandy–Walker syndrome and chromosomal abnormalities. Congenit. Anom. (Kyoto). 47(4), 113–118. https://doi.org/10.1111/j.1741-4520.2007.00158.x (2007).
Van Bon, B. W. et al. The 2q23.1 microdeletion syndrome: Clinical and behavioural phenotype. Eur. J. Hum. Genet. 18(2), 163–170. https://doi.org/10.1038/ejhg.2009.152 (2010) (Erratum in: Eur J Hum Genet. 2010 Feb;18(2):170. Moloney, Susan [added]. Erratum in: Eur J Hum Genet. 2010 Oct;18(10):1171. PMID: 19809484; PMCID: PMC2987180).
Ihara, K. et al. Identification of mutations in the c-mpl gene in congenital amegakaryocytic thrombocytopenia. Proc. Natl. Acad. Sci. U. S. A. 96(6), 3132–3136. https://doi.org/10.1073/pnas.96.6.3132 (1999).
Stambolliu, E. et al. The most common comorbidities in Dandy–Walker syndrome patients: A systematic review of case reports. J. Child Neurol. 32(10), 886–902. https://doi.org/10.1177/0883073817712589 (2017) (Epub 2017 Jun 21).
Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: {614631}: {12/14/2020}: https://www.omim.org/entry/616052#references (accessed 20 Jan 2023).
Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: {300502}: {01/13/2021}. https://www.omim.org/entry/312170 (accessed 20 Jan 2023).
Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: {606871}: {02/11/2014}. https://www.omim.org/entry/613730. (accessed 20 Jan 2023).
Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: {120328}: {07/21/2022}. World Wide Web URL:. https://www.omim.org/entry/267750 (accessed 20 Jan 2023).
Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: {612123}: {04/21/2017}. World Wide Web URL. https://www.omim.org/entry/251950 (accessed 20 Jan 2023).
Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: {603557}: {06/30/2009}. World Wide Web URL. https://www.omim.org/entry/601382 (accessed 20 Jan 2023).
Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: {604592}: {07/13/2018}. World Wide Web URL: https://www.omim.org/entry/259700 (accessed 20 Jan 2023).
Ben Hamouda, H. et al. Association of severe autosomal recessive osteopetrosis and Dandy–Walker syndrome with agenesis of the corpus callosum. Acta Orthop. Belg. 67(5), 528–532 (2001).
Bosemani, T. et al. Congenital abnormalities of the posterior fossa. Radiographics 35(1), 200–220. https://doi.org/10.1148/rg.351140038 (2015).
Dogan, Y. et al. Intracranial ultrasound abnormalities and fetal cytomegalovirus infection: Report of 8 cases and review of the literature. Fetal Diagn. Ther. 30(2), 141–149. https://doi.org/10.1159/000330636 (2011) (Epub 2011 Sep 29).
Puvabanditsin, S. et al. Not a Dandy–Walker malformation but congenital cytomegalovirus infection. HK J. Paediatr. 13(1), 56–59 (2008).
Husain, M. A. & Bunyan, M. A. Consanguineous marriages in a Saudi population and the effect of inbreeding on prenatal and postnatal mortality. Ann. Trop. Paediatr. 17(2), 155–160. https://doi.org/10.1080/02724936.1997.11747879 (1997).
Monies, D. et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum. Genet. 136(8), 921–939. https://doi.org/10.1007/s00439-017-1821-8 (2017) (Epub 2017 Jun 9).
Whitehead, M. T. et al. Refining the neuroimaging definition of the Dandy–Walker phenotype. AJNR Am. J. Neuroradiol. 43(10), 1488–1493. https://doi.org/10.3174/ajnr.A7659 (2022) (Epub 2022 Sep 22).
Madkhali, A. M. et al. Framework for obstetrics and gynecology department change management in response to COVID 19 pandemic: A tertiary center experience. Ann. Thorac. Med. 16(1), 57 (2021).
Salomon, L. J. et al. Practice guidelines for performance of the routine mid-trimester fetal ultrasound scan. Ultrasound Obstet. Gynecol. 37(1), 116–126 (2011).
Klein, O., Pierre-Kahn, A., Boddaert, N., Parisot, D. & Brunelle, F. Dandy–Walker malformation: Prenatal diagnosis and prognosis. Childs Nerv. Syst. 19(7–8), 484–489. https://doi.org/10.1007/s00381-003-0782-5 (2003) (Epub 2003 Jul 16).
Tortori-Donati, P. et al. Cystic malformations of the posterior cranial fossa originating from a defect of the posterior membranous area. Mega cisterna magna and persisting Blake’s pouch: Two separate entities. Childs Nerv. Syst. 12(6), 303–308. https://doi.org/10.1007/BF00301017 (1996).
Zakzouk, S., El-Sayed, Y. & Bafaqeeh, S. A. Consanguinity and hereditary hearing impairment among Saudi population. Ann. Saudi Med. 13(5), 447–450 (1993).
Funding
For this study, no specific funding was obtained.
Author information
Authors and Affiliations
Contributions
H.A. had the idea for this article, performed a literature search, and drafted and finalized the manuscript. S.A. analyzed the data and drafted and finalized the result section of the manuscript. S.A. and F.A. drafted the genetic paragraph of the manuscript. S.A., N.A. and N.A. collected the data. D.B. performed a literature review and critically revised the manuscript. All authors discussed the results and contributed to the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alsehli, H., Alshahrani, S.M., Alzahrani, S. et al. Fetal and neonatal outcomes of posterior fossa anomalies: a retrospective cohort study. Sci Rep 14, 8411 (2024). https://doi.org/10.1038/s41598-024-59163-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-59163-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
