
Abstract
N6-methyladenosine (m6A) is a form of posttranscriptional modification that plays important roles in cancer including oral squamous cell carcinoma (OSCC). Most studies to date have focused on a limited number of regulators and oncogenic pathways, thus failing to provide comprehensive insight into the dynamic effects of m6A modification. In addition, the role of m6A modification in shaping immune cell infiltration in OSCC has yet to be clarified. This study was designed to assess m6A modification dynamics in OSCC and to understand how such modifications influence clinical immunotherapeutic treatment outcomes. m6A modification patterns linked with 23 m6A regulators were analyzed in 437 OSCC patients from TCGA and GEO cohorts. These patterns were then quantified through m6A score based on algorithms derived from a principal component analysis (PCA) approach. The m6A modification patterns of OSCC samples were grouped into two clusters based on the m6A regulators expression, and immune cell infiltration was linked with the 5-year survival outcomes of patients in these clusters. 1575 genes associated with OSCC patient prognosis were identified and used to re-cluster these samples into two groups. Patients in clusters exhibiting higher levels of m6A regulator expression exhibited poorer overall survival (OS), whereas patients with high m6A scores survived for longer (p < 0.001). The overall mortality rates in the groups of patients with low and high m6A scores were 55% and 40%, respectively, and the m6A score distributions in clusters of patients grouped by m6A modification patterns and gene expression further supported the link between a high m6A score and better prognostic outcomes. Immunophenoscore (IPS) values for patients in different m6A score groups suggested that the use of PD-1-specific antibodies or CTLA-4 inhibitors alone or in combination would yield superior treatment outcomes in patients in the high-m6A score group relative to the low-m6A score group. m6A modification patterns are relevant to heterogeneity in OSCC. Detailed analyses of m6A modification patterns may thus offer novel insight regarding immune cell infiltration within the OSCC tumor microenvironment, guiding novel efforts to provide patients with more effective immunotherapeutic interventions.
Similar content being viewed by others

Introduction
Oral squamous cell carcinoma (OSCC) accounts for over 90% of all oral malignancies, and the 5-year overall survival (OS) rate for affected patients is just 60%1. While there have been advances in the diagnosis and treatment of OSCC patients in recent years, this has failed to translate to a pronounced increase in OS, with tumor immune evasion playing an important role in poor patient outcomes2. The advent of immune checkpoint inhibitors (ICIs) has offered a new opportunity to treat OSCC, with anti-PD-1/PD-L1 immunotherapeutic regimens having demonstrated efficacy in advanced head and neck squamous cell carcinoma cases3. However, only an estimated 20–40% of patients ultimately benefit from ICI administration4. Treating OSCC thus remains a difficult clinical challenge, and the identification of reliable biomarkers associated with patient responses to immunotherapeutic treatment and prognostic outcomes has the opportunity to reduce the morbidity of OSCC by providing patients with more personalized and effective pharmacological tools.
The N6-methyladenosine (m6A) modification of mRNA is the most common posttranscriptional modification in eukaryotic cells, shaping a range of physiological and pathogenic processes5. A diverse array of methyltransferases, demethylases, and m6A binding proteins (respectively known as “writers”, “erasers”, and “readers) shape the m6A methylation landscape in a dynamic manner. When these m6A regulatory proteins are dysregulated, this can alter the expression of oncogenes in a manner that may ultimately be conducive to oncogenic transformation6. For example, in tumors from acute myeloid leukemia (AML) patients, significant increases in the expression of METTL3, METTL14, and YTHDF2 have been reported7. In lung adenocarcinoma, METTL3 can promote the enhanced translation of oncogenic factors including BRD4, EGFR, and TAZ in cooperation with EIF38,9. Other m6A regulators that have been studied to date include IGF2BP1, YTHDF1, YTHDF2, and FTO6. High-level alterations in global m6A abundance have been linked to tumor progression, metastasis, chemoresistance, and recurrence10. However, a majority of studies conducted to date have only focused on a limited number of m6A regulators and oncogenic pathways without fully exploring the complex and dynamic m6A modification landscape, thus failing to provide a comprehensive overview of how this form of posttranscriptional modification shapes pro-tumorigenic processes. There have also been several reports that m6A modification plays a role in shaping the composition of the tumor-associated immune microenvironment and related immune response induction11,12, although the precise mechanisms underlying such activity are poorly understood, particularly in OSCC. As such, further detailed studies of a variety of m6A regulatory proteins may offer new insight regarding the role that m6A modification plays in the pathogenesis of OSCC. These analyses may further enable the identification of distinct subgroups of OSCC patients with specific tumor characteristics and immunophenotypes, thereby supporting efforts to define novel biomarkers that can predict patient responses to immunotherapeutic treatment.
The present study was developed to conduct a comprehensive analysis of OSCC-related patterns of m6A modification by integrating data from the Cancer Genome Atlas (TCGA) and Gene-Expression Omnibus (GEO) databases. Together, these analyses offered new insight into the dynamic landscape of m6A modification patterns and their relationship with intratumoral immune cell infiltration. In addition, these results were used to establish a scoring system to enable the quantification of m6A methylation levels in individual patient samples based on patterns of differentially expressed genes (DEGs) under different patterns. Immunophenoscore (IPS) values were further leveraged to guide the selection of appropriate immunotherapeutic interventions.
Results
Patterns of m6A regulator gene expression in OSCC
An initial analysis was used to explore patterns of m6A regulator gene expression in OSCC patients and normal tissue samples (Fig. 1A), revealing that all of these genes were expressed at higher levels in OSCC tumor tissue samples consistent with the enhancement of m6A modification activity in OSCC. These differences were significant for all m6A regulator genes other than RBM15B and YTHDC2. The m6A regulator that exhibited the lowest levels of expression was IGF2BP1, whereas HNRNPA2B1 expression levels were highest. Next, the mutational status of these m6A regulators was assessed in OSCC samples, revealing that 65/506 samples (12.85%) harbored mutations in m6A regulator genes (Fig. 1B). These mutations were spread across 13 m6A regulator genes, of which LRPPRC exhibited the highest mutational frequency. Patients harboring LRPPRC mutations also expressed higher levels of IGF2BP2 (Fig. 1C). These mutation and expression data are consistent with a model wherein interactions among m6A regulators, rather than individual regulatory proteins, ultimately shape the development and pathogenesis of OSCC such that combination treatment strategies will be critical to effectively remediate dysregulated m6A modification patterns in this oncogenic context. Copy number variations (CNVs) were also observed for all 23 m6A regulator genes in this patient cohort (Fig. 1D). Copy number amplifications were the primary CNVs observed for 11 of these genes (IGF2BP2, FMR1, YTHDC1, RBMX, VIRMA, YTHDF1, METTL14, LRPPRC, IGF2BP3, IGF2BP1, and ALKBH5), whereas the remainder primarily presented with copy number losses. The locations of these m6A CNVs were also noted on specific chromosomes (Fig. 1E). Together, these results suggest that OSCC tumors exhibit increased m6A regulator gene expression relative to normal tissues, and that mutations in these m6A regulator genes are a common finding in OSCC consistent with the important role that m6A methylation modification plays as a driver of tumor malignancy and progression.

Landscape of genetic and expression variation of m6A regulators in OSCC. (A) Expression of 23 m6A regulator genes between normal tissues and OSCC tissues. Tumor, red; Normal, blue. Top edge of box: upper quartile; bottom edge of box: lower quartile; Internal horizontal line: median. Dots above and below: outliers. *p < 0.05; **p < 0.01; ***p < 0.001. (B) Mutation frequency of 23 m6A regulator genes in 506 OSCC samples from TCGA database. Each column represented a sample. The upper barplot showed TMB; the lower barplot showed conversion fraction in each sample; the right barplot showed the proportion of each variant type. Number on the right represented the mutation frequency of each regulator gene. (C) Differential expression of IGF2BP2 in LRPPRC wild and LRPPRC mutant samples. LRPPRC mutant, red; LRPPRC wild, blue. Top edge of box: upper quartile; bottom edge of box: lower quartile; Internal horizontal line: median. Dots above and below: outliers. The upper number represented p value. (D) The CNV frequency of 23 m6A regulator genes in TCGA-HNSC cohort. The height of the column represented the CNV frequency. The deletion frequency, blue dot; the amplification frequency, red dot. (E) The CNV location of 23 m6A regulator genes on chromosomes.
Analyses of the prognostic relevance of individual m6A regulators
To gain further insight into the association between m6A regulator genes and patient outcomes, survival outcome data were extracted after pooling the TCGA and GSE41613 datasets13. As the METTL16 and RBMX genes were not included in the GEO dataset, only the remaining 21 m6A genes were subject to subsequent analyses. Univariate Cox regression models (Table 1) indicated that RBM15, HNRNPC, LRPPRC, HNRNPA2B1, IGF2BP2, and ALKBH5 were associated with a high risk of worse patient survival (p < 0.05), while only two genes (RBM15B, YTHDC2) were associated with lower risk. Patients were then stratified into two groups based on whether they expressed high or low levels of each of these genes, after which Kaplan–Meier OS curves were generated. Of the 15 m6A regulator genes associated with patient OS in these analyses, HNRNPA2B1, IGF2BP2, IGF2BP3, LPPPRC, VIRMA, and ZC3H13 were found to be associated with poor prognostic outcomes (Fig. 2A, Fig. S1). To more fully explore the interconnected relationships among these genes and the prognostic relevance of these m6A regulators, a network diagram was constructed based on correlative relationships (p < 0.0001) (Fig. 2B). The expression of m6A regulators included in the same functional category was found to be positively correlated, and positive correlations were also observed among these m6A writers, readers, and erasers. These data thus indicate that m6A regulators can be used to predict prognostic outcomes in individuals with OSCC, with interactions among these three classes of regulators potentially playing a critical role in key oncogenic processes.

Correlation of m6A regulators with prognosis. (A) Kaplan–Meier survival analysis of m6A regulators between the high-expression group and the low-expression group using clinical information of OSCC patients in TCGA and GSE41613 cohort. High-expression, red curve; Low-expression, blue curve. P value less than 0.05 was statistically significant. (B) The interaction between m6A regulators in OSCC. Writers, readers and erasers were marked with red, orange and gray, respectively. The circle size represented the effect of each regulator on prognosis, and the range of values calculated by Cox test was p < 1e−04, p < 0.001, p < 0.01, p < 0.05 and p < 1, respectively. Green in the circle, favorable factors of prognosis; Purple in the circle, risk factors of prognosis. Curves linking regulators showed their interactions, with thickness showing the correlation strength. Positive correlation with p < 0.0001, pink curve; Negative correlation with p < 0.0001, blue curve. The figure was generated using R software (V 4.1.2, https://www.r-project.org/).
m6A methylation modification pattern characteristics
Next, the R “ConsensusCluster” package was used to classify patient tumor samples into two clusters based on the patterns of m6A regulator gene expression in these samples (Fig. 3A). PCA scatter plots revealed that samples within each of these clusters, designated cluster A and cluster B, were effectively grouped together (Fig. 3B). A heatmap analysis revealed that patterns of m6A regulator gene expression were markedly increased in cluster B (Fig. 3C). A non-significant trend towards better 5-year OS was observed in cluster A relative to cluster B (Fig. S2). These results were consistent with the ability of m6A regulators including VIRMA, ZC3H13, HNRNPA2B1, IGF2BP2, IGF2BP3, and LPPPRC to regulate mRNA m6A methylation patterns in OSCC in a manner that favors cancer progression. A GSVA approach was further used to explore differences in biological activity in these two sample clusters (Fig. 3D). Samples in cluster A exhibited pronounced metabolic pathway enrichment, whereas samples in cluster B were enriched for pathways associated with proliferation and DNA repair including the nonhomologous end joining, homologous recombination and mismatch repair, cell cycle, DNA replication, and aminoacyl tRNA biosynthesis pathways. These results further emphasize the differences in the biological characteristics of tumor samples from patients in these two m6A regulator-based subgroups. Intratumoral immune cell infiltration was next examined in these two clusters (Fig. 3E), revealing significant enrichment for various immune cell types in samples from cluster A, consistent with the better 5-year survival outcomes in this group that may be indicative of more robust antitumor immunity. Based on this combination of immune cell infiltration and prognostic results, cluster A was designated an immune-inflammatory phenotype, while cluster B was designated an immune-desert phenotype. These results further support the ability of m6A methylation modification patterns to impact antitumor immune responses.

Characteristics of m6A methylation modification patterns. (A) Consensus clustering analysis of m6A regulators. Consensus clustering matrix for k = 2. Cluster A, 1; Cluster B, 2. (B) PCA analysis of m6A regulators. Samples in cluster A, blue dots; Samples in cluster B, yellow dots. (C) Unsupervised clustering of m6A regulators in OSCC patients from TCGA and GSE41613 cohort. Fustat, gender, age, project and m6Acluster were used as patient annotations. High expression, red; low expression, blue. (D) The heatmap of GSVA showing KEGG pathways in each m6A modification patterns. Red represented activated pathways and blue represented inhibited pathways. KEGG pathways14,15,16 were downloaded from the website (http://www.gsea-msigdb.org/). (E) ssGSEA of immune cells infiltration in individual m6A modification patterns. Cluster A, bule; Cluster B, yellow. Top edge of box: upper quartile; bottom edge of box: lower quartile; Internal horizontal line: median. Dots above and below: outliers. *p < 0.05; **p < 0.01; ***p < 0.001; ns no statistical significance.
Identification of prognosis-related DEGs
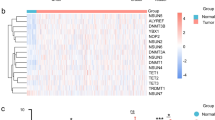
Next, DEGs associated with m6A modification patterns were identified, yielding 7075 total genes. GO enrichment analyses of these DEGs were then performed for three major categories of GO terms (Fig. 4A), and KEGG enrichment analyses revealed these genes to be associated with oncogenesis and infection-related pathways (Fig. 4B). Univariate Cox regression analysis screening then identified 1575 prognosis-related genes, of which 4% (64/1575) were associated with lower risk, while the remainder were associated with increased risk (p < 0.05). The top 10 low- and high-risk genes are presented in Table S1. These 1575 genes were then used to repeat the clustering of OSCC patient samples into two clusters (gene cluster A and gene cluster B) (Fig. 4C). Heatmap analyses indicated that these two gene clusters exhibited distinct gene expression signatures (Fig. 4D). Kaplan–Meier analyses revealed that patients in gene cluster B exhibited increased OS as compared to patients in gene cluster A (p < 0.001) (Fig. 4E). Patients in gene cluster A also exhibited significant increases in the expression of all m6A regulator genes (Fig. 4F). These data confirmed that m6A methylation modifications are associated with antitumor immunity.

DEGs associated with prognosis. (A) Functional annotation of DEGs by GO enrichment analysis. The color depth of the barplot represented q value and the length of the barplot represented number of enriched genes. (B) Functional annotation of DEGs by KEGG enrichment analysis. The color depth of the barplot represented q value and the length of the barplot represented number of enriched genes. (C) Consensus clustering analysis of prognosis related genes. Consensus clustering matrix for k = 2. Gene cluster A, 1; Gene cluster B, 2. (D) Unsupervised clustering of prognosis related genes in OSCC patients from TCGA and GSE41613 cohort. Fustat, gender, age, project, m6Acluster and gene cluster were used as patient annotations. High expression, red; low expression, blue. (E) Kaplan–Meier survival analysis of OSCC patients in different gene clusters. Gene cluster A, blue curve; Gene cluster B, yellow curve. Log-rank p < 0.001 showed a significant survival difference between two gene clusters. (F) Differential expression of m6A regulators between gene cluster A and gene cluster B. Gene cluster A, blue; Gene cluster B, yellow. Top edge of box: upper quartile; bottom edge of box: lower quartile; internal horizontal line: median. Dots above and below: outliers. *p < 0.05; **p < 0.01; ***p < 0.001.
Higher m6A score values are linked with better prognostic outcomes
To better classify OSCC patients and to mitigate heterogeneity among these patients, a scoring system was next established to quantify patterns of m6A methylation termed the m6A score, which was based on these 1575 prognosis-associated genes. An optimal m6A score cutoff was established with the “survminer” package, with patients then being stratified into high- and low-m6A score groups. Individuals in the high-m6A score group exhibited significantly better survival than patients in the low-m6A score group (p < 0.001) (Fig. 5A). Box-plot analyses revealed that the median m6A score values were higher in living patients than in deceased patients (Fig. 5B). The overall mortality rates in the low- and high-m6A score groups were 55% and 40%, respectively (Fig. 5C). These data indicate that higher m6A scores are related to better prognostic outcomes. Further analyses were conducted comparing correlations between OS, m6A score values, and clinical T stage (Fig. S3). No significant differences were observed for stages T1–2 or stages T3–4. Moreover, score distributions were calculated for patients in different clusters generated based on m6A methylation modification patterns and gene clusters, revealing that in both settings the patient clusters that exhibited higher survival rates also exhibited higher m6A scores (Fig. 5D,E). Sankey diagrams were further used to visualize these results (Fig. 5F). In general, patients in the high-m6A score group were primarily individuals from gene cluster B with better prognostic outcomes, as well as some patients in m6A cluster A with longer OS. As such, these findings indicate that m6A score can be used to predict OSCC patient clinical outcomes.

High m6A score associated with better prognosis. (A) Kaplan–Meier survival analysis of OSCC patients in different m6A score groups. Low-m6A score group, blue curve; High-m6A score group, red curve. Log-rank p < 0.001 showed a significant survival difference between two groups. (B) Differences in m6A score between alive and dead patients in TCGA and GSE41613 cohort. Alive, blue; Dead, red. Top edge of box: upper quartile; bottom edge of box: lower quartile; internal horizontal line: median. Dots above and below: outliers. The upper number represented p value. (C) The proportion of alive and dead patients in the low-m6A score group and the high-m6A score group. Alive, blue; Dead, red. Numbers in the barplots represented percentages. (D) Differences in m6A score between m6A clusters. Cluster A, blue; Cluster B, yellow. Top edge of box: upper quartile; bottom edge of box: lower quartile; internal horizontal line: median. Dots represented samples and the upper number represented p value. (E) Differences in m6A score between gene clusters. Gene cluster A, blue; Gene cluster B, yellow. Top edge of box: upper quartile; bottom edge of box: lower quartile; internal horizontal line: median. Dots represented samples and the upper number represented p value. (F) Sankey diagram showing the correlations among m6A clusters, gene clusters, m6A score and fustat.
The relationship between mutational burden, immunity, and tumor m6A modification
An increased tumor mutational burden (TMB) entails neoantigen production such that these antigens can be recognized by T cells, thereby promoting immune response activation17. Next, the OS of patients with differing TMB frequencies was compared (Fig. 6A). In contrast to literature-based expectations, the survival of patients in the low TMB group was actually higher than that of patients in the high TMB group. No significant differences in TMB were observed when comparing the low- and high-m6A score groups (Fig. S4). This suggests that there may not be any significant relationship between TMB and m6A modification with respect to OSCC patient outcomes. The R “maftools” package was then used to explore differences in somatic mutations between individuals in the low- and high-m6A score groups based on the ten most frequently mutated genes (Table S2). Similar mutational frequencies for these ten genes were observed in both groups (89.51% vs. 88.30%) (Fig. 6B,C), although a 1.3-fold higher TP53 mutation rate was observed in the low-m6A score group relative to the high-m6A score group (72% vs. 57%), and the CDKN2A mutation rate in the high-m6A score group was 1.7-fold higher than in the low m6A score group (25% vs. 15%). As such, while overall TMB was not related to patient prognosis, a relationship was observed between TP53 and CDKN2A mutation status and m6A modification-related differences in patient prognosis.

Mutation and immunity in tumor m6A modification patterns. (A) Kaplan–Meier survival analysis of OSCC patients in low TMB group and high TMB group. Low TMB group, blue curve; High TMB group, red curve. Log-rank p < 0.001 showed a significant survival difference between two groups. (B,C) TMB of top ten high-frequency mutated genes in 162 samples from low-m6Ascore group (B) and 171 samples from high-m6Ascore group (C). Each column represented a sample. The upper barplot showed TMB; the right barplot showed the proportion of each variant type. Numbers on the right represented the mutation frequency of genes. (D) Correlations between m6A score and immune cells infiltration using Spearman analysis. Negative correlation was marked with blue and positive correlation with red. *p < 0.05. (E) Correlations between m6A score and IPS. Low-m6A score group, blue; High-m6A score group, red. The upper number represented p value. p value less than 0.05 was statistically significant.
Correlations between m6A score and immune cell infiltration were next examined (Fig. 6D), revealing that 11 immune cell types were positively correlated with m6A score, whereas activated CD4+ T cells, NK T cells, NK cells, and Th2 cells were negatively correlated with m6A score. The expression of immune checkpoint genes was also examined (Fig. S5), but no significant differences in PD-L1 or CTLA-4 expression were observed. To explore the efficacy of immunotherapeutic treatment, patient IPS scores were examined (Fig. 6E). These scores suggested that individuals in the high-m6A score group would experience superior outcomes relative to patients in the low-m6A score group irrespective of whether they were treated with PD-1-specific antibodies, CTLA-4 inhibitors, or a combination of both of these ICIs. In addition, the IPS distributions in the high-m6A score group were more concentrated when patients were treated with PD-1-specific antibodies alone or in combination with CTLA-4 inhibitors as compared to CTLA-4 inhibitors alone, while the combination of PD-1-specific antibodies and CTLA-4 inhibitors was not more effective. These findings suggest that m6A score can predict OSCC patient immunotherapy responses as compared to traditional analyses of TMB, with patients with higher m6A scores being more sensitive to ICIs targeting PD-1 or CTLA-4 as compared to individuals with low m6A scores. However, individual heterogeneity was found to significantly impact the predicted efficacy of CTLA-4 inhibitor treatment.
Discussion
Many recent studies have examined the relationship between m6A modifications and cancer, underscoring the importance of such posttranscriptional modifications in OSCC. However, these prior studies have focused only on specific individual regulators such as METTL318,19,20, METTL1421, ALKBH522, FTO23,24, and HNRNPA2B125. How other m6A-related regulatory proteins shape the pathogenesis of OSCC remains to be established. In addition, the different classes of m6A readers, writers, and erasers do not function independently of one another, instead interacting in a dynamic manner to govern patterns of m6A methylation. To date, technological limitations have hampered efforts to comprehensively evaluate these different factors. As such, a bioinformatics approach was herein used to examine as many regulatory factors as possible with the goal of better understanding the importance of different m6A regulatory mechanisms in OSCC, thereby providing a foundation for future work.
In these analyses, RBM15, HNRNPC, LRPPRC, HNRNPA2B1, IGF2BP2, and ALKBH5 were identified as high-risk genes, whereas HNRNPA2B1, IGF2BP2, IGF2BP3, LPPPRC, VIRMA, and ZC3H13 were related to poorer prognostic outcomes, suggesting that HNRNPA2B1, IGF2BP2, and LPPPRC are particularly reliable predictors of negative patient outcomes. Higher HNRNPA2B1 levels in individuals with OSCC have previously been reported to be associated with poorer OS25. HNRNPA2B1 can also contribute to the progression of a range of solid and hematological tumor types. Jiang et al.26 further determined that HNRNPA2B1 was upregulated in myeloma and linked to poorer outcomes through its ability to stabilize the ILF3 mRNA and promote AKT3 upregulation. IGF2BP2 has also been reported to be related to patient outcomes in individuals with colorectal cancer27, hepatocellular carcinoma28, pancreatic cancer29, gastric cancer30, and OSCC31, while LPPPRC is an independent predictor of prognostic outcomes in many cancers. While these results support the present findings, research focused on HNRNPA2B1 and IGF2BP2 in OSCC has been limited, and no studies have assessed LPPPRC in this oncogenic context. As such, more work is needed to fully explore the prognostic relevance of these genes in OSCC.
Substantial heterogeneity is evident when comparing tumor cells among individuals with the same form of cancer, and this heterogeneity is also evident within tumors in a given patient32. Tumor complexity is often explained using a model outlining eight major hallmarks of cancer33, including the acquired capabilities for sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing/accessing vasculature, activating invasion and metastasis, reprogramming cellular metabolism, and avoiding immune destruction. In this study, OSCC patients were stratified into two groups based on patterns of m6A regulator expression, and the characteristics of patients in these clusters were assessed. While abundant immune cell infiltration was evident in cluster A together with the significant enrichment of metabolic pathways, cluster B exhibited a lack of immune cell infiltration together with enrichment for pathways associated with DNA repair and cellular proliferation. This suggests that high levels of m6A modification are related to certain hallmarks of cancer including resistance to cell death, supporting replicative immortality and the ability to evade immune-mediated destruction as observed for samples in m6A cluster B. While all OSCC tumors in this study are likely to exhibit these eight hallmarks of cancer to varying degrees, the actual associated phenotypes may vary among individuals, and m6A may shape these processes throughout the development and progression of OSCC given that the abilities to avoid apoptotic death and proliferate indefinitely may arise during different stages of oncogenesis. Different modification patterns may also impact prognostic outcomes given that individuals in gene cluster A exhibited the upregulation of all m6A regulator genes and such upregulation was related to poorer patient OS.
Univariate Cox regression analyses were further used to analyze DEGs identified by comparing these two patient clusters, focusing specifically on the ten highest and lowest risk genes. While their roles in OSCC remain to be studied in detail, some prior work does highlight potential mechanisms through which these genes may shape cancer patient outcomes. For example, in Ewing sarcoma, RING1B can repress SCN8A, thereby contributing to oncogenic progression in a manner independent of fusion oncoproteins34. Overexpression of CEACAM21, which is a member of the carcinoembryonic antigen family, has been detected in high-grade serous ovarian cancer in immune-activated tissues relative to immune-silent tissues35. SZT2 can suppress mTORC1 activity, thereby limiting the inhibitor effects of PI3Kα inhibitor and improving breast cancer-associated therapeutic efficacy36. In OSCC prognostic predictive models, NAGK has been identified as a low-risk gene37, while many high-risk genes have been studied in detail as oncogenes in other cancers38,39,40,41,42,43,44,45. Many of the top 10 low-risk genes identified herein have not been described previously in OSCC, and may thus offer novel predictive utility. In light of individual heterogeneity, m6A scores were utilized as a quantitative tool to compare patterns of m6A methylation among samples based on these prognostic genes. The resultant m6A scoring model has not previously been reported in OSCC, and was found to predict OSCC patient clinical outcomes such that higher m6A scores were linked to a better prognosis.
The majority of studies exploring m6A modification to date have analyzed oncogenic pathways without also examining the effects of such modifications on host antitumor immunity. However, a limited number of studies have examined the mechanisms through which specific m6A regulators can impact immune cell populations. For example, depleting METTL3 can enhance RIG-I-mediated innate immune responses to certain viruses46, while YTHDF2 is an m6A reader protein that regulates the maturation and homeostasis of NK cells, functioning as a positive regulator of antitumor immune responses47. YTHDF1 is capable of recognizing and enhancing the translation of m6A-modified lysosomal cathepsins in dendritic cells, thereby suppressing antigen cross-presentation and CD8+ T cell activation, ultimately inhibiting antitumor immunity48. In this study, significant differences in immune cell infiltration were evident when comparing patients in clusters A and B, and in KEGG analyses over 200 DEGs were enriched in the herpes simplex virus 1 infection pathway, supporting a link between differences in m6A modification and immune cell infiltration. High and low m6A score values were also related to different immune cell populations, emphasizing the important relationship between m6A modification and the composition of immune cells within the local microenvironmental landscape.
Important predictors of ICI responses identified to date include high levels of microsatellite instability, TMB, and the expression of CTLA-4 and PD-L1 by tumor cells49. The m6A scores generated herein were also identified as predictors of immunotherapeutic efficacy, with individuals in the high-m6A score group exhibiting greater predicted sensitivity to treatment with CTLA-4 inhibitors and PD-1-specific antibodies as compared to individuals in the low m6A score group. ICI treatment can facilitate tumor clearance by activating T cells and enabling them to more effectively recognize and respond to tumor-associated antigens. Higher TMB levels are associated with the generation of more neoantigens, thus providing these T cells with more opportunities to detect tumor cells, thus contributing to enhanced ICI efficacy17. In this study, however, patients with low TMB exhibited better OS outcomes, potentially because they did not undergo immunotherapeutic treatment. In patients not administered ICIs, higher TMB is associated with a poorer prognosis in many cancers50. While these neoantigens can aid in the establishment of spontaneous antitumor immunity, these immune responses are generally not sufficient or durable enough to fully eliminate tumors48. As such, even though no significant differences in TMB were observed in the two groups in this study, ICI treatment responses may ultimately be shaped by differences in immune cell infiltration within the local tumor microenvironment in the context of different patterns of m6A modification. The overall mutation rates in frequently mutated genes were largely similar irrespective of patient m6A scores in this study, although mutational frequencies for TP53 and CDKN2A differed significantly between patients with low and high m6A scores. TP53 encodes the p53 tumor suppressor protein, and its expression is subject to control mediated by both promoter methylation and post-transcriptional m6A mRNA modification such that the silencing of m6A methyltransferases can significantly dysregulate p53 signaling activity through changes in gene expression and alternative splicing5. The mutation or deletion of TP53 is a major mechanism by which tumors evade immune detection through alterations in the local immune microenvironment via increases in the recruitment of regulatory T cells and the downregulation of MHC-I presentation51. How CDKN2A mutations influence ICI responses in different tumors remains somewhat controversial. In renal cell carcinoma patients, for example, mutations in CDKN2A have been linked to reductions in ICI-related clinical benefit52, whereas they reportedly had no effect in melanoma53. No prior studies have explored the association between CDKN2A mutations and ICI responses in OSCC. As such, these results may suggest that TP53 mutations impact the immune microenvironment through mechanisms related to differences in m6A methylation patterns, thereby contributing to differences in how patients respond to ICI treatment. No significant differences in PD-L1 or CTLA-4 expression were observed between patient groups in this study, indicating that m6A score may be a more reliable predictor of immunotherapeutic efficacy. As the analyzed datasets did not provide information on microsatellite instability, the relationship between this parameter, m6A modification patterns, and OSCC patient outcomes warrants further study.
Conclusions
In summary, these data highlight the important role that m6A modifications play in shaping prognostic outcomes in individuals with OSCC. Differences in these m6A modification patterns are important contributors to the observed heterogeneity among patients, and further in-depth analyses of these m6A patterns in individual tumors may thus offer new insight into the pathogenesis of OSCC and associated immune cell infiltration within the local tumor microenvironment. These data also offer new insight with the potential to improve clinical responses to immunotherapeutic treatment through the identification of specific tumor immunophenotypes, thereby aiding in the personalized selection of appropriate checkpoint inhibitor regimens with the highest chances of achieving beneficial outcomes in a given patient.
Methods
OSCC dataset analyses
Transcriptomic mRNA expression data and corresponding clinical details and somatic mutation data for all OSCC patients included in the TCGA database (https://portal.gdc.cancer.gov/) were downloaded. Diseases included in this dataset included tumors of “other and ill-defined sites in lip, oral cavity and pharynx”, “oropharynx”, “other and unspecified parts of tongue”, “floor of mouth”, “other and unspecified parts of mouth”, “base of tongue”, “gum”, “lip” and “palate”. In total, transcriptomic data in the FPKM (fragments per kilobase per million) format from 340 cancer patients and 32 normal control patients were obtained and transformed into the TPM (transcripts per kilobase million) format using the R “limma” package for targets with known gene symbols. The GSE41613 dataset from the GEO database (https://www.ncbi.nlm.nih.gov/geo/) was also selected for analysis as it contained enough OSCC patients. Clinical information for patients with OSCC in the TCGA and GSE41613 cohorts were combined for univariate Cox analyses and Kaplan–Meier survival analyses, with samples lacking corresponding being excluded from these analyses. Gene-level copy number data for tumor patients were obtained from the following source: https://xena.ucsc.edu/.
m6A regulator-based consensus clustering
After combining data for patients in the selected TCGA and GEO cohorts, the R “ConsensusClusterPlus” package was used to group samples into two m6A modification pattern-based clusters according to m6A expression levels. In total this clustering was performed based on the expression of 23 m6A regulators, including eight writers (METTL3, METTL14, METTL16, RBM15, RBM15B, WTAP, VIRMA, and ZC3H13), two erasers (ALKBH5 and FTO) and 13 readers (YTHDC1, YTHDC2, YTHDF1, YTHDF2, YTHDF3, IGF2BP1, IGF2BP2, IGF2BP3, HNRNPA2B1, HNRNPC, FMR1, LRPPRC, and RBMX).
Gene set variation analysis (GSVA)
Differences in biological activity in these different m6A OSCC patient clusters were explored based on a GSVA enrichment analysis approach performed by the R “GSVA” package and the KEGG pathway set (“c2.cp.kegg.v7.5.1.symbols”) downloaded from http://www.gsea-msigdb.org/. An adjusted p < 0.05 was the threshold for significance.
Single-sample gene-set enrichment analysis (ssGSEA)
Immune cell infiltration in tumor tissue samples for patients in different m6A clusters was explored through a ssGSEA approach. Markers for 23 types of immune cells were analyzed based on prior literature evidence54. Enrichment scores were thus generated corresponding to the relative abundance of different populations of infiltrating immune cells.
Functional enrichment analyses
DEGs were identified based on the intersection of gene expression datasets in the two defined m6A clusters using the R “limma” package, with p < 0.001 as the significance threshold. GO and KEGG functional enrichment analyses of these DEGs were then performed, with p < 0.05 as the significance threshold.
Prognostic m6A-related scoring model development
An m6A-related scoring model was developed as a tool to predict OSCC patient prognostic outcomes54. Initially, samples were stratified into multiple groups based on DEGs expression patterns using an unsupervised clustering approach. Univariate Cox regression analyses were then used to identify genes significantly associated with patient prognosis (p < 0.05). A principal component analysis (PCA) was then used to extract two principal components (PC1 and PC2) as independent variables from these m6A clusters. The m6A scoring formula was then developed as follows: m6Ascore = ∑(PC1i + PC2i), where “i” corresponds to significant prognostic genes.
Statistical analysis
All statistical analyses were performed using R (V 4.1.2, https://www.r-project.org/) packages downloaded from http://www.bioconductor.org.
Ethics approval and consent to participate
The patient data in this work were acquired from the publicly available datasets whose informed consent of patients were complete.
Data availability
All data used in this work can be acquired from the Gene-Expression Omnibus https://www.ncbi.nlm.nih.gov/geo/, the Cancer Genome Atlas https://portal.gdc.cancer.gov/ and https://xena.ucsc.edu/.
Abbreviations
- CNV:
-
Copy number variation
- DEGs:
-
Differentially expressed genes
- FPKM:
-
Fragments per kilobase per million
- GEO:
-
Gene-Expression Omnibus
- GSVA:
-
Gene set variation analysis
- ICIs:
-
Immune checkpoint inhibitors
- IPS:
-
Immunophenoscore
- m6A:
-
N6-Methyladenosine
- OS:
-
Overall survival
- OSCC:
-
Oral squamous cell carcinoma
- PCA:
-
Principal component analysis
- PD-L1:
-
Anti-PD-1/programmed death-ligand 1
- ssGSEA:
-
Single-sample gene-set enrichment analysis
- TCGA:
-
The Cancer Genome Atlas
- TMB:
-
Tumor mutation burden
- TPM:
-
Transcripts per kilobase million
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. https://doi.org/10.3322/caac.21492 (2018).
Zanoni, D. K. et al. Survival outcomes after treatment of cancer of the oral cavity (1985–2015). Oral Oncol. 90, 115–121. https://doi.org/10.1016/j.oraloncology.2019.02.001 (2019).
Lin, W., Chen, M., Hong, L., Zhao, H. & Chen, Q. Crosstalk between PD-1/PD-L1 blockade and its combinatorial therapies in tumor immune microenvironment: A focus on HNSCC. Front. Oncol. 8, 532. https://doi.org/10.3389/fonc.2018.00532 (2018).
Doroshow, D. B. et al. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 18, 345–362. https://doi.org/10.1038/s41571-021-00473-5 (2021).
Dominissini, D. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. https://doi.org/10.1038/nature11112 (2012).
He, L. et al. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 18, 176. https://doi.org/10.1186/s12943-019-1109-9 (2019).
Paris, J. et al. Targeting the RNA m(6)A reader YTHDF2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell 25, 137-148.e136. https://doi.org/10.1016/j.stem.2019.03.021 (2019).
Choe, J. et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 561, 556–560. https://doi.org/10.1038/s41586-018-0538-8 (2018).
Lin, S., Choe, J., Du, P., Triboulet, R. & Gregory, R. I. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 62, 335–345. https://doi.org/10.1016/j.molcel.2016.03.021 (2016).
Huang, H., Weng, H. & Chen, J. m(6)A modification in coding and non-coding RNAs: Roles and therapeutic implications in cancer. Cancer Cell 37, 270–288. https://doi.org/10.1016/j.ccell.2020.02.004 (2020).
Chen, Y. G. et al. N6-methyladenosine modification controls circular RNA immunity. Mol. Cell 76, 96-109.e109. https://doi.org/10.1016/j.molcel.2019.07.016 (2019).
Yang, S. et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat. Commun. 10, 2782. https://doi.org/10.1038/s41467-019-10669-0 (2019).
Lohavanichbutr, P. et al. A 13-gene signature prognostic of HPV-negative OSCC: discovery and external validation. Clin. Cancer Res. 19, 1197–1203. https://doi.org/10.1158/1078-0432.Ccr-12-2647 (2013).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587-d592. https://doi.org/10.1093/nar/gkac963 (2023).
Snyder, A. et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199. https://doi.org/10.1056/NEJMoa1406498 (2014).
Xu, T. et al. Methyltransferase-like 3-induced N6-methyladenosine upregulation promotes oral squamous cell carcinoma by through p38. Oral Dis. https://doi.org/10.1111/odi.14016 (2021).
Liu, L. et al. METTL3 promotes tumorigenesis and metastasis through BMI1 m(6)A methylation in oral squamous cell carcinoma. Mol. Ther. 28, 2177–2190. https://doi.org/10.1016/j.ymthe.2020.06.024 (2020).
Ai, Y. et al. METTL3 Intensifies the progress of oral squamous cell carcinoma via modulating the m6A amount of PRMT5 and PD-L1. J. Immunol. Res. 2021, 6149558. https://doi.org/10.1155/2021/6149558 (2021).
Wang, F. et al. N6-methyladenosine methyltransferase METTL14-mediated autophagy in malignant development of oral squamous cell carcinoma. Front. Oncol. 11, 738406. https://doi.org/10.3389/fonc.2021.738406 (2021).
Shriwas, O. et al. DDX3 modulates cisplatin resistance in OSCC through ALKBH5-mediated m(6)A-demethylation of FOXM1 and NANOG. Apoptosis 25, 233–246. https://doi.org/10.1007/s10495-020-01591-8 (2020).
Li, X. et al. Fat mass and obesity-associated protein regulates tumorigenesis of arecoline-promoted human oral carcinoma. Cancer Med. 10, 6402–6415. https://doi.org/10.1002/cam4.4188 (2021).
Wang, F. et al. N6-methyladenosine demethyltransferase FTO-mediated autophagy in malignant development of oral squamous cell carcinoma. Oncogene 40, 3885–3898. https://doi.org/10.1038/s41388-021-01820-7 (2021).
Zhu, F., Yang, T., Yao, M., Shen, T. & Fang, C. HNRNPA2B1, as a m(6)A reader, promotes tumorigenesis and metastasis of oral squamous cell carcinoma. Front. Oncol. 11, 716921. https://doi.org/10.3389/fonc.2021.716921 (2021).
Jiang, F. et al. HNRNPA2B1 promotes multiple myeloma progression by increasing AKT3 expression via m6A-dependent stabilization of ILF3 mRNA. J. Hematol. Oncol. 14, 54. https://doi.org/10.1186/s13045-021-01066-6 (2021).
Hou, P. et al. LINC00460/DHX9/IGF2BP2 complex promotes colorectal cancer proliferation and metastasis by mediating HMGA1 mRNA stability depending on m6A modification. J. Exp. Clin. Cancer Res. CR 40, 52. https://doi.org/10.1186/s13046-021-01857-2 (2021).
Pu, J. et al. IGF2BP2 promotes liver cancer growth through an m6A-FEN1-dependent mechanism. Front. Oncol. 10, 578816. https://doi.org/10.3389/fonc.2020.578816 (2020).
Xu, X., Yu, Y., Zong, K., Lv, P. & Gu, Y. Up-regulation of IGF2BP2 by multiple mechanisms in pancreatic cancer promotes cancer proliferation by activating the PI3K/Akt signaling pathway. J. Exp. Clin. Cancer Res. CR 38, 497. https://doi.org/10.1186/s13046-019-1470-y (2019).
Shen, H. et al. ZEB1-induced LINC01559 expedites cell proliferation, migration and EMT process in gastric cancer through recruiting IGF2BP2 to stabilize ZEB1 expression. Cell Death Dis. 12, 349. https://doi.org/10.1038/s41419-021-03571-5 (2021).
Xu, L. et al. m(6)A methyltransferase METTL3 promotes oral squamous cell carcinoma progression through enhancement of IGF2BP2-mediated SLC7A11 mRNA stability. Am. J. Cancer Res. 11, 5282–5298 (2021).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70. https://doi.org/10.1016/s0092-8674(00)81683-9 (2000).
Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 12, 31–46. https://doi.org/10.1158/2159-8290.Cd-21-1059 (2022).
Hernandez-Muñoz, I. et al. RING1B contributes to Ewing sarcoma development by repressing the NaV1.6 sodium channel and the NF-κB pathway, independently of the fusion oncoprotein. Oncotarget 7, 46283–46300. https://doi.org/10.18632/oncotarget.10092 (2016).
Kreuzinger, C. et al. A complex network of tumor microenvironment in human high-grade serous ovarian cancer. Clin. Cancer Res. 23, 7621–7632. https://doi.org/10.1158/1078-0432.Ccr-17-1159 (2017).
Cai, Y. et al. Genomic alterations in PIK3CA-mutated breast cancer result in mTORC1 activation and limit the sensitivity to PI3Kα inhibitors. Can. Res. 81, 2470–2480. https://doi.org/10.1158/0008-5472.Can-20-3232 (2021).
Huang, Z. D. et al. Construction of prognostic risk prediction model of oral squamous cell carcinoma based on nine survival-associated metabolic genes. Front. Physiol. 12, 609770. https://doi.org/10.3389/fphys.2021.609770 (2021).
Chen, W. et al. Replication the association of 2q32.2-q32.3 and 14q32.11 with hepatocellular carcinoma. Gene 561, 63–67. https://doi.org/10.1016/j.gene.2015.02.006 (2015).
Dong, L. M. et al. LINC00511/miRNA-143-3p modulates apoptosis and malignant phenotype of bladder carcinoma cells via PCMT1. Front. Cell Dev. Biol. 9, 650999. https://doi.org/10.3389/fcell.2021.650999 (2021).
Planells-Palop, V. et al. Human germ/stem cell-specific gene TEX19 influences cancer cell proliferation and cancer prognosis. Mol. Cancer 16, 84. https://doi.org/10.1186/s12943-017-0653-4 (2017).
Xu, H. et al. GRPr-mediated photothermal and thermodynamic dual-therapy for prostate cancer with synergistic anti-apoptosis mechanism. Nanoscale 13, 4249–4261. https://doi.org/10.1039/d0nr07196j (2021).
Wang, J. et al. The CCDC43-ADRM1 axis regulated by YY1, promotes proliferation and metastasis of gastric cancer. Cancer Lett. 482, 90–101. https://doi.org/10.1016/j.canlet.2020.03.026 (2020).
Zhao, R. et al. HOTTIP predicts poor survival in gastric cancer patients and contributes to cisplatin resistance by sponging miR-216a-5p. Front. Cell Dev. Biol. 8, 348. https://doi.org/10.3389/fcell.2020.00348 (2020).
Kim, J., Park, J. Y., Shin, S. J., Lim, B. J. & Go, H. Neo-Fs index: A novel immunohistochemical biomarker panel predicts survival and response to anti-angiogenetic agents in clear cell renal cell carcinoma. Cancers https://doi.org/10.3390/cancers13061199 (2021).
Wu, T., Zhang, S. Y., Dong, W. J., Wang, M. & Sun, Y. B. The potential influence of long non-coding RNA PRKG1-AS1 on oral squamous cell carcinoma: A comprehensive study based on bioinformatics and in vitro validation. J. Oral Pathol. Med. 49, 409–416. https://doi.org/10.1111/jop.12980 (2020).
Li, N. et al. METTL3 regulates viral m6A RNA modification and host cell innate immune responses during SARS-CoV-2 infection. Cell Rep. 35, 109091. https://doi.org/10.1016/j.celrep.2021.109091 (2021).
Ma, S. et al. The RNA m6A reader YTHDF2 controls NK cell antitumor and antiviral immunity. J. Exp. Med. https://doi.org/10.1084/jem.20210279 (2021).
Han, D. et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature 566, 270–274. https://doi.org/10.1038/s41586-019-0916-x (2019).
Adib, E. et al. CDKN2A alterations and response to immunotherapy in solid tumors. Clin. Cancer Res. 27, 4025–4035. https://doi.org/10.1158/1078-0432.Ccr-21-0575 (2021).
Valero, C. et al. The association between tumor mutational burden and prognosis is dependent on treatment context. Nat. Genet. 53, 11–15. https://doi.org/10.1038/s41588-020-00752-4 (2021).
Blagih, J., Buck, M. D. & Vousden, K. H. p53, cancer and the immune response. J. Cell Sci. https://doi.org/10.1242/jcs.237453 (2020).
Braun, D. A. et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 26, 909–918. https://doi.org/10.1038/s41591-020-0839-y (2020).
Morrison, C. et al. Predicting response to checkpoint inhibitors in melanoma beyond PD-L1 and mutational burden. J. Immunother. Cancer 6, 32. https://doi.org/10.1186/s40425-018-0344-8 (2018).
Zhang, B. et al. m(6)A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol. Cancer 19, 53. https://doi.org/10.1186/s12943-020-01170-0 (2020).
Funding
This work was supported by the National Nature Science Foundation of China (Grant No. 81970941), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD, 2018-87), and the Special Fund of Jiangsu Provincial Key Research and Development Project (Social Development) (Grant No. BE2021723).
Author information
Authors and Affiliations
Contributions
L.P., Y.F. and J.H.Y. were involved in the overall design of experiments and interpretation of results. L.P. and H.S. performed the computational analysis. K.Y.W., W.H.X. and H.N.T. collected all the data. Y.F. and J.H.Y. applied the funding and designed the experiments. The manuscript was written by L.P. with contributions by all co-authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pan, L., She, H., Wang, K. et al. Characterization of the m6A regulator-mediated methylation modification patterns in oral squamous cell carcinoma. Sci Rep 13, 6617 (2023). https://doi.org/10.1038/s41598-023-33891-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-33891-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
