
Abstract
The nasal microbiota is a key contributor to animal health, and characterizing the nasal microbiota composition is an important step towards elucidating the role of its different members. Efforts to characterize the nasal microbiota composition of domestic pigs and other farm animals frequently report the presence of bacteria that are typically found in the gut, including many anaerobes from the Bacteroidales and Clostridiales orders. However, the in vivo role of these gut-microbiota associated taxa is currently unclear. Here, we tackled this issue by examining the prevalence, origin, and activity of these taxa in the nasal microbiota of piglets. First, analysis of the nasal microbiota of farm piglets sampled in this study, as well as various publicly available data sets, revealed that gut-microbiota associated taxa indeed constitute a substantial fraction of the pig nasal microbiota that is highly variable across individual animals. Second, comparison of herd-matched nasal and rectal samples at amplicon sequencing variant (ASV) level showed that these taxa are largely shared in the nasal and rectal microbiota, suggesting a common origin driven presumably by the transfer of fecal matter. Third, surgical sampling of the inner nasal tract showed that gut-microbiota associated taxa are found throughout the nasal cavity, indicating that these taxa do not stem from contaminations introduced during sampling with conventional nasal swabs. Finally, analysis of cDNA from the 16S rRNA gene in these nasal samples indicated that gut-microbiota associated taxa are indeed active in the pig nasal cavity. This study shows that gut-microbiota associated taxa are not only present, but also active, in the nasal cavity of domestic pigs, and paves the way for future efforts to elucidate the function of these taxa within the nasal microbiota.
Similar content being viewed by others

Introduction
The network of microorganisms inhabiting the bodies of animals is known as the microbiota1,2,3. The microbiota has been shown to play a pivotal role in various aspects of host health, for example by providing critical support in immune system maturation, nutrient utilization2,3, and defense against pathogen invasion3,4. In the case of respiratory pathogens, one of the first lines of defense is the nasal microbiota5, and many recent studies have focused on characterizing the commensal nasal microbiota and its relationship with respiratory pathogens in humans5,6 and various animal species7,8,9,10,11. These studies have identified a variety of taxa that are frequently found in the nasal microbiota of different host species, including members from different genera such as Moraxella, Lactobacillus, Streptococcus, Haemophilus/Glaesserella, and Staphylococcus6,7,8,9,10,11.
Surprisingly, these studies also frequently detected microorganisms in the nasal cavity that are normally associated with the gut microbiota, in particular many anaerobic bacteria from the Bacteroidales and Clostridiales orders. These gut-microbiota associated taxa can be found in human nasal microbiota, where anaerobic Gram-negative bacteria such as Prevotella and Veillonella are frequently detected6, but are particularly prevalent in the nasal microbiota of pigs7. Considering that the respiratory tract is unlikely to have anaerobic niches12, there is ongoing discussion about the in vivo role of these gut-microbiota associated taxa in the nasal microbiota. Recent in vivo studies in piglets have shown that these taxa are differently abundant under different health-status scenarios11,13,14,15,16,17 and are variable through age stages12,18,19, pointing towards a functional role. In contrast, other studies have attributed the presence of these gut-microbiota associated taxa to contamination from fecal material and/or soil12,20, which could be explained by the rooting behavior of pigs12,21,22.
In this study, we investigate the presence of these gut-microbiota associated anaerobic taxa in the nasal microbiota of piglets. Specifically, we focus on three questions: (1) how prevalent are these gut-microbiota associated taxa in the nasal microbiota of domestic pigs? (2) What is their source (i.e. do these microorganisms truly originate from the gut)? and (3) are these taxa active in the aerobic nasal environment? We tackle these questions using a combination of 16SrRNA amplicon sequencing of DNA (total communities) and cDNA retrotranscribed from RNA (active communities) in matched in vivo samples obtained from individual animals. We confirm that gut-microbiota associated taxa indeed represent a substantial fraction of the pig nasal microbiota across a wide range of samples from this study and literature. Comparison of Amplicon Sequencing Variants (ASVs) in matched rectal/nasal samples suggests a shared pool of these taxa in both body sites, pointing to a common source. Moreover, surgical sampling of the inner nasal tract indicates that these gut-microbiota associated taxa are not introduced during sampling but are truly located in the pig nose. Finally, comparison of total and active microbial communities suggests that these taxa are active throughout the nasal cavity of pigs. Overall, this work sheds light on the role of gut-microbiota associated taxa in the nasal microbiota of pigs and supports the notion that these taxa are not only present, but also active.
Results
Characterizing the gut-microbiota associated fraction in the pig nasal microbiota
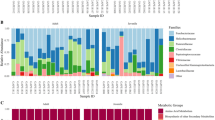
As the starting point of this study, we aimed to characterize the fraction of gut-microbiota associated taxa found in the nasal microbial communities of piglets. Towards this end, we sampled the nasal cavity and rectum of 24 healthy animals from three different commercial farms located in Spain without a history of respiratory disease outbreaks and used 16S rRNA gene sequencing to determine the microbiota composition (Fig. 1A). We obtained a total number of 9,103 different ASVs (mean of 132,051.44 read counts per sample) after processing the raw reads. To determine the fraction of gut-microbiota associated microorganisms in these samples, we focused on two orders (Bacteroidales/Clostridiales) which constitute the most abundant taxa in the gut microbiota and defined these as “gut-microbiota associated taxa”. We found that Bacteroidales and Clostridiales represented a substantial fraction of the pig nasal microbiota in most animals, accounting for 9.43 ± 3.8% and 20 ± 7.7% of the total composition, respectively (mean ± SD across samples) (Fig. 1B, left). As expected, this fraction was higher in the respective rectal swabs (Fig. 1B, right). We also determined this gut-microbiota associated fraction in the pig nasal microbiota in an alternative way by identifying taxa that are prevalent in a reference data set of gut microbiota (obtained from about 300 animals23) and obtained very similar results (supplementary Text 1 and supplementary Fig. 1). Consistent with previous reports7,11,12, in the nasal samples, Prevotella and Bacteroides together with Ruminococcaceae and Lachnospiraceae, were the most prevalent taxa within Bacteroidales and Clostridiales, respectively. Reassuringly, other reported nasal colonizers such as Moraxella, Acinetobacter, and Enhydrobacter genera from Pseudomonadales, Lactobacillus and Streptococcus genera from Lactobacillales and Glaesserella from Pasteurellales were also highly abundant in the nasal samples (supplementary Fig. 2).

Detection of gut-microbiota associated taxa in nasal and rectal swabs from healthy 2–3 week-old piglets from three farms. (A) Schematic of microbiota sampling and sequencing approaches (created with BioRender.com). (B) Summed relative abundance of Bacteroidales (blue) and Clostridiales (green) taxa in nasal (left) and rectal (right) swabs of 24 individual animals. Summed abundance of other taxa (order level) with > 1% mean relative abundance across samples are shown as “Other” (pink). Orders with < 1% relative abundance are summed in the category “Low abundant”. (C) Relative abundances in nasal and rectal samples (mean across 24 individuals) of genera in nasal microbiota with > 0.5% mean relative abundance across animals. Square brackets in taxonomical assignations indicate contested names in the reference Greengenes database used (13.8).
To assess whether this substantial fraction of gut-microbiota associated taxa is a unique feature of these particular samples, we quantified the fraction of Bacteroidales and Clostridiales in 11 publicly available data sets of pig nasal microbiota samples (11,15,20,24,25,26,27,28,29,30, see supplementary Table 1). We found that across different countries of origin, pig ages, and sequenced regions, Bacteroidales and Clostridiales represent a substantial (albeit variable between individual animals) fraction of the nasal microbiota (supplementary Fig. 3, supplementary Table 2). Taken together, these results confirm that the pig nasal microbiota has a substantial fraction of commonly gut-microbiota associated taxa.
Identifying the source of gut-microbiota associated taxa in the pig nasal microbiota
The most probable source of gut-microbiota associated taxa in the pig is the gut microbiota itself, and in support of this hypothesis we found that the most abundant genera belonging to either Bacteroidales or Clostridiales in rectal samples also tended to be highly abundant in matched nasal samples (Fig. 1C). On the other hand, most genera previously reported as nasal colonizers exhibited low abundances in rectal samples (Fig. 1C). However, it is conceivable that although the gut-microbiota associated taxa in these two body sites belong to the same genera, pig nose and gut may nevertheless be inhabited by distinct strains with different niche preferences.
To address this question, we examined the individual ASVs (as a proxy for strain identity31) found in nasal and rectal swabs within each farm. Specifically, for each farm we identified the most abundant Bacteroidales and Clostridiales ASVs in nasal and rectal samples and determined their overlap (the 100 most relatively abundant ASVs in nasal and rectal samples were selected, Fig. 2A, supplementary Table 3). We found that 39%, 14% and 30% of these most abundant ASVs belonging to Bacteroidales, were shared between nose and rectal samples in farms 1, 2 and 3, respectively. In the case of Clostridiales, these proportions were 36%, 9% and 39%. Importantly, we found that the ASVs shared between both body sites were often more highly abundant than the site-specific ones (especially in farms 1 and 3). Notably, only few of these shared ASVs were also found across farms (dark gray circles in Fig. 2A), suggesting that our observations are not simply the result of biases due to high sequence conservation in these taxa. Moreover, we did not observe any biases for certain bacterial families or genera within the body-site specific ASVs. For example, we found ASVs classified as Prevotella, Bacteroides, Veillonella and Oscillospira among the site-specific ASVs in both nasal and rectal samples (supplementary Table 3), and phylogenetic analysis of these most abundant Bacteroidales and Clostridiales sequences, showed little clustering across body sites and farms (supplementary Fig. 4). In contrast to Bacteroidales and Clostridiales, ASVs belonging to families of known nasal colonizers such as Moraxellaceae, Pasteurellaceae and Weeksellaceae showed a much lower degree of overlap between body sites (Fig. 2B). Among the exceptions we found Lactobacillaceae and Streptococcaceae, whose ASVs still exhibited substantial overlap between the two body sites. Thus, the analysis of nasal and rectal microbiota at ASV level suggests a shared origin of the gut-microbiota associated taxa (presumably environmental e.g. through transfer of fecal matter) found in the pig nasal and rectal microbiota.

Nasal and rectal overlap of gut-microbiota associated taxa (A) and nasal colonizers (B) most prevalent ASVs per sampled farm. For gut-microbiota associated taxa the 100 most abundant ASVs in nasal and rectal samples were selected. For other nasal colonizers’ families (B), the top 20 ASVs were considered. Each dot corresponds to one ASV (mean abundance of 8 animals from each herd). Turquoise: ASV is among the most abundant ASVs in nasal but not rectal swabs. Yellow: ASV is among the most abundant ASVs in rectal but not nasal swabs. Pink: ASV is among the most abundant ASVs in both sites. Dark gray: ASV is among the most abundant ASVs in both sites, and also shared across the three sampled farms.
Surgical deep sampling of the pig nasal cavity to identify gut-microbiota associated taxa at different depths
The shared gut-microbiota associated taxa in nasal and rectal swabs described above are consistent with the hypothesis that these taxa enter the pig nasal cavity through the transfer of fecal matter driven by the animals’ rooting behavior. To further validate that these taxa were indeed resident in the pig nasal cavity and did not stem from contaminations introduced by the sampling through the nostrils, we performed additional in-depth sampling (Fig. 3A). Specifically, we surgically opened the nasal cavity of five animals post-mortem to enable sampling of its (normally inaccessible) deep and middle parts (as detailed in the “Methods” section), avoiding a possible swab contamination from the skin surrounding the nostril openings. Additionally, standard nasal swabs, swabs of the external nasal area, and rectal swabs of each animal were taken for comparison. We observed a gradient in microbial load (highest to lowest, as determined by 16S rRNA gene qPCR) from external to deep internal samples, which was consistent with the number of total reads and abundance of contaminant sequences from negative control samples (supplementary Fig. 5). Moreover, the deep nasal microbiota exhibited lower richness compared to samples from external nose (Chao1 index p < 0.05, supplementary Fig. 6A), and was identified as a distinct community (as determined by beta diversity analysis) compared to the external nares (Jaccard and Bray–Curtis PERMANOVA p < 0.05, supplementary Fig. 6A, supplementary Table 4), when deep nasal samples were compared pairwise with standard and external swab samples. Comparison of the composition at different nasal cavity locations within the same animal revealed that those taxa which were found at all sampling sites (of which there were only few in any given animal) tended to constitute the bulk of the observed microbiota (supplementary Fig. 7, see supplementary text 2 for detailed compositional analysis). This dominance of few taxa was found both at the genus level, as well as at the level of individual ASVs, suggesting that the nasal microbiota of individual animals is consistently composed of few dominating strains across the whole nasal cavity. Importantly, nasal swabs as well as matched surgical nasal samples showed a variable, but substantial, fraction of gut-microbiota associated taxa (Fig. 3B and supplementary Fig. 6B), many of which were found consistently throughout the nasal cavity in each animal (Fig. 3C). Thus, these findings suggest that gut-microbiota associated taxa do reside in the pig nasal cavity and do not stem from contaminations introduced by the sampling procedure (i.e. sampling through the nostril using swabs) itself.

Characterization of surgical microbiota samples. (A) Collection of nasal samples at four sites. External and standard nasal sampling; deep and middle nose samples after longitudinal surgical cuts of piglet heads (see “Methods”). (B) Summed relative abundance of Bacteroidales (blue) and Clostridiales (green) taxa in the different types of nasal swabs of the 5 individual animals. Summed abundance of orders with > 1% mean relative abundance across samples are shown as “Other” (pink). Taxa with < 1% relative abundance are summed in the category “Low abundant”. (C) Most prevalent genera across the nose of the 5 sampled piglets. Genera are ordered from top to bottom by prevalence (present in most samples) and global relative abundance. Gut-microbiota associated genera are labelled in blue (Bacteroidales) and green (Clostridiales). Numbered column on the right: mean relative abundance across all samples. Sample sites are marked as E(xternal), S(tandard), M(iddle), D(eep).
Assessing the activity of gut-microbiota associated taxa by quantifying the 16S rRNA transcripts
The results described above suggested that gut-microbiota associated taxa are present throughout the whole pig nasal cavity. Next, we wanted to determine whether these taxa (which include many obligate anaerobic species) are also active in the aerobic environment of the pig nose. Towards this end, we quantified 16S rRNA gene transcripts (a proxy for protein synthesis potential and thus indirect measure of cellular activity,32,33,34) in the nasal microbiota samples described above. We found that gut-microbiota associated taxa constituted a similar portion in these RNA derived samples as in the respective DNA samples described above. Specifically, in nasal samples taken from 24 animals across 3 farms, Bacteroidales and Clostridiales orders accounted for a mean 7.1% ± 6.2 and 22.4% ± 14.1, respectively (Fig. 4A and Supplementary Fig. 8A). In the surgical samples taken at different sites in the nasal cavity, Bacteroidales accounted for 10.3% ± 8.8 (Deep), 8.2% ± 10.4 (Middle), 1.6% ± 3.2 (Standard) and 0.1% ± 0.1 (External); and Clostridiales for 3.6% ± 2.8 (Deep), 3.9% ± 3.6 (Middle), 3.2% ± 5.6 (Standard) and 2.9% ± 4.6 (External) (Fig. 4A and Supplementary Fig. 8B). Moreover, we found that gut-microbiota associated taxa had similar RNA to DNA ratios as the ratios for reported nasal colonizers when examined globally (summing across different taxa) in individual animals (Fig. 4B), or when examining different genera (Fig. 4C) and families (supplementary Fig. 9). To identify individual families belonging to gut-microbiota associated taxa that deviate from this general trend, we finally compared their RNA/DNA ratio distributions with those of nasal colonizers in the farm animal samples (supplementary Fig. 10). With some exceptions (e.g. lower RNA/DNA ratios for Bacteroidales (unclass.) and higher ratios for Clostridiaceae), RNA/DNA ratio distributions were largely not significantly different for most gut-microbiota associated taxa compared to reported nasal colonizers. Taken together, these results suggest that gut-microbiota associated taxa are not only present in the pig nasal environment, but also active.

RNA-based quantification of nasal microbiota activity. (A) Summed relative abundance of Bacteroidales (blue) and Clostridiales (green) taxa in the two sets of nasal samples (nasal swabs from 24 farm animals, left; surgical samples from 5 animals, right; see “Methods”). Other taxa (order level) with > 1% mean relative abundance are shown as “Other” (pink). Taxa with < 1% relative abundance are summed in the category “Low abundant”). (B) Relative abundance in RNA/DNA samples of Bacteroidales (blue), Clostridiales (green), and nasal colonizers (Lactobacillaceae, Moraxellaceae, Pasteurellaceae, Streptococcaceae and [Weeksellaceae], pink). Each dot corresponds to the abundances of the mentioned taxa in one individual animal. Open circles: invasive nasal samples from 5 animals (deep and middle nasal cavity, see Fig. 3). Filled circles: standard nasal swabs from 24 animals across 3 farms. (C) Relative abundance in DNA and RNA samples (as determined by 16S rRNA sequencing of DNA and cDNA, see main text) of the most relatively abundant genera, selected as those > 0.1% mean abundance in DNA or RNA farm samples. Gut-microbiota associated taxa (Bacteroidales and Clostridiales) are labelled in blue and green, respectively. Genera from other orders are shown in pink. Open circles: deep nasal samples obtained surgically from 5 animals (deep and middle nasal cavity, see Fig. 3). Filled circles: standard nasal swabs from 24 animals across 3 farms. Shown are mean values across all samples.
Discussion
In this study, we aimed to elucidate the role of typically gut-microbiota associated taxa in the nasal cavity of domestic pigs. Specifically, we asked three questions: (1) how prevalent are these taxa in vivo and across different anatomical sites in the nasal cavity?; (2) do these taxa indeed stem from the gut microbiota?; and (3) are these taxa active in the pig nasal cavity? To answer these questions, we used a combination of regular swab and surgical deep sampling of the pig nasal microbiota and inferred its composition/activity by 16S rRNA gene DNA and cDNA sequencing, respectively. These efforts yielded three key findings.
First, we found that gut-microbiota associated taxa constitute a substantial fraction of the nasal microbiota both across individual animals, as well as across different sites within the nasal cavity. This finding is in agreement with previous studies, which sampled the pig nasal microbiota, and is also consistent with other studies relying on sampling of the lower respiratory tract of pigs, where Clostridium and Prevotella genera were prevalent7,13,35. Thus, our results confirm that gut-microbiota associated taxa are part of the swine nasal respiratory microbiota and are not a sampling artifact. Nevertheless, the sampling of environments that do not have high microbial biomass, including many anatomical sites in the respiratory tract36,37, can pose technical challenges such as the false detection of transient environmental microbes37. Although we controlled for some potential sources of contamination (sequencing blank controls and getting undisturbed nasal samples) more studies are needed to validate the inhabitants of the pig nasal microbiota.
Second, our analysis of matched nasal and rectal microbiota samples from different farms showed a large overlap between gut-microbiota associated taxa from these two sites at ASV level. This finding suggests a common source of these taxa in both anatomic sites, for example in the form of solid fecal matter that enters the nasal cavity12,20, or through the air within a farm. In support of this hypothesis, a recent study found that gut-microbiota associated taxa are highly abundant in the air of pig farms, as well as in the nasal microbiota of pig farmers (compared to non-exposed individuals)38,39, suggesting substantial flow of bacterial material within animal farms. In contrast to gut-microbiota associated taxa, many other dominant taxa in the nasal microbiota, such as Moraxellaceae, Pasteurellaceae, and Weeksellaceae, showed a low degree of overlap between the two body sites, indicating that they may be professional nasal colonizers with reduced ability to colonize other (i.e. anaerobic) niches. Interesting exceptions were the Lactobacillus and Streptococcus genera, which also overlapped substantially at ASV level between these two body sites. Given that both Lactobacillus and Streptococcus are facultative anaerobes, it is tempting to speculate whether these genera may in fact predominantly reside in the gut, but are also able to colonize the nasal cavity (and possibly other sites) if presented with the opportunity.
Third, our quantification of 16S rRNA expression showed that gut-microbiota associated taxa are indeed active in the pig nasal cavity. To our knowledge, this is the first study which quantified pig nasal microbiota activity in vivo, and it suggests that these gut-microbiota associated taxa are not merely inactive transient members in the nasal cavity. Notably, recent metatranscriptomics analyses did report expression of some gut-microbiota associated taxa (e.g. Prevotella) in nasal samples taken from children40 and adult patients with asthma41, suggesting that these taxa may also be active in the human nasal cavity. One caveat is that we quantified 16S rRNA gene transcripts to determine the activity of gut-microbiota associated taxa in the pig nasal microbiota, which is an imperfect measure of cellular activity32. Future efforts could examine the activity of these taxa with methods that quantify metabolic activity more directly, for example by using fluorescently-tagged metabolic probes42,43,44 or metaproteomics45.
This study has several limitations. First, we sampled animals at an early age (i.e. pre-weaning stage at 2–3 weeks of age). Our choice was motivated by the fact that pigs at this age are most susceptible to respiratory pathogens, while the nasal microbiota is still rather unstable11,46. Nevertheless, our analysis of other published studies did reveal that gut-microbiota associated taxa are also prevalent at later stages in the animals’ life20,25,29, and future studies may examine to which extent the findings presented herein also hold in adult pigs and other species.
Second, although our comparison of abundant 16S DNA ASVs does indicate a shared origin of gut-microbiota associated taxa in the nasal and gut microbiota, our analysis was restricted to one section of the 16S DNA (variable regions V3-V4). Therefore, strains that share this DNA region, but differ in other parts of their 16S DNA (or other parts of their genome), cannot be distinguished with the approach chosen here. Given that we only identified few ubiquitous ASVs (that is, ASVs conserved across all three tested farms) within these taxa, we argue that our approach has the necessary resolution to detect strain differences. Nevertheless, to further validate that gut-microbiota associated strains are indeed shared by the nasal and gut microbiota, and to identify the species these strains belong to, future studies may use either full-length 16S DNA sequencing, or metagenomics, of matched nasal and gut microbiota samples.
Third, although our surgical samples suggest that gut-microbiota associated taxa are present throughout the pig nasal cavity, the methods used in this study are not able to resolve whether these taxa are located in any specific anatomical niches. Scanning electron microscopy imaging of the upper respiratory tract of piglets has recently suggested the absence of anaerobic crypts12, but it is conceivable that there may still be anaerobic substructures e.g. within or under the mucosal layer. Such potential substructures or locations may explain why strictly anaerobic bacterial taxa survive and are active in a basically aerobic environment such as nasal cavity. To resolve these questions, future efforts may use in vivo imaging methods like these47,48 to resolve the microstructure of the nasal microbiota in more detail.
Fourth, in this study we analyzed various nasal microbiota samples, but did not specifically address the impact of environmental factors. For example, while we found that gut-microbiota associated taxa are prevalent in the pig nasal microbiota across many studies (see Supplementary Fig. 3), their relative abundance varied widely across these samples. Moreover, the degree of overlap of gut-microbiota associated ASVs between nasal and gut microbiota samples varied between the three different farms (i.e. with farm 2 showing a much lower overlap, an observation for which we currently do not have an explanation). Further epidemiological studies may use this work as a starting point to assess the impact of environmental factors such as feeding strategies, housing conditions, or sanitation protocols, on the presence of gut-microbiota associated taxa in the nasal microbiota.
Finally, while our results do suggest that gut-microbiota associated taxa are indeed active in the pig nasal cavity, we did not examine the in vivo function of these taxa within the pig nasal microbiota. Recent in vivo studies11,13,14,15,16,17,49 have shown that the presence of these taxa is associated with disease outcomes and dysbiosis in various farm animals, but also in humans41. For example, in humans with chronic rhinosinusitis, Prevotella (genus belonging to Bacteroidales) was associated with increased inflammatory severity50. Future studies may use the results presented here as a starting point to elucidate the mechanistic underpinnings of these associations.
In conclusion, this study suggests that gut-microbiota associated taxa are indeed present and active in the nasal cavity of domestic pigs. These findings may serve as a starting point for future research aiming at elucidating the in vivo function of these taxa within the nasal microbiota.
Methods
Animal experimentation and ethics approval
Animal experimentation was performed following proper veterinary practices, in accordance with European (Directive 2010/63/EU) and Spanish (Real Decreto 53/2013) regulation, and in compliance with the ARRIVE guidelines (https://arriveguidelines.org/about). Sampling in farms was done with the approval of the Ethics Commission in Animal Experimentation of the Generalitat de Catalunya (Protocol number 11213). For the surgical sampling of five animals’ different nasal depths, euthanasia was performed following good veterinary practices. According to European (Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes) and Spanish (Real Decreto 53/2013) normative, this latter procedure did not require specific approval by an Ethical Committee (Chapter I, Article 3. 1 of 2010/63/EU).
Sample collection
Farm samples
Matched nasal and rectal swabs were taken from 24 healthy 2–3-week-old piglets from three commercial farms from Spain without reported respiratory diseases (termed Farm 1, Farm 2, Farm 3). After sampling, swabs were placed into sterile tubes filled with 1000µL DNA/RNA shield (Zymo Research) and transported to the laboratory under refrigeration.
Surgical samples
Deep surgical samples of the nasal cavity were obtained as follows: five healthy piglets of 2–3 weeks of life were moved to IRTA-CReSA facilities and euthanized by means of an overdose of sodium pentobarbital (Dolethal). Four types of nasal swabs per piglet were taken (labeled standard, external, middle, and deep) as described below. First, a nasal swab from one nostril was taken (“standard” swab, Fig. 3A). Second, an external swab was taken by introducing the swab superficially in the second nostril. Afterwards, deep and middle swabs were taken (at positions outlined in Fig. 3A) from the second nostril after longitudinally cutting and separating the skin of each piglet’s head (to prevent contaminations from the skin surface) and subsequent cutting the skull, directly from the nasal turbinate without touching any other part of the piglet nose to avoid contamination and after removing the cartilaginous nasal wall. Additionally, rectal swabs were obtained from the same animals. Sample swabs were collected in sterile plastic tubes filled with 800 µL of DNA/RNA shield (Zymo Research) to ensure the stability and preservation of the genetic material. Negative controls (sterile DNA/RNA shield without a swap) were included in each extraction. Samples were stored at – 20 °C until extraction.
DNA/RNA extraction
Metagenomic DNA and RNA were extracted starting from 350 µL of DNA/RNA shield swab sample (previously vortexed) and following a modified protocol of ZymoBIOMICS DNA/RNA Miniprep kit (Zymo Research) in which the lysis was performed only chemically using 700 µL of ZymoBIOMICS DNA/RNA lysis buffer (2 volumes of lysis buffer per 1 volume of sample). The RNA fraction of the samples was treated with 80 µL of DNAse I (included in the kit) at room temperature for 20 min. Elution of both DNA and RNA was done in 50 µL of elution buffer. DNA and RNA concentration was measured using BioDrop DUO (BioDrop Ltd). DNA and RNA were stored at – 80 °C until sequencing.
16S rRNA gene sequencing
16S rRNA gene libraries were prepared from the total extracted DNA and cDNA from RNA samples and sequenced at Servei de Genòmica, Universitat Autònoma de Barcelona (Illumina pair-end 2X300 bp, MS-102-2003 MiSeq Re-agent Kit v2, 500 cycle), using Illumina recommended primers for variable regions V3V4 of 16S rRNA gene (fwd 5′TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG, rev 5′ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC). The size of the amplicons was verified on a Bioanalyzer DNA 1000 chip (Agilent), as expected amplicon length was approximately 460 bp. Finally, the sequences were sorted into samples and used as input for bioinformatic analysis.
Bioinformatic analysis of 16S rRNA sequencing data
The microbiota composition of the samples was analyzed with quantitative insights into microbial ecology (QIIME) 2 software version 2022.251. The detailed pipeline followed from raw reads to obtain tables of abundances at ASV and other taxonomic levels, the diversity analyses and the statistical tests to determine the differential abundances, can be found at https://zenodo.org/record/8013997 (Zenodo ID 8013997). Briefly, the pre-processing of the reads was done separately for each sequencing run. At first, raw demultiplexed reads with quality (fastq) were imported and their quality was evaluated with qiime2 demux plugin. Primers from the variable region V3-V4 were extracted using qiime2 cutadapt plugin52 Sequences that did not contain primers and thus, were not a sequencing product, were removed from the analysis. DADA2 software package53 was used under the parameters detailed in the pipeline to quality filter, paired-end merge, remove chimeras and sort reads into ASVs. Contaminant artifactual amplicons from non-prokaryotic origin were identified with qiime2 quality control plugin54 by aligning with VSEARCH55 all ASVs against Greengenes database Vs. 13.856 clustered with 88% identity (available at https://docs.qiime2.org/2022.2/data-resources/). Unmatched sequences were filtered out from the analysis. Additionally, ASVs identified as Archea, Mitochondria or Chloroplast (which also contain 16S rRNA), were also discarded. The alignment of the remaining sequences was performed with MAFFT57, and the hypervariable positions were masked58 with qiime2 alignment plugin. Finally, ASVs found in the negative control of the deep nose dataset were removed from the analysis (21 and 41 for DNA and RNA controls, respectively, Supplementary table 7). The taxonomic classification of the ASVs was performed using a naïve Bayes classifier with scikit-learn Python module for machine learning59. In order to increase the classifier accuracy60, it was previously trained against prokaryotic 16S rRNA gene V3-V4 region, extracted from Greengenes database (13.8 version) clustered at 99% sequence identity. This qiime2 feature classifier artifact can be found at https://zenodo.org/record/8013997 (Zenodo ID 8013997). Square brackets in taxonomical assignations indicate contested names in the reference Greengenes database used (13.8).
In order to normalize uneven sampling depths61, the diversity analyses of the samples from the surgical sampling (supplementary text 2) were performed at a normalized depth of 2291, corresponding to the lowest depth sample. The alpha diversity of the samples was estimated with Chao162 and Shannon63 indexes. Significant differences were found with pairwise non-parametric t-tests (999 random permutations) using qiime2 diversity alpha-group-significance plugin64. Beta diversity was calculated with Jaccard65 and Bray–Curtis66 dissimilarity indexes for the qualitative and quantitative analyses, respectively. Qiime2 core-metrics plugin67,68 was used to compute principal coordinate (PCoA) analysis. The percentage of explanation of the variables under study was estimated with the Adonis function from the Vegan package, in R software69. The significance of beta diversity analyses was calculated by PERMANOVA pairwise test (999 random permutations) using qiime2 diversity beta-group-significance plugin70. Differently abundant taxa between groups were found with ANCOM-BC algorithm71. For all the stated analyses, the significance threshold p value was set to 0.05. To perform the phylogenetic analyses, the ASVs of interest were multiple aligned with MAFFT57, from where the phylogeny was built with the IQtree72 online tool (available in: http://iqtree.cibiv.univie.ac.at/) using automatic selection of the substitution model, with 1,000 bootstrap alignments and iterations. Output microbiota data processing and plot generation was performed using R script language version 4.2.2 in RStudio environment version 2022.07.073, using the packages qiime2r74, reshape275, ggplot276, tidyverse77, and ggtree78, as well as MATLAB (version 2021A).
Quantification of total 16S rRNA gene concentration
Total 16S rRNA gene concentrations (as a proxy for microbial load in swabs) were quantified as follows. Briefly, the reaction was prepared in a volume of 20 µL consisting in 2 µL of the template DNA and 18 µL of Femto Bacterial qPCR Premix, which includes a primer mix targeting the 16S rRNA (Femto Bacterial DNA Quantification Kit, Zymo Research). The PCR reaction was performed using a 7500 Fast Real-Time PCR System (ThermoFisher Scientific) at 95 °C for 10 min, 40 cycles of 95 °C for 30 s, 50 °C for 30 s, 72 °C for 1 min, followed by a melting curve and a final extension of 72 °C for 7 min. Each run contained two replicates per sample, a standard curve of 6 points (2–0.00002 ng), a negative and a positive extraction control as well as PCR negative controls. Graphpad 8.3 (538) Prism software (Dotmatics, San Diego CA) was used to analyze the data obtained from the 16s rRNA qPCR of DNA extracted from the swabs.
Data availability
The raw data used in this study are publicly available at NCBI’s SRA database under BioProject ID PRJNA981084. Processed data sets are available as supplementary tables 1–7 in individual files as described below: Supplementary Table 1—List of publicly available data sets used in this study. Supplementary Table 2—Relative abundance of gut-microbiota associated taxa (i.e. Clostridiales and Bacteroidales orders) across individual samples in 11 publicly available data sets used in this study. Supplementary Table 3—Most relatively abundant gut-microbiota associated ASVs in nasal and rectal samples (top 100 in each body site) in each of the three sampled farms. Supplementary Table 4—ANCOM-BC statistically significant (corrected q-value < 0.05) differences between sampling sites in nasal cavity at ASV, order, family, genus and species taxonomic levels. Supplementary Table 5—Relative abundance of all identified taxa at ASV, order, family, genus and species level across the two groups of samples used in this study. Supplementary Table 6—Sample metadata file containing identifiers and description of the samples used in this study. Supplementary Table 7—Taxonomic classification and abundance across samples of the amplicon sequence variants detected in the DNA and RNA blank negative controls. Supplementary text and figures are provided as a single document containing: Supplementary text 1—identification of gut-microbiota associated taxa in the pig nasal microbiota. Supplementary text 2—compositional analysis of deep pig nasal microbiota samples. Supplementary Figs. 1–10.
References
Whipps, J., Lewis, K. & Cooke, R. Mycoparasitism and plant disease control. In Fungi in Biological Control Systems (ed. Burge, M.) 161–187 (Manchester University Press, 1988).
Lloyd-Price, J., Abu-Ali, G. & Huttenhower, C. The healthy human microbiome. Genome Med. 8(1), 51. https://doi.org/10.1186/s13073-016-0307-y (2016).
Pickard, J. M., Zeng, M. Y., Caruso, R. & Núñez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 279(1), 70–89. https://doi.org/10.1111/imr.12567 (2017).
Cho, D. Y., Hunter, R. C. & Ramakrishnan, V. R. The microbiome and chronic rhinosinusitis. Immunol. Allergy Clin. N. Am. 40(2), 251–263. https://doi.org/10.1016/j.iac.2019.12.009 (2020).
Kumpitsch, C., Koskinen, K., Schöpf, V. & Moissl-Eichinger, C. The microbiome of the upper respiratory tract in health and disease. BMC Biol. 17(1), 87. https://doi.org/10.1186/s12915-019-0703-z (2019).
Bomar, L., Brugger, S. D. & Lemon, K. P. Bacterial microbiota of the nasal passages across the span of human life. Curr. Opin. Microbiol. 41, 8–14. https://doi.org/10.1016/j.mib.2017.10.023 (2018).
Pirolo, M., Espinosa-Gongora, C., Bogaert, D. & Guardabassi, L. The porcine respiratory microbiome: Recent insights and future challenges. Anim. Microbiome 3(1), 9. https://doi.org/10.1186/s42523-020-00070-4 (2021).
Bond, S., McMullen, C., Timsit, E. & Léguillette, R. Topography of the respiratory, oral, and guttural pouch bacterial and fungal microbiotas in horses. J. Vet. Intern. Med. 37(1), 349–360. https://doi.org/10.1111/jvim.16612 (2023).
McDaneld, T. G., Kuehn, L. A. & Keele, J. W. Microbiome of the upper nasal cavity of beef calves prior to weaning12. J. Anim. Sci. 97(6), 2368–2375. https://doi.org/10.1093/jas/skz119 (2019).
Nicola, I. et al. Characterization of the upper and lower respiratory tract microbiota in Piedmontese calves. Microbiome 5(1), 152. https://doi.org/10.1186/s40168-017-0372-5 (2017).
Correa-Fiz, F., Fraile, L. & Aragon, V. Piglet nasal microbiota at weaning may influence the development of Glässer’s disease during the rearing period. BMC Genom. 17, 404. https://doi.org/10.1186/s12864-016-2700-8 (2016).
Pena-Cortes, L. C., LeVeque, R. M., Funk, J. A., Marsh, T. L. & Mulks, M. H. Development of the tonsil microbiome in pigs and effects of stress on the microbiome. Front. Vet. Sci. 5, 220. https://doi.org/10.3389/fvets.2018.00220 (2018).
Huang, T. et al. Microbial communities in swine lungs and their association with lung lesions. Microb. Biotechnol. 12(2), 289–304. https://doi.org/10.1111/1751-7915.13353 (2019).
Jiang, N. et al. Illumina MiSeq sequencing investigation of microbiota in bronchoalveolar lavage fluid and cecum of the swine infected with PRRSV. Curr. Microbiol. 76(2), 222–230. https://doi.org/10.1007/s00284-018-1613-y (2019).
Wang, Q. et al. Comparison of oropharyngeal microbiota in healthy piglets and piglets with respiratory disease. Front. Microbiol. 9, 3218. https://doi.org/10.3389/fmicb.2018.03218 (2018).
Megahed, A. et al. Impacts of environmental complexity on respiratory and gut microbiome community structure and diversity in growing pigs. Sci. Rep. 9(1), 13773. https://doi.org/10.1038/s41598-019-50187-z (2019).
Blanco-Fuertes, M., Correa-Fiz, F., Fraile, L., Sibila, M. & Aragon, V. Altered nasal microbiota composition associated with development of polyserositis by Mycoplasma hyorhinis. Pathogens. 10(5), 603. https://doi.org/10.3390/pathogens10050603 (2021).
Slifierz, M. J., Friendship, R. M. & Weese, J. S. Longitudinal study of the early-life fecal and nasal microbiotas of the domestic pig. BMC Microbiol. 15(1), 184. https://doi.org/10.1186/s12866-015-0512-7 (2015).
Pena-Cortes, L. C., LeVeque, R. M., Funk, J., Marsh, T. L. & Mulks, M. H. Development of the tonsillar microbiome in pigs from newborn through weaning. BMC Microbiol. 18(1), 35. https://doi.org/10.1186/s12866-018-1176-x (2018).
Espinosa-Gongora, C., Larsen, N., Schønning, K., Fredholm, M. & Guardabassi, L. Differential analysis of the nasal microbiome of pig carriers or non-carriers of Staphylococcus aureus. PLoS One. 11(8), e0160331. https://doi.org/10.1371/journal.pone.0160331 (2016).
Soave, O. & Brand, C. D. Coprophagy in animals: A review. Cornell Vet. 81(4), 357–364 (1991).
Sansom, B. F. & Gleed, P. T. The ingestion of sow’s faeces by suckling piglets. Br. J. Nutr. 46(3), 451–456. https://doi.org/10.1079/bjn19810053 (1981).
Xiao, L. et al. A reference gene catalogue of the pig gut microbiome. Nat. Microbiol. 1, 16161. https://doi.org/10.1038/nmicrobiol.2016.161 (2016).
Correa-Fiz, F., Gonçalves-Dos-Santos, J. M., Illas, F. & Aragon, V. Antimicrobial removal on piglets promotes health and higher bacterial diversity in the nasal microbiota. Sci. Rep. 9(1), 6545. https://doi.org/10.1038/s41598-019-43022-y (2019).
Chrun, T., Leng, J., La Ragione, R. M., Graham, S. P. & Tchilian, E. Changes in the nasal microbiota of pigs following single or co-infection with porcine reproductive and respiratory syndrome and swine influenza a viruses. Pathogens 10(10), 1225. https://doi.org/10.3390/pathogens10101225 (2021).
Correa-Fiz, F. et al. Feed additives for the control of post-weaning Streptococcus suis disease and the effect on the faecal and nasal microbiota. Sci. Rep. 10(1), 20354. https://doi.org/10.1038/s41598-020-77313-6 (2020).
Mou, K. T. et al. Shifts in the nasal microbiota of swine in response to different dosing regimens of oxytetracycline administration. Vet. Microbiol. 237, 108386. https://doi.org/10.1016/j.vetmic.2019.108386 (2019).
Patel, S. et al. Differential analysis of longitudinal methicillin-resistant Staphylococcus aureus colonization in relation to microbial shifts in the nasal microbiome of neonatal piglets. mSystems. 6(4), e0015221. https://doi.org/10.1128/mSystems.00152-21 (2021).
Strube, M. L., Hansen, J. E., Rasmussen, S. & Pedersen, K. A detailed investigation of the porcine skin and nose microbiome using universal and Staphylococcus specific primers. Sci. Rep. 8(1), 12751. https://doi.org/10.1038/s41598-018-30689-y (2018).
Blanco-Fuertes, M., Correa-Fiz, F., López-Serrano, S., Sibila, M. & Aragon, V. Sow vaccination against virulent Glaesserella parasuis shapes the nasal microbiota of their offspring. Sci. Rep. 12(1), 3357. https://doi.org/10.1038/s41598-022-07382-2 (2022).
Callahan, B. J., McMurdie, P. J. & Holmes, S. P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11(12), 2639–2643. https://doi.org/10.1038/ismej.2017.119 (2017).
Blazewicz, S. J., Barnard, R. L., Daly, R. A. & Firestone, M. K. Evaluating rRNA as an indicator of microbial activity in environmental communities: Limitations and uses. ISME J. 7(11), 2061–2068. https://doi.org/10.1038/ismej.2013.102 (2013).
Shaffer, J. P. et al. A comparison of DNA/RNA extraction protocols for high-throughput sequencing of microbial communities. Biotechniques 70(3), 149–159. https://doi.org/10.2144/btn-2020-0153 (2021).
Ya, W., Kelsey, N. T., Yan, Y. et al. Characterizing microbial community viability with RNA-based high-throughput sequencing, PREPRINT (Version 1) available at Research Square. https://doi.org/10.21203/rs.3.rs-1870950/v1 (2022).
Pirolo, M. et al. Bacterial topography of the upper and lower respiratory tract in pigs. Anim. Microbiome 5(1), 5. https://doi.org/10.1186/s42523-023-00226-y (2023).
Dickson, R. P., Erb-Downward, J. R., Martinez, F. J. & Huffnagle, G. B. The microbiome and the respiratory tract. Annu. Rev. Physiol. 78, 481–504. https://doi.org/10.1146/annurev-physiol-021115-105238 (2016).
Kennedy, K. M. et al. Questioning the fetal microbiome illustrates pitfalls of low-biomass microbial studies. Nature 613(7945), 639–649. https://doi.org/10.1038/s41586-022-05546-8 (2023).
Kraemer, J. G., Aebi, S., Oppliger, A. & Hilty, M. The indoor-air microbiota of pig farms drives the composition of the pig farmers’ nasal microbiota in a season-dependent and farm-specific manner. Appl. Environ. Microbiol. 85(9), e0303818. https://doi.org/10.1128/AEM.03038-18 (2019).
Kraemer, J. G., Ramette, A., Aebi, S., Oppliger, A. & Hilty, M. Influence of pig farming on the human nasal microbiota: Key role of airborne microbial communities. Appl. Environ. Microbiol. 84(6), e0247017. https://doi.org/10.1128/AEM.02470-17 (2018).
Rajagopala, S. V. et al. Metatranscriptomics to characterize respiratory virome, microbiome, and host response directly from clinical samples. Cell Rep. Methods 1(6), 100091. https://doi.org/10.1016/j.crmeth.2021.100091 (2021).
Castro-Nallar, E. et al. Integrating microbial and host transcriptomics to characterize asthma-associated microbial communities. BMC Med. Genom. 8, 50. https://doi.org/10.1186/s12920-015-0121-1 (2015).
Campisciano, G. & Biffi, S. Microbiota in vivo imaging approaches to study host-microbe interactions in preclinical and clinical setting. Heliyon 8(12), e12511. https://doi.org/10.1016/j.heliyon.2022.e12511 (2022).
Geva-Zatorsky, N. et al. In vivo imaging and tracking of host-microbiota interactions via metabolic labeling of gut anaerobic bacteria. Nat. Med. 21(9), 1091–1100. https://doi.org/10.1038/nm.3929 (2015).
Lin, L. et al. Imaging the in vivo growth patterns of bacteria in human gut Microbiota. Gut Microbes 13(1), 1960134. https://doi.org/10.1080/19490976.2021.1960134 (2021).
Kim, Y. S. et al. Antibiotic-dependent relationships between the nasal microbiome and secreted proteome in nasal polyps. Allergy Asthma Immunol. Res. 13(4), 589–608. https://doi.org/10.4168/aair.2021.13.4.589 (2021).
Nowland, T. L., Plush, K. J., Barton, M. & Kirkwood, R. N. Development and function of the intestinal microbiome and potential implications for pig production. Anim. Basel 9(3), 76. https://doi.org/10.3390/ani9030076 (2019).
Mark Welch, J. L., Rossetti, B. J., Rieken, C. W., Dewhirst, F. E. & Borisy, G. G. Biogeography of a human oral microbiome at the micron scale. Proc. Natl. Acad. Sci. U. S. A. 113(6), E791–E800. https://doi.org/10.1073/pnas.1522149113 (2016).
Mark Welch, J. L., Ramírez-Puebla, S. T. & Borisy, G. G. Oral microbiome geography: Micron-scale habitat and niche. Cell Host Microbe 28(2), 160–168. https://doi.org/10.1016/j.chom.2020.07.009 (2020).
Obregon-Gutierrez, P., Aragon, V. & Correa-Fiz, F. Sow contact is a major driver in the development of the nasal microbiota of piglets. Pathogens 10(6), 697. https://doi.org/10.3390/pathogens10060697 (2021).
Kuhar, H. N. et al. Relative abundance of nasal microbiota in chronic rhinosinusitis by structured histopathology. Int. Forum Allergy Rhinol. 8(12), 1430–1437. https://doi.org/10.1002/alr.22192 (2018).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37(8), 852–857. https://doi.org/10.1038/s41587-019-0209-9 (2019).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17(1), 10–12. https://doi.org/10.14806/ej.17.1.200 (2011).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13(7), 581–583. https://doi.org/10.1038/nmeth.3869 (2016).
Camacho, C. et al. BLAST+: Architecture and applications. BMC Bioinform. 10, 421. https://doi.org/10.1186/1471-2105-10-421 (2009).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 4, e2584. https://doi.org/10.7717/peerj.2584 (2016).
McDonald, D. et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6(3), 610–618. https://doi.org/10.1038/ismej.2011.139 (2012).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30(4), 772–780. https://doi.org/10.1093/molbev/mst010 (2013).
DJ Lane. 16s/23s rrna sequencing. In Nucleic Acid Techniques in Bacterial Systematics (eds. Stackebrandt, E, & Goodfellow, M. ) 115–175 (Wiley, 1991).
Fabian, P. et al. Scikit-learn: Machine learning in python. JMLR 2011, 2825–2830 (2011).
Werner, J. J. et al. Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J. 6(1), 94–103. https://doi.org/10.1038/ismej.2011.82 (2012).
Weiss, S. et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 5(1), 27. https://doi.org/10.1186/s40168-017-0237-y (2017).
Eren, M. I., Chao, A., Hwang, W. H. & Colwell, R. K. Estimating the richness of a population when the maximum number of classes is fixed: A nonparametric solution to an archaeological problem. PLoS One 7(5), e34179. https://doi.org/10.1371/journal.pone.0034179 (2012).
Shannon, C. & Weaver, W. The mathematical theory of communication. Bell Syst. Tech. J. 27, 379–423. https://doi.org/10.1002/j.1538-7305.1948.tb01338.x (1948).
William, H. K. & Wallis, W. A. Use of ranks in one-criterion variance analysis. JASA 47(260), 583–621 (1952).
Jaccard, P. Nouvelles Recherches sur la Distribution Florale (Rouge, 1908).
Sørensen, T. J. A Method of Establishing Groups of Equal Amplitude in Plant Sociology Based on Similarity of Species Content and Its Application to Analyses of the Vegetation on Danish Commons. In I kommission hos E. Munksgaard; København, Demark (1948).
Nathan, H., Per-Gunnar, M., Yoel, S. & Mark, T. An algorithm for the principal component analysis of large data sets. arXiv:1007.5510 (2010).
Pierre, L. & Louis, L. Numerical Ecology 499 (Elsevier, 2012).
Jari Oksanen, F. et al. Stevens, Eduard Szoecs and Helene Wagner. Vegan: Community Ecology Package. R Package Version 2.5-7. https://CRAN.R-project.org/package=vegan (2020).
Anderson, M. J. A new Method for non-parametric multivariate analysis of variance. Austral. Ecol. 26, 32–46. https://doi.org/10.1111/j.1442-9993.2001.01070.pp.x (2001).
Lin, H. & Peddada, S. D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 11(1), 3514. https://doi.org/10.1038/s41467-020-17041-7 (2020).
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44(W1), W232–W235. https://doi.org/10.1093/nar/gkw256 (2016).
RStudio|Open Source & Professional Software for Data Science Teams. https://rstudio.com/ (2023).
Bisanz, J. Tutorial: Integrating QIIME2 and R for data visualization and analysis using qiime2R v0.99.6 (2021).
Wickham, H. Reshaping data with the reshape package. J. Stat. Softw. 21(12), 1–20 (2007).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, 2009). https://doi.org/10.1007/978-0-387-98141-3.
Wickham, H. et al. Welcome to the tidyverse. J. Open Source Softw. 4, 1686 (2019).
Yu, G., Smith, D., Zhu, H., Guan, Y. & Lam, T. T. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36. https://doi.org/10.1111/2041-210X.12628 (2017).
Acknowledgements
The authors are grateful to Nuria Galofré-Milà and Eva Huerta for their technical support. The authors are also grateful to the Centres de recerca de Catalunya (CERCA) Program.
Funding
This work was supported by funding from the Spanish Ministry of Research and Innovation (RYC2021-033035-I to KK, PID2019-106233RB-I00/AEI/https://doi.org/10.13039/501100011033 to VA and FCF). LBL and POG are supported by FPI(PRE2020-096048) and FPU (FPU19/02126) fellowships, respectively.
Author information
Authors and Affiliations
Contributions
Conceived and designed the study: KK, FCF, VA. Performed experiments: LBL, MS, JS. Performed analyses: POG, LBL, KK. Supervised the analyses: KK and FCF. Wrote the manuscript with contributions from all authors: POG, KK.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Obregon-Gutierrez, P., Bonillo-Lopez, L., Correa-Fiz, F. et al. Gut-associated microbes are present and active in the pig nasal cavity. Sci Rep 14, 8470 (2024). https://doi.org/10.1038/s41598-024-58681-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-58681-9
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
