
Abstract
In this study, we aimed to identify molecular markers associated with type II alveolar epithelial cell injury in acute lung injury (ALI) models using bioinformatics methods. The objective was to provide new insights for the diagnosis and treatment of ALI/ARDS. We downloaded RNA SEQ datasets (GSE109913, GSE179418, and GSE119123) from the Gene Expression Omnibus (GEO) and used R language package to screen differentially expressed genes (DEGs). DEGs were annotated using Gene Ontology (GO), and their pathways were analyzed using Kyoto Encyclopedia of Genes and Genomes (KEGG). DEGs were imported into the STRING database and analyzed using Cytoscape software to determine the protein network of DEGs and calculate the top 10 nodes for the hub genes. Finally, potential therapeutic drugs for the hub genes were predicted using the DGIdb database. We identified 78 DEGs, including 70 up-regulated genes and 8 down-regulated genes. GO analysis revealed that the DEGs were mainly involved in biological processes such as granulocyte migration, response to bacterial-derived molecules, and cytokine-mediated signaling pathways. Additionally, they had cytokine activity, chemokine activity, and receptor ligand activity, and functioned in related receptor binding, CXCR chemokine receptor binding, G protein-coupled receptor binding, and other molecular functions. KEGG analysis indicated that the DEGs were mainly involved in TNF signaling pathway, IL-17 signaling pathway, NF-κB signal pathway, chemokine signal pathway, cytokine-cytokine receptor interaction signal pathway, and others. We identified eight hub genes, including IRF7, IFIT1, IFIT3, PSMB8, PSMB9, BST2, OASL2, and ZBP1, which were all up-regulated genes. We identified several hub genes of type II alveolar epithelial cells in ALI mouse models using bioinformatics analysis. These results provide new targets for understanding and treating of ALI.
Similar content being viewed by others

Introduction
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are common causes of respiratory failure in critically ill patients, and their pathogenesis is not fully understood1. The pathological manifestations of ALI/ARDS are characterized by damage to pulmonary capillary endothelial cells and alveolar epithelial cells. This damage leads to an excessive production of inflammatory factors within lung tissue, resulting in respiratory distress, refractory hypoxemia, and non-cardiogenic pulmonary edema2. Regarding the pathogenesis of ALI/ARDS, the most widely discussed factors include the overactivation of the inflammatory response, increased permeability of both alveolar epithelium and vascular endothelium, as well as a decrease in the clearance of alveolar fluid in affected patients. However, further details and insights into this condition are still under investigation3. Despite progress in improving their diagnosis and treatment, the mortality rate of ALI/ARDS remains high, ranging from 30 to 40%4.
The alveolar epithelium, comprising both type II alveolar epithelial cells (ATII) and type I alveolar epithelial cells (ATI), governs fluid and ion transport, serving a pivotal function in preserving lung homeostasis. Additionally, these cells engage in fusion with the endothelial cells of capillaries, collectively forming a barrier crucial for lung ventilation5. However, various factors can induce damage to the epithelial cells during the early stage of ALI/ARDS, leading to disruption of the barrier function6. ATII cells play a pivotal role primarily in overseeing the proliferation and differentiation of ATI cells and in the recovery of lung epithelial function. However, they also possess the capacity to activate alveolar macrophages, which can exacerbate lung damage. Throughout this process, ATII cells can attract circulating immune cells, leading to the release of various mediators aimed at eliminating pathogens7. Consequently, the repair of damaged alveolar epithelial cells, especially the proliferation and differentiation of ATII cells, holds significant importance for the prognosis of ALI/ARDS8. Despite the identification of several biomarkers that can predict the severity of damage to alveolar epithelial cells and vascular endothelial cells in ARDS patients, these markers lack uniformity and specificity9. Hence, it is imperative to investigate related molecular markers specific to ATII cells in order to enhance our understanding of ARDS and explore novel therapeutic options.
Bioinformatics is an interdisciplinary field that combines computer science, information technology, novel mathematical algorithms, and statistical methods. It specializes in the analysis of biological experimental data, uncovering the hidden biological significance within the data. Moreover, it aims to develop novel data analysis tools for the acquisition and management of diverse information10. In comparison to other frequently used statistical methods, bioinformatics is known for its comprehensive and efficient approach. Recent advancements in gene sequencing at the mRNA level have opened up new avenues for investigating the mechanisms and treatment of diseases. The combination of chip technology and bioinformatics analysis further enables the exploration of diseases at the genetic level. While this method has been extensively employed for screening tumor targets at the genome level, there has been a limited number of bioinformatics studies focused on ALI/ARDS. Through these bioinformatics investigations, previous researchers have identified a multitude of genes associated with ALI/ARDS, with many of them being linked to inflammatory mechanisms11,12. Additionally, other studies have highlighted the significance of the body's immune response in relation to the prognosis of ALI/ARDS13,14. In this study, we employed bioinformatics analysis to explore the pivotal genes and molecular markers linked to ATII injury in various ALI models. We retrieved the necessary dataset from GEO, conducted data sorting and analysis using the R language, and identified the shared Differentially Expressed Genes (DEGs) in different ALI models. By subjecting these DEGs to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses, our aim was to uncover common patterns in gene expression. This approach enables us to potentially identify novel strategies for improving patient outcomes in the context of ARDS.
Materials and methods
Data sources
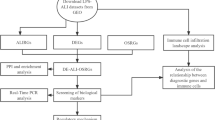
The Gene Expression Omnibus (GEO) is a gene expression database established by the National Center for Biotechnology Information (NCBI) in 2000. This database contains gene expression data submitted by research institutions worldwide, including gene chip and high-throughput sequencing data. We utilized GEO to retrieve the data needed for our study. We used “ATII”, “acute lung injury” and “ATII and acute lung injury”as key words to retrieve data sets. The screening criteria for selecting data were as follows: (1) model: acute lung injury mouse models, (2) data type: high-throughput sequencing and single-cell sequencing, (3) cell type: ATII cells, and (4) sample size: each dataset includes at least three samples in both the control and experimental groups. By setting these conditions, we aimed to obtain high-quality and relevant data for our study. The data set is screened again based on processing time.
Research method
Identify differentially expressed genes (DEGs)
R language is a statistical programming language known for its capabilities in statistical computation, data mining, and data visualization. RStudio provides a supportive environment for executing code, creating visualizations, and more. In our study, we employed the R software to preprocess the data, which encompassed batch correction and standardization tasks accomplished through the use of the DESeq2 software package.To identify the differentially expressed genes (DEGs), we utilized the limma software package and removed overlapping genes from the datasets to obtain a final list of DEGs. The screening criteria for differential expression were set at an adjusted p-value < 0.05 and an absolute value of logarithmic fold change (|LogFC|) > 115. These criteria ensured that only significant and biologically relevant genes were included in subsequent enrichment and protein network analyses.
Enrichment analysis of DEGs
To gain insights into the biological functions and pathways associated with the differentially expressed genes (DEGs), we performed Gene Ontology (GO) enrichment analysis (biological process, cellular component, and molecular function) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis using the clusterProfiler package in R software. We used the “ggplot2” package to generate graphical representations of the analysis results. The primary parameters were configured as follows: an enrichment significance threshold of (p-value) < 0.05 and a corrected p-value (q-value) < 0.05. Using these criteria, we were able to identify significantly enriched components within the analysis module.
Protein–protein interaction (PPI) network and identification of hub genes
To construct a protein–protein interaction (PPI) network for the differentially expressed genes (DEGs), we used the Search Tool for the Retrieval of Interacting Genes (STRING) database (http://string-db.org/). We uploaded the list of Differentially Expressed Genes (DEGs) into the STRING database, specifying the species as “Mouse,” and set the significance threshold at 0.4. Subsequently, the interaction network diagram of DEGs was automatically generated by the database. We then exported the node file for visualization using Cytoscape software16.
The CytoHubba plug-in and the MCODE plug-in are widely employed analytical tools within the software. In our analysis, we opted for the degree scoring algorithm in CytoHubba, as it is the most commonly used algorithm for identifying key genes. We set the criteria to select the top 10 key genes based on the degree ranking. Additionally, the MCODE plug-in was utilized to detect the most densely interacting module within the Protein–Protein Interaction (PPI) network. This was accomplished by calculating scores for each node. These identified core subnetworks, and the intersection between them, were identified as the hub genes17.
Identify drug candidates
DGIdb database is a valuable resource for predicting potential drugs for identified hub genes. By integrating drug-gene interaction data, it allows users to search for potential drugs that can target specific genes of interest18. The database provides information on FDA-approved drugs, experimental drugs, and investigational drugs, as well as their indications, target genes, and drug-gene interaction types19. Users can search for drugs based on gene symbols or drug names, and filter the results based on various criteria such as drug type, drug status, and interaction type. The database also provides links to other resources such as PubMed and ClinicalTrials.gov for further information on drugs and their clinical applications.
Results
Acquisition of data sets
According to the 2012 Berlin definition of ARDS, respiratory dysfunction must occur within one week of a known insult20. Therefore, we selected mouse models of ALI from datasets with time points within 72 h. Ultimately, we identified three gene chip datasets from the GEO database platform: GSE109913, which contains three samples of lung injury caused by lipopolysaccharide infection and control samples; GSE179418, which contains three samples of lung injury caused by Escherichia coli infection; and GSE119123, which contains five samples of lung injury caused by influenza virus. Each dataset includes an equal number of control samples. The detailed informationis illustrated in Table 1.
Differentially expressed genes of ATII in 3 datasets
The R programming language package was utilized to analyze the aforementioned single cell RNA sequencing (RNA-seq) datasets. Using Venn diagrams, a total of 82 genes were identified, with 70 being up-regulated and 8 being down-regulated (Fig. 1A–C). Among them, four genes exhibited disparate expression patterns across different datasets and were thus excluded. Ultimately, 78 genes were identified as differentially expressed genes (DEGs) and were selected for further investigation.

Expression of differential genes in GSE109913, GSE179418 and GSE119123. (A) All differentially expressed genes; (B) up-regulated genes; (C) down-regulated genes.
Go enrichment and KEGG pathway enrichment analysis of DEGs
The Go analysis was performed on the biological processes (BP), cellular components (CC), and molecular functions (MF) of the DEGs. The results revealed that the CC of these DEGs were primarily composed of host cell components, proteinome core complexes, and endopeptidase complexes, etc. The BP were significantly enriched in granulocyte migration, response to bacterial-derived molecules, cytokine-mediated signaling pathways, and others. These DEGs were also found to have cytokine activity, chemokine activity, and receptor ligand activity, and they can participate in related receptor binding, CXCR chemokine receptor binding, and G protein-coupled receptor binding, among others. Additionally, KEGG analysis indicated that the DEGs were mainly involved in TNF signaling pathway, IL-17 signaling pathway, NF-κB signaling pathway, chemokine signaling pathway, and cytokine receptor interaction signaling pathway (as shown in Figs. 2 and 3). The present study suggests that these pathways play a crucial role in the development of human ALI/ARDS. The NF-κB signaling pathway is intricately involved in various processes, including inflammatory responses, immune responses, apoptosis regulation, and stress responses. In the context of acute lung injury inflammation, NF-κB serves as a key transcription factor that, upon activation, promotes the expression of relevant inflammatory mediators. Additionally, it has the capacity to regulate the expression of genes associated with ALI21. Recent literature highlights the cytokine storm as a pivotal factor in inducing ARDS, with IL-17, TNF-α, and IL-6 being extensively discussed22. IL-17 plays a role in recruiting neutrophils, stimulating the release of various inflammatory cytokines, and amplifying the inflammatory response. Reducing the expression of TNF-α can benefit patients dealing with the disease. Chemokines also play a critical role in these processes, as they are produced by neutrophils and macrophages, and contribute to cell aggregation while maintaining local inflammation homeostasis. Considering these pathways, blocking the activity of relevant factors may be a viable approach for the treatment of ALI/ARDS.

Gene ontology (GO) analysis (BP, CC, and MF) of DEGs. (A) Barplot, the abscissa is the number of enriched genes; (B) Bubble, the abscissa GeneRatio represents the proportion of enriched genes to the total number of genes.

The KEGG enrichment analysis of DEGs. (A) Barplot, the abscissa is the number of enriched genes; (B) Bubble, the abscissa GeneRatio represents the proportion of enriched genes to the total number of genes.
Protein–protein interaction (PPI) network and identification of hub genes
Through the use of the STRING database and Cytoscape software, we identified the top 10 genes with the highest degree and the subnetwork with the highest MCODE score. The intersection of these results yielded eight hub genes: IFIT1, IFIT3, IRF7, PSMB8, PSMB9, BST2, OASL2, and ZBP1 (Table 2). These genes were identified as critical players in the PPI network and could potentially serve as therapeutic targets for ARDS. The visualization of this subnetwork is shown in Fig. 4.

Protein–protein interaction (PPI). (A) Top 10 genes with the highest interaction degrees in PPInetwork analysis; (B) Cluster 1; (C) Hub genes.
Identify drug candidates
Through the use of the DGIdb online database, we were able to identify drugs that are potentially relevant for the genes PSMB8 and PSMB9. However, we were unable to find any relevant drugs for the other hub genes. Table 3 displays the results of the drug prediction analysis for PSMB8 and PSMB9.
Discussion and conclusions
In recent years, significant progress has been made in the research of ALI/ARDS, including epidemiology, pathogenesis, and pathophysiology. Studies on optimizing mechanical ventilation modes and fluid management have also brought benefits to clinical treatment. However, specific and effective therapeutic drugs for ALI/ARDS have not yet been identified23. With the rapid development of modern biotechnology, bioinformatics has gained more attention as researchers explore therapeutic options for ALI/ARDS at the molecular and cellular levels24. Numerous studies have shown that multiple pathogenic factors induce gene alterations during ALI/ARDS25. In this study, 78 co-expressed DEGs were screened from three ATII single-cell RNA sequencing datasets of ALI mouse models induced by three different pathogens. Finally, eight hub genes, including IRF7, IFIT1, IFIT3, PSMB8, PSMB9, BST2, OASL2, and ZBP1, were identified using bioinformatics methods. These hub genes are closely related to ATII injury during ALI, as indicated by protein network analysis.
A literature review shows that IRF7, IFIT1, IFIT3, BST2, and OASL2 are related to the immune response of the body. During lung injury, these genes are induced by interferon and participate in the IRF3–IFNAR–STAT1–IFIT1 signal pathway of pulmonary epithelial cells. They work together to regulate the activation of inflammatory cells and induce the death of infected cells26. The expression of IRF7 is increased by viral infection, tumor necrosis factor-α (TNF-α), and the inflammatory cytokine IL-1β. IRF7 also induces plasmacytoid dendritic cells and monocytes to produce the inflammatory cytokine IL-6, which participates in the occurrence and development of ALI/ARDS27. Proteomic studies of bronchoalveolar lavage fluid and ATII cells in ALI mouse models confirmed the important role of IRF726. Excessive activation of IRF7 promotes the development of ALI/ARDS caused by Influenza A Avirus (IAV), and reducing IRF7 activity at local infection sites may be a new method to treat ALI/ARDS in IAV28. Studies have shown that IFN-induced protein with tetrapeptide repeats 3 (IFIT3) protects against lung injury caused by viral infection29,30. IFIT1 and IFIT3 induced by interferon can be used as relevant marker proteins of M1 polarization of pulmonary macrophages, and a useful marker of potential inflammatory diseases31. Bone marrow stromal cell antigen 2 (BST2) activates the NF-κB signal pathway, promoting the production of proinflammatory factors such as TNF-α, IL-1β, and IL-632. As a member of the 2′-5′ oligoadenylate synthetase (OAS) family, OASL2 encodes an important antiviral protein and promotes the cleavage of viruses or infected cells33. At present, the role of OASL2 in ALI/ARDS has not been reported. Our results show that the expression of OASL2 is increased in different ALI models, suggesting that OASL2 may be a potential target worth further exploration in ALI/ARDS.
Proteasome subunit beta type-8 (PSMB8) and Proteasome subunit beta type-9 (PSMB9) are mainly expressed in monocytes and T lymphocytes, encoding proteasome β subunit. They are responsible for the degradation of proteasome after ubiquitination, promoting abnormal proliferation and anti-apoptosis of cancer cells34. The expression of PSMB8 and PSMB9 is significantly down-regulated in tumors35, but in inflammatory diseases, PSMB8 is highly expressed. By recognizing and degrading pathway repressor proteins in the NF-κB pathway, PSMB8 can promote the release of inflammatory mediators36. Selective inhibitors of PSMB8 can block and reduce the inflammatory reaction37. In the study of IAV-induced lung epithelial cell injury, researchers found that PSMB8 gene was up-regulated and inhibition of PSMB8 reduced the replication of influenza virus and attenuated lung epithelial injury38. Similarly, when analyzing the lung tissue and single-cell transcriptome results of patients with COVID19 infection, the results also showed that the expression of PSMB8 and PSMB9 was increased and related to the polarization of pulmonary macrophages39. Our results, together with others, indicate that PSMB8 and PSMB9 may play an important role in ALI/ARDS, providing a new target for treatment.
Z-DNA binding protein 1 (ZBP1), mostly expressed in CD8 + T lymphocytes, plays an important role in immune defense40. Previous reports revealed that ZBP1 could regulate the activation of the Nod-like receptor with pyrin domain-containing 3 inflammasome (NLRP3) through the RIPK3-caspase-8 axis, and promote the secretion of IL-1β and interleukin-18 (IL-18). Moreover, it could stimulate the apoptosis of necrotic cells at the infection site through pyroptosis41. Additionally, in an IAV-induced lung injury model of mice, ZBP1 could activate the NF-κB signaling and promote pro-inflammatory cytokines, resulting in the formation of neutrophil extracellular traps42. Our results showed that ZBP1 was highly expressed in three different ALI models, suggesting a prominent role in ALI/ARDS. However, the particular role and mechanism of ZBP1 still need further study.
Analyzing the final hub genes, we found that they all play an important role in regulating the immune system, which provides new ideas for the treatment of ALI/ARDS. Through online drug prediction, we obtained drugs primarily targeting genes PSMB8 and PSMB9, which are proteasome inhibitors mainly used in tumors and immune-related diseases43. An experiment demonstrated that bortezomib could improve lung function in an acute pancreatitis model of mice and reduce other complications44. Studies of other drugs are mainly used for tumor diseases such as hematological malignancies45, glioblastoma46, etc. Currently, there are no relevant reports on ALI/ARDS, so further research is needed in this field.
In summary, using bioinformatics methods, we screened and analyzed the common characteristics of differentially expressed genes of ATII in ALI/ARDS caused by different pathogens. The results of this study provide a basis for further exploring the pathogenesis, prognosis evaluation, and new drug targets of ALI/ARDS. The predicted related drugs need to be further investigated in animal experiments and clinical studies.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article.
References
Huppert, L. A., Matthay, M. A. & Ware, L. B. Pathogenesis of acute respiratory distress syndrome. Semin. Respir. Crit. Care Med. 40(1), 31–39 (2019).
Qingchun, L. et al. Validation of novel hub genes and molecular mechanisms in acute lung injury using an integrative bioinformatics approach. Acta Biochim. Biophys. Sin. 53(3), 342–353 (2021).
Sinha, P. & Bos, L. D. Pathophysiology of the acute respiratory distress syndrome: Insights from clinical studies. Crit. Care Clin. 37(4), 795–815 (2021).
Liu, Y. et al. Hypoxia-inducible factor-1: A potential target to treat acute lung injury. Oxid. Med. Cell Longev. 2020, 8871476 (2020).
Seddigh, P. et al. Quantitative analysis of proteome modulations in alveolar epithelial type II cells in response to pulmonary aspergillus fumigatus infection. Mol. Cell Proteom. 16(12), 2184–2198 (2017).
Yang, Q. et al. Resolvin conjugates in tissue regeneration 1 promote alveolar fluid clearance by activating alveolar epithelial sodium channels and Na, K-ATPase in lipopolysaccharide-induced acute lung injury. J. Pharmacol. Exp. Ther. 379(2), 156–165 (2021).
Aspal, M. & Zemans, R. L. Mechanisms of ATII-to-ATI cell differentiation during lung regeneration. Int. J. Mol. Sci. 21, 9 (2020).
Frye, M. et al. Interfering with VE- PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J. Exp. Med. 212, 2267–2287 (2015).
Kumar, Y. et al. Molecular analysis of serum and bronchoalveolar lavage in a mouse model of influenza reveals markers of disease severity that can be clinically useful in humans. PLoS One. 9, 2 (2014).
Reilly, J. P. et al. Low to moderate air pollutant exposure and acute respiratory distress syndrome after severe trauma. Am. J. Respir. Crit. Care Med. 199, 62–70 (2018).
Qu, L. et al. Caveolin-1 identified as a key mediator of acute lung injury using bioinformatics and functional research. Cell Death Dis. 13(8), 686 (2022).
Liang, Q. et al. Validation of novel hub genes and molecular mechanisms in acute lung injury using an integrative bioinformatics approach. Acta Biochim. Biophys. Sin. (Shanghai) 53(3), 342–353 (2021).
Hosseini, A. et al. Innate and adaptive immune responses against coronavirus. Biomed. Pharmacother. 132, 110859 (2020).
Jing, H., Chen, X. & Wang, D. Identification of biomarkers associated with diagnosis of acute lung injury based on bioinformatics and machine learning. Med. (Baltim.) 102(33), e34840 (2023).
Liu, X., Lou, L. & Zhou, L. Molecular mechanisms of cardiac injury associated with myocardial SARS-CoV-2 infection. Front. Cardiovasc. Med. 8, 643958 (2022).
Szklarczyk, D. et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47(D1), D607–D613 (2019).
Chin, C. H. et al. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8(Suppl 4), S11 (2014).
Cotto, K. C. et al. DGIdb 3.0: A redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 46(D1), D1068–D1073 (2018).
Freshour, S. L. et al. Integration of the drug-gene interaction database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 49(D1), D1144–D1151 (2021).
The ARDS Definition Task Force. Acute respiratory distress syndrome: The Berlin definition. JAMA 307, 23 (2012).
Pan, W. Z., Du, J., Zhang, L. Y. & Ma, J. H. The roles of NF-kB in the development of lung injury after one-lung ventilation. Eur. Rev. Med. Pharmacol. Sci. 22(21), 7414–7422 (2018).
Ragab, D., Salah Eldin, H., Taeimah, M., Khattab, R. & Salem, R. The COVID-19 cytokine storm; what we know so far. Front. Immunol. 16(11), 1446 (2020).
Wang, G. et al. Parp-1 inhibitor attenuates lps-induced acute lung injury through inhibiting nf-kappab-mediated inflammatory response. PloS One 8(11), e79757 (2013).
Liu, D. et al. Proteomic analysis of lung tissue in a rat acute lung injury model: Identification of prdx1 as a promoter of inflammation. Mediat. Inflamm. 4, 69358–69365 (2014).
Confalonieri, M., Salton, F. & Fabiano, F. Acute respiratory distress syndrome. Eur. Respir. Rev. 26(144), 160116 (2017).
Yap, G. L. R. & Sachaphibulkij, K. Annexin-A1 promotes RIG-I-dependent signaling and apoptosis via regulation of the IRF3-IFNAR-STAT1-IFIT1 pathway in A549 lung epithelial cells. Cell Death Dis. 11(6), 463 (2020).
Ning, S., Pagano, J. S. & Barber, G. N. IRF7: Activation, regulation, modification and function. Genes Immun. 12, 399–414 (2011).
Lei, Y. et al. Attenuation of interferon regulatory factor 7 activity in local infectious sites of trachea and lung for preventing the development of acute lung injury caused by influenza A virus. Immunology 157(1), 37–51 (2019).
Hsu, Y. L., Shi, S. F., Wu, W. L., Ho, L. J. & Lai, J. H. Protective roles of interferon-induced protein with tetratricopeptide repeats 3 (IFIT3) in dengue virus infection of human lung epithelial cells. PLoS One 8(11), e79518 (2013).
Ternette, N., Wright, C., Kramer, H. B., Altun, M. & Kessler, B. M. Label-free quantitative proteomics reveals regulation of interferon-induced protein with tetratricopeptide repeats 3 (IFIT3) and 5’-3’-exoribonuclease 2 (XRN2) during respiratory syncytial virus infection. Virol. J. 8(1), 442 (2011).
Cheng, H. et al. Proteomic identification of interferon-induced proteins with tetratricopeptide repeats as markers of M1 macrophage polarization. J. Proteome Res. 17(4), 1485–1499 (2018).
Rui, P. G. et al. Retroviral retention activates a Syk-dependent HemITAM in human tetherin. Cell Host Microbe 16(3), 291–303 (2014).
Ghosh, A. et al. Oligoadenylate-synthetase-family protein OASL inhibits activity of the DNA Sensor cGAS during DNA virus infection to limit interferon production. Immunity 50(1), 51–63 (2019).
Shen, M., Schmitt, S., Buac, D. & Dou, Q. P. Targeting the ubiquitin-proteasome system for cancer therapy. Expert Opin. Ther. Targets 17, 1091–1108 (2013).
Guo, J. Y., Jing, Z. Q., Li, X. J. & Liu, L. Y. Bioinformatic analysis identifying PSMB 1/2/3/4/6/8/9/10 as prognostic indicators in clear cell renal cell carcinoma. Int. J. Med. Sci. 19(5), 796–812 (2022).
Basler, M. et al. Co-inhibition of immunoproteasome subunits LMP2 and LMP7 is required to block autoimmunity. EMBO Rep. 19(12), e46512 (2018).
Vachharajani, N. et al. Prevention of colitis-associated cancer by selective targeting of immunoproteasome subunit LMP7. Oncotarget 8(31), 50447–50459 (2017).
More, S. et al. Long non-coding RNA PSMB8-AS1 regulates influenza virus replication. RNA Biol. 16(3), 340–353 (2019).
Desterke, C., Turhan, A. G., Bennaceur-Griscelli, A. & Griscelli, F. HLA-dependent heterogeneity and macrophage immunoproteasome activation during lung COVID-19 disease. J. Transl. Med. 19(1), 290 (2021).
Xuefeng, Z. et al. ZBP1(DAI/DLM-1) promotes osteogenic differentiation while inhibiting adipogenic differentiation in mesenchymal stem cells through a positive feedback loop of Wnt/β-catenin signaling. Bone Res. 8(2), 219–228 (2020).
Kesavardhana, S. et al. ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J. Exp. Med. 214, 12 (2017).
Momota, M. et al. ZBP1 governs the inflammasome-independent IL-1α and neutrophil inflammation that play a dual role in anti-influenza virus immunity. Int. Immunol. 32, 3 (2020).
Salvini, M. et al. Pharmacokinetic drug evaluation of ixazomib citrate for the treatment of multiple myeloma. Expert Opin. Drug Metabol. Toxicol. 14(1), 91–99 (2018).
Zhu, Q. et al. Dynamic changes of proteasome and protective effect of bortezomib, a proteasome inhibitor, in mice with acute pancreatitis. Biochem. Biophys. Res. Commun. 505(1), 126–133 (2018).
Clemens, J. et al. Bortezomib, carfilzomib and ixazomib do not mediate relevant transporter-based drug-drug interactions. Oncology. 14(3), 3185–3192 (2017).
Wen, P. Y. et al. Glioblastoma in adults: A Society for Neuro-Oncology(SNO) and European Society of Neuro-Oncology(EANO)consensus review on current management and future directions. Neuro Oncol. 22(8), 1073–1113 (2020).
Funding
This work was supported by National Natural Science Foundation of China (No. 81571882).
Author information
Authors and Affiliations
Contributions
W.L. designed the study, X.Y. and J.W. performed experiments and analyzed data. The manuscript was drafted and edited by X.Y. and W.L. All authors approved the submitted work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, X., Wang, J. & Liu, W. Molecular markers of type II alveolar epithelial cells in acute lung injury by bioinformatics analysis. Sci Rep 13, 17797 (2023). https://doi.org/10.1038/s41598-023-45129-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-45129-9
This article is cited by
-
Highly Expressed Z-DNA Binding Protein 1 in Esophageal Cancer Promotes Tumor Growth
Digestive Diseases and Sciences (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
