
Abstract
Plant resins are rich in bioactive compounds with high medicinal values. However, the chemistry and anti-inflammatory activity of the resins produced by trees of the genus Eucalyptus were scarcely investigated. The inflammatory targets cyclooxygenase-1 (COX-1), COX-2, TNF-, NF-B, and NO were significantly inhibited by the methanolic extract of Eucalyptus maculata kino resin (EME) and its CH2Cl2 soluble fraction (MCF). Sakuranetin (C1), (E)-cinnamic acid (C2), kaempferol 7- methyl ether (C3), 7-O-methyl aromadendrin (C4), and 1,6- dicinnamoyl-O-α-D-glucopyranoside (C5) were isolated from MCF. Three compounds (C1, C2, and C4) showed potent in vitro COX-1 inhibition, while C5 inhibited COX-2, TNF-α, NF-κB, and NO significantly. An in-silico study revealed that C5 had the highest binding affinity to the active site in COX-2 with binding energy score (S) of -14.85 kcal/mol, better than celecoxib (COX-2 inhibitor). In conclusion, 1,6-dicinnamoyl-O-α-D-glucopyranoside (C5) could be investigated further in the search for anti-inflammatory agents.
Similar content being viewed by others
Introduction
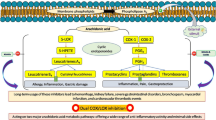
Inflammation is a complex, dynamic response to cell injury, infection, trauma, or toxins that can last for a few days (acute inflammation) or for a longer duration (chronic inflammation). The overproduction of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1b (IL-1b), and IL-6, as well as inflammatory targets, secreted by immune cells and macrophages, all play an important role in mediating the inflammatory reactions1. Also, there is a close link between inflammation and oxidative stress, as one activates the other2. Moreover, inflammation triggered by oxidative stress can cause various ailments, such as cancer, rheumatoid arthritis, asthma, and diabetes.3 Several polyphenols are known to exert anti-inflammatory effects through modulation of different signaling pathways such as arachidonic acid (AA) metabolism, nuclear factor-kappa B (NFKB), and TNF-α4. In the AA-dependent pathway, the anti-inflammatory effect of plant polyphenols is related to their ability to inhibit cyclooxygenase (COX), which converts AA into prostaglandins.
Medicinal plants are considered a valuable source of potential anti-inflammatory agents5. Evidence to support this anti-inflammatory effect is lacking, as most of these plants have not been subjected to chemical, pharmacological, or toxicological studies in order to investigate their bioactive compounds6. Eucalyptus is one of the most important genera in the family Myrtaceae, which includes 132 genera and 5950 species. Eucalyptus maculata Hook. is indigenous to Australia and is cultivated in Egypt. E. maculata is an evergreen tree that reaches up to 60 m in height, with fragrant white flowers and small brown to green fruit. It has smooth mottled pinkish grey or bluish grey, often dimpled bark that is shed in small, irregular flakes7. Eucalyptus kino resin, also known as "blood gum," has a very dark color like blood and is exuded by eucalyptus trees (Angophora Corymbia, and Eucalyptus spp.). It is known to contain high levels of potentially useful polyphenolic compounds8,9. Previous work on Eucalyptus kino resin resulted in the isolation of (E)-cinnamic acid, sakuranetin, 7-O-methyl aromadendrin, and 1,6-dicinnamoyl-O-α-d-glucopyranoside9, in addition to p-coumaric acid, 1-O-cinnamoyl 6-O-p-coumaroylglucose, and 7-methyl-aromadendrin-4′-O-(6′′-trans-p-coumaroyl)-β-D-glucopyranoside8. Also, the methanolic extract of the kino resin (EME), had previously demonstrated promising in vivo anti-inflammatory and in vitro antioxidant activities10. Sakuranetin, 7-O-methyl aromadendrin, and 1,6-dicinnamoyl-O-α -D-glucopyranoside showed antioxidant and hepatoprotective effects11. 7-O-methyl aromadendrin had cytotoxicity against MCF-7 and C32 cell lines12, it stimulated glucose uptake and improved insulin resistance13. Sakuranetin also possessed antioxidant, antibacterial, anti-inflammatory, antiparasitic, antimutagenic, and antiallergic characteristics, and it exhibited antiviral activity against human rhinovirus 3 and influenza B virus14. These findings prompted the authors to further fractionate EME to identify and isolate the active anti-inflammatory agent(s) from the kino resin. Furthermore, a molecular docking study was performed to study the binding mode of the active compounds to the active site of COX-2.
Results and discussion
Identification of the bioactive compounds from E. maculata exudate.
Five compounds (C1–C5) (Fig. 1) were isolated from the bioactive methylene chloride fraction of E. maculata. The isolated compounds were identified by TLC comparison with the previously isolated compounds or reference standards, spectroscopic analysis (Suppl. Data S1), and comparison with the reported data in literature as sakuranetin [(2S)-5,4'-dihydroxy-7-methoxyflavanone] (C1)9,15, (E)-cinnamic acid (C2)9,16, kaempferol 7- methyl ether (C3) (isolated for the first time from MEEM15,17), 7-O-methyl aromadendrin (C4)9,15, and 1,6-dicinnamoyl-O-α-D-glucopyranoside (C5)9 (Fig. 1). The trans-form of C2 was deduced from the large coupling constant of the olefinic protons with J = 16 Hz of Hα, β.

Chemical structures of compounds isolated from E. maculata resin.
In vitro anti-inflammatory activity of selected isolated compounds
The effect on COX-1 and COX-2
Aspirin is one of the most famous non-steroidal anti-inflammatory drugs. It carries out its anti-inflammatory effect through the rough inhibition of two enzymes: COX-1 and COX-2. Its action is associated with gastric bleeding, mainly due to the inhibition of COX-1. So, our main target was the development of more selective COX-2 inhibitors like celecoxib (celebrex®)18. COX-1 and COX-2 were estimated according to the reported method19 and were compared to indomethacin and celecoxib (selective COX-2 and selective COX-1 inhibitors, respectively). The 50% inhibitory concentration (IC50) values were calculated using the concentration-inhibition response curve and were presented as mean ± standard deviation (SD). All enzyme inhibition activities were performed in triplicate and were examined using the sample t-test using the SPSS 26.0 program at (p < 0.05) (Table 1).
(E)-Cinnamic acid (C2) and sakuranetin (C1) exhibited remarkable, significant inhibition of COX-1 with IC50 of 0.16 ± 0.10 and 0.19 ± 0.01 respectively, meanwhile, Indomethacin (IC50 0.23 ± 0.00 µg/mL) was not statistically different from 7-O-methyl aromadendrin (C4) (IC50 of 0.22 ± 0.01 µg/mL) at (p < 0.05). Zhang et al.15 previously reported that sakuranetin and 7-O-methyl aromadendrin possessed moderate inhibitory effects on COX-1.
Concerning COX-2 inhibitory activity, 1,6-dicinnamoyl-O-α-D-glucopyranoside (C5) showed the most significant effect with IC50 of 1.16 ± 0.01 µg/mL, compared to celecoxib (IC50 1.2 ± 0.00 µg/mL) at (p < 0.05). In addition, 7-O-methyl aromadendrin (C4) and sakuranetin (C1) showed moderate inhibitory activity towards COX-2. Sakuranetin was previously reported to suppress the synthesis of COX-220. In contrast, Zhang et.al (2006)15 stated that 7-O-methyl aromadendrin and sakuranetin had no inhibitory activity against COX-2.
In vitro inhibitory activities of the isolated compounds (C1, C2, C4, and C5) against TNF-R2, NF-кB, NO production
TNF-R2, NF-K B, and NO were measured by ELISA kit for compounds (C1, C2, C4, C5). The 50% inhibitory concentration (IC50) values were calculated from the concentration-inhibition response curve.
C5 (IC50 5.17 ± 0.07 ng/mL), exhibited higher binding affinity to TNF-R2 (TNF-Receptors) than certolizumab with IC50 (6.70 ± 0.07 ng/mL) at (p < 0.05). (Table 1). Concerning inhibition of NF-KB, C5 had a significantly lower IC50 of 6.07 ± 0.07 Pg/mL than curcumin standard (IC50 7.80 ± 0.03 Pg/mL) at (p < 0.05). Moreover, C1, C2, and C4 showed significantly moderate suppression of NF-KB (Table 1). These results were in accordance with Lee et al.21, reporting that 7-O-methyl aromadendrin exhibited anti-inflammatory activity by suppressing the nuclear translocation of NF-KB. A previous study showed that sakuranetin could treat inflammation through the reduction of NF-KB14 while (E)-cinnamic acid showed moderate suppression of NF-KB and TNF-R222. Compared to standard 7-nitroindazole, nitric oxide synthase inhibitor23 (IC50 = 49.2 ± 0.80 µg/mL), C5 significantly inhibited NO with IC50 of 38 ± 0.04 µg/mL at (p < 0.05). (Table 1).
Molecular docking
Based on the previous promising in vitro biological evaluation results, compounds (C1, C2, C4, and C5) isolated from MCF were selected for molecular docking studies into the binding site of COX-2 enzyme to develop an insight into the putative intermolecular interactions and explore the possible binding pattern behind the inhibitory activities of these compounds. The choice of COX-2 for performing the docking study was mainly because it is a vital key enzyme in inflammation and is considered a rate-limiting enzyme that catalyzes prostaglandin production, responsible for the formation of inflammatory mediators24,25,26,27,28. The docking study was performed using Molecular Operating Environment software (MOE, 2016.0802). The X-ray crystal structures of COX-2 (PDB ID: 1CX2) with its co-crystallized ligand S58 [4-(5-(4-bromophenyl)-3-(trifluoromethyl)-1H-pyrazol-1yl) benzene sulfonamide] were obtained from the Protein Data Bank (PDB) (Fig. 2).

Co-crystallized ligand S58 [4-(5-(4-bromophenyl)-3-(trifluoromethyl)-1H-pyrazol-1yl) benzenesulfonamide].
The docking poses were chosen based on the top-scored conformation with the optimum binding interactions found by the MOE search algorithm and scoring function. Binding affinities to the binding pockets enzyme were also determined by binding energy scores, the creation of binding interactions with neighboring amino acid residues, and the relative alignment of docked poses in relation to co-crystallized ligands.
The proposed docking algorithm was validated by re-docking the co-crystallized ligands S58 into the binding site. The initial poses generated from the PDB were retrieved with a root mean square deviation (RMSD) of 1.53 Å and a docking score of − 14.08 kcal/mol, for COX-2 (Fig. 3). These results indicated that the docking protocol could reliably predict docking poses for the tested compounds. It was reported that values less than 1.5 or 2 Å were indicators of a successful and reliable docking protocol29.

An overlay and binding pattern of S58 into COX-2 active site (PDB 1CX2) 2D (A,C) 3D (B,D) co-crystalized ligand (Brown), S58 (Purple).
Docking to COX-2 active site
With a binding energy score (S) of − 14.08 kcal/mol and (RMSD) of 1.53, the co-crystallized ligand S58 bound to the active site of the COX-2 enzyme displayed two hydrogen bonds of 3.46 and 3.40 Å between the sulfonamide oxygen and His90 and Arg513, respectively. Furthermore, two hydrogen bonds of 3.03 and 3.36 Å were observed between pyrazole nitrogen and the trifluoromethyl group with Tyr355 and Arg120, respectively. Another hydrophobic contact of 3.85 Å formed between the sulfonamide phenyl ring and Ser353 (Fig. 3A–D). Molecular docking studies of the target (C1) with a binding energy score (S) of − 11.87 kcal/mol and (RMSD) of 0.78 indicated that the 4-hydroxyphenyl ring and the chroman aromatic ring form two hydrophobic interactions of 4.24 and 3.82 Å with Ser353 and Tyr355, respectively. Also, the hydroxyl group forms a hydrogen bond of 2.91 Å with Tyr355 (Fig. 4A,B). Moreover, the orientation of (C1) in the binding pocket was quite similar to that of the native ligand (Fig. 4C,D). Molecular docking studies of the target (C2) with a binding energy score (S) of − 8.16 and (RMSD) of 1.19 revealed that the (E)-cinnamic acid carbonyl group participated in H-bonding of 3.43 Å with His90 (Fig. 4E,F). Furthermore, the orientations of (C2) and the co-crystalized ligand S58 inside the binding pocket of COX-2 were quite similar (Fig. 4G,H). Examination of the best-docked pose of (C4) with (S) of − 12.17 and (RMSD) of 1.38 demonstrated that it was perfectly positioned in the active site of the COX-2 enzyme. It was lodged in the active site through a hydrogen bond of 2.93 Å between chroman ring oxygen and hydrogen bond donor Tyr355. In addition, two hydrophobic interactions between the 4-hydroxy phenyl ring of 4.24 and 4.54 Å with Tyr355 and Lue 4.53, respectively. On the same track, chroman aromatic rings form hydrophobic interaction of 3.81 Å with Ser353 (Fig. 4I,J). Interestingly, their orientation in the binding pocket was quite similar to that of the co-crystallized ligand S58, occupying the same position and spatial area as depicted in (Fig. 4 K,L). With regard to (C5), the best-docked position is being examined with (S) of − 14.85 and (RMSD) of 1.56. Three significant H-bonds were observed between the Arg513 and His90 and the 3,4,5-trihydroxytetrahydro-2H-pyrans of 2.56, 3.55, and 3.43 Å, respectively. The complex formed was further stabilized by the hydrophobic interaction of 4.71 Å between the cinnamate aromatic ring and Tyr355 (Fig. 4M,N). Interestingly, its placement in the binding pocket was very similar to that of the co-crystallized ligand S58 in terms of position and spatial orientation, as depicted in (Fig. 4O,P) (Table 2).




Docking and binding pattern into COX-2 active site (PDB 1CX2) of Target compound C1, 2D (A), 3D (B), an overlay of the docked pose of compound C1 with co-crystalized ligand S58 2D (C), 3D (D) S58 (purple), C1 (yellow). Regarding compound C2, 2D (E), 3D (F), an overlay of the docked pose of compound C2 with co-crystalized ligand S58 2D (G), 3D (H) S58 (purple), C2 (yellow). Concerning C4, 2D (I), 3D (J), an overlay of the docked pose of compound C4 with co-crystalized ligand S58 2D (K), 3D (L) S58 (purple), C4 (yellow). Finally target compound C5, 2D (M), 3D (N), an overlay of the docked pose of compound C5 with co-crystalized ligand S58 2D (O), 3D (P) S58 (purple), C5 (yellow).
Based on earlier docking experiments, it is possible to explain why compound (C2) has the lowest in vitro activity and why compounds (C5), (C4), and (C1) have higher in vitro activity. Compound C5 displayed the most effective through non-covalent bonding interactions with three essential amino acids Arg513, His90, and Tyr355, as does the cocrystallized ligand S58, whereas C4 exhibits less activity than (C5) because it interacts with only two amino acids: Ser353 and Tyr355. Additionally, (C4) has an advantage over (C1) since it can interact with Ser353, Tyr355, and Leu531, but (C1) lacks the interaction with Leu531 and forms interaction only with Ser353 and Tyr355. Furthermore, the least active one, (C2), only interacts with His90, so minimal activity is predictable.
Further, an in silico study was performed in order to predict the pharmacokinetics and toxicity of selected isolated compounds using the Graph-Based Signatures (pkCSM) program30 (Table 3). The outcomes for intestinal absorption showed high levels for each isolated compound tested. Additionally, C5 is sparingly soluble in water, whereas the other separated compounds have considerable water solubility. In terms of distribution, C2 and C5 exhibit good volumes of distribution compared with C1 and C4. Concerning metabolism, all compounds inhibited the majority of CYP450 isoenzymes. With respect to excretion, an acceptable total clearance was achieved by C2 and C5. Finally, compounds C2, C4, and C5 are free from toxicity (Table 3).
Conclusion
The current study revealed that E. maculata resin is a promising Egyptian natural medicine, rich in phenolics and flavonoids, and could be considered a new therapeutic candidate for inflammation. Moreover, 1,6-Dicinnamoyl-O-α-D-glucopyranoside (C5) showed the best binding activity towards the COX-2 active site. This binding looks similar to or better than celecoxib which is considered the first drug of choice for COX-2 inhibition, indicating the importance of this compound as a natural product for the treatment of inflammation. Moreover, further biological and clinical studies are required on the standardized bioactive extract; EME to verify its possible use as adjuvant therapy in the management of inflammation and its disorders. The continuous need for new natural therapeutic agents requires many hard efforts to open new opportunities from available traditional medicine resources. This can be reached only by understanding their chemical constituents and their medicinal actions, which might be a hard task as a result of the many hundreds of compounds in each formula31. Fortunately, the in silico tools have not only accelerated the novel natural molecules' discovery but also explained new activities for known compounds32.
Methods
Plant material
The kino resin of E. maculata, cultivated in the Zoo Garden, Giza, Egypt was scratched from the stem in April 2019. It was pulverized and placed in sealed bottles. The plant was identified by Dr. M. Gibali, Senior Taxonomist in the Department of Botany, National Research Center, Giza, Egypt and Agriculture Engineer Mrs. Therese Labib, Senior Botanist, Orman botanic garden, Giza, Egypt. Voucher specimens of E. maculata (Sp. # EM 2.7.2019) was deposited at the Department of Pharmacognosy, Faculty of Pharmacy, Cairo University, Egypt. The study complies with local, national, and international guidelines and no specific consent was required for the collection of kino resin from the plant.
Chemicals for phytochemical study
All solvents used were of AR grade. Pre-coated TLC plates of silica gel GF254 (Merck, Germany) were used for TLC analysis. TLC analysis was performed using CHCl3-MeOH (9.5:05) as a solvent system (S1) and the plates were observed under UV light (254 and 366 nm). Kaempferol 7-methyl ether was purchased from Aktin Laboratories, Phytochemicals Division of Aktin Chemicals, Inc. (Chengdu, China). Silica gel 60 (mesh size 230–400) and silica gel H for vacuum liquid chromatography (VLC) were purchased from Merck (Germany).
In vitro anti-inflammatory activity
Cyclooxygenases (COX-1 and COX-2) were measured by an enzyme immunoassay (EIA) kit (Cayman Chemical, Ann Arbor, MI, USA) according to the manufacturer’s instructions. TNF-R2, NF-KB, and NO were measured by ELISA kit (#79756, BPS Bioscience, USA), (#CSB-EL015761HU, Cusabio, China).
General experimental procedures
1H-NMR and 13C-NMR spectra of the isolated compounds were recorded on a Bruker High-Performance Digital FT-NMR-spectrophotometer (Avance III HD), Bremen, Germany. Chemical shift values were recorded in δ ppm. The solvents used are CDCl3 and CD3OD (Cambridge isotope laboratory, USA). The obtained data were processed using Mestrenova NMR processor software, version 6.0.2-5475.
Extraction of E. maculata resin
The air-dried powdered resin (140 g) of E. maculata exudate (EME) was extracted with methanol (3 × 100 mL) using a sonicator (Soltec Co., 230/240 V, 50/60 Hz, Italy) for 15 min. The extract was concentrated under reduced pressure to a constant weight to yield dry methanol residue; (MEEM, 130 g), then it was kept in a desiccator to dry over anhydrous CaCl2.
The methanolic extract of E. maculata exudate (MEEM, 120 g) was suspended in distilled water (300 mL) and partitioned with CH2Cl2 (4 × 100 mL) at room temperature to give methylene chloride fraction (MCF, 7 g) on evaporation.
Isolation of the major compounds from methylene chloride fraction
The MCF (7 g) was dissolved in the least volume of methanol, mixed well with about 20 g of silica gel 60 and left at room temperature to dry, then applied onto the top of a VLC packed to a glass column with silica gel H (15 × 6 cm i.d., 210 g). Gradient elution with increasing polarity (5% increments) was carried out starting with 100% n-hexane, n-hexane/CH2Cl2, CH2Cl2, and finally CH2Cl2/EtOAc mixture, up to 100% EtOAc and finally washing with 100% methanol. Fractions, 200 mL each, were separately concentrated and monitored by TLC. The developed plates were sprayed with p-anisaldehyde/H2SO4 spray reagent and then heated at 115 °C. Similar fractions were pooled together. Different collective fractions were subjected to chromatographic separation and purification techniques to obtain five compounds (C1–C5). Fraction I (125 mg; 90–95% CH2Cl2 in n-hexane). It was further purified on Si gel 60 column (15 × 1.5 cm) using n-hexane–ethyl acetate (9.5:0.5, v/v) to give yellowish white powder of compound C1 (70 mg). Fraction II (140 mg; 95% CH2Cl2 in EtOAc) showed two major spots at Rƒ = 0.56 and 0.43, respectively (TLC, CH2Cl2-MeOH; 9.5:0.5). It was subjected to further purification on another Si gel 60 column (25 × 1.5 cm) using CH2Cl2-MeOH (9.7:0.3 v/v) to give compound C1 (25 mg) and compound C2 as a white amorphous powder (40 mg). Fraction III (1.2 g; 85% CH2Cl2 in EtOAc) was further purified on Si gel 60 column (35 × 3 cm) using CH2Cl2-MeOH (9:1, v/v) to give subfraction (440 mg) that showed one major spot at Rƒ = 0.39 (TLC, CH2Cl2-MeOH; 9:1). It was subjected to further purification on Si gel 60 column (25 × 1.5 cm, 230–400 mesh) using CH2Cl2/MeOH (9.5:0.5, v/v) to give yellow powder of compound C3 (30 mg).
Fraction IV (1.2 g; 80% CH2Cl2 in EtOAc) showed one major spot at Rƒ = 0.44 (TLC, CH2Cl2-MeOH; 9:1). It was further purified on another Si gel 60 column (35 × 3 cm) using CH2Cl2-MeOH (9.9: 0.1, v/v) to give yellowish white powder of compound C4 (300 mg). Fraction V (100 mg; 40% CH2Cl2 in EtOAc) showed one major spot at Rƒ = 0.58 (TLC, CH2Cl2-MeOH; 9.5:0.5). It was further purified on another Si gel 60 column (15 × 1.5 cm) column using CH2Cl2-MeOH (9.9:0.1, v/v) to give white powder of compound C5 (45 mg).
1H NMR and 13C-NMR spectra of the isolated compounds
Compound C1 (70 mg), was obtained as yellowish white powder, with Rf 0.78 (S1), and it was identified as sakuranetin 9,15.
1H NMR (400 MHz, CDCl3) δH 12.00 (s, OH), 7.26 (2H, d, J = 8.2 Hz, H-2', H-6'), 6.81 (2H, d, J = 8.3 Hz, H-3', H-5'), 5.99 (2H, d, J = 2.2 Hz, H-6, H-8), 5.28 (1H, dd, J = 13.0, 2.8 Hz, H-2), 3.73 (s, 3H, OMe), 3.02 (1H, dd, J = 17.2, 13.0 Hz, H3ax), 2.71 (1H, dd, J = 17.2, 2.9 Hz, H3eq). 13C-NMR (100 MHz, CDCl3): δc 80.9 (C-2), 44.6 (C-3), 198.1(C-4), 165.3 (C-5), 96.3 (C-6), 169.6 (C-7), 95.5 (C-8), 164.7 (C-9), 104.4 (C-10), 56.9 (OCH3), 129.3 (C-1'), 130.9 (C-2'), 116.8 (C-3'), 159.1 (C-4'), 116.8 (C-5'), 129.3 (C-6').
Compound C2 (40 mg), was isolated as white amorphous powder, with Rf 0.56 (S1), and it was identified as Trans- cinnamic acid 9,16.
1H-NMR (400 MHz, MeOH-d4) δH 7.69 (1H, d, J = 16 Hz) and 6.5 (1H, d, J = 16 Hz) that could be attributed to trans olefinic protons of α, β unsaturated ketone attached to C-1, 7.43 (2H, m, H2/6) and 7.62–7.6 (3H, m, H 3, 4, 5).
Compound C3 (30 mg), was obtained as yellow powder, with Rf 0.53 (S1), and it was identified as Kaempferol -7- methyl ether 15,17.
1H-NMR (400 MHz, MeOH-d4) δH 6.33 (1H, d, J = 2.1 Hz, H-6), 6.62 (1H, d, J = 2.1 Hz, H-8), 8.14 (2H, d, J = 8.9 Hz, H-2'/6'), 6.93 (2H, d, J = 8.9 Hz, H-3'/5'), 3.91 (3H, s, Me-7). 13C-NMR (100 MHz, MeOH-d4): δc 146.93 (C-2), 135.93 (C-3), 176.08 (C-4), 160.01 (C-5), 97.46 (C-6), 165.98 (C-7), 91.43 (C-8), 159.35 (C-9), 104.47 (C-10), 55.03 (OCH3), 122.24 (C-1'), 129.93 (C-2', 6'), 115.24 (C-3',5'), 156.64 (C-4').
Compound C4 (30 mg), was isolated as yellowish white powder, with Rf 0.49 (S1), and it was identified as 7-O-methyl aromadendrin 9,15.
1H-NMR (400 MHz, MeOH-d4) δH 5.02 (1H, d, J = 11.5 Hz, H-2), 4.59 (1H, d, J = 11.5 Hz, H-3), 6.05 (1H, d, J = 2.2 Hz, H-6), 6.09 (1H, d, J = 2.2 Hz, H-8), 7.37 (1H, d, J = 8.5 Hz, H-2'/6'), 6.85 (1H, d, J = 8.6 Hz, H-3'/5'), 3.82 (3H, s, Me-7).
Compound C5 (25 mg), was obtained as white powder, Rf 0.43 (S1), and it was identified as 1,6-dicinnamoyl-O-α-D-glucopyranoside9.
1H-NMR (500 MHz, CHCl3- d)
Cinnamoyl moieties
δH 7.8, 7.71 (2H, d, J = 16, 2*Hα), 7.53 (4H, m, 2*H2, 2*H6), 7.38 (6H, m, 2*H-3, H4, H5), 6.49 (2H, d, J = 16, 2Hβ).
α-glucose
δH 5.69 (1H,d, J = 3.9), 4.54 (1H, d, J = 12, H6ʹa), 4.43 (1H, dd, J = 12, 4.5, H6ʹb), 3.74 (1H, m, H-5ʹ), 3.57 (2H, m, H-2ʹ, H-3ʹ), 3.5 (1H, m, H-4ʹ).
13C-NMR (125 MHz, CHCl3-d)
Cinnamoyl moieties
δC 165.47, 167.23 (2xC = O), 146.46, 145.37 (2xC-α), 133.3, 133.87 (2xC-1), 130.39, 130.15 (2xC-3), 128.59 (2x*C-4), 128.54 (2xC-5), 127.89 (2xC-2), 127.82 (2xC-6), 117.05, 116.56 (2xC-β).
α-glucose
δC 94.03 (C-1), 72.14 (C-2), 76.2 (C-3), 69.43 (C-4), 74.57 (C-5), 63.16 (C-6).
In silico study for the interaction of the identified compounds
Computer-aided docking experiments were performed using Molecular Operating Environment software (MOE 2016.0802, Chemical Computing Group, Montreal, Canada)33. Crystal coordinates from the X-ray crystal structure of COX-2 (PDB ID code: 1CX2, with the inhibitor S58 bound in the active site) were obtained from RCSB protein data bank and processed consequently with the MOE program. Redundant chains, water molecules, and any surfactants were discarded, explicit hydrogen atoms were added to the receptor complex structure and partial charges were calculated. The preparation was completed with a structure preparation module employing protonated 3D function. The co-crystal ligands were extracted from their corresponding proteins and used as reference molecules for the validation study29.
The target compounds were constructed using the builder module of MOE. The compounds were then collected in a database and prepared by adding hydrogens, calculating partial charges, and energy minimizing using Force field MMFF94x.
Docking procedure
The MOE-Site Finder was used to generate the active site of the receptor, and the MOE-Dock was used to dock the ligands within the active site. As a placement method, we used Triangle Matcher, London as a scoring function, and 10 retained poses as parameters. All receptor-ligand complexes were examined further to determine binding interactions and the optimum docking pose. The best-docked complex, which is thought to represent protein–ligand interactions, was chosen based on docking score, ligand alignment at the active site that was similar to the reference ligands, and retention of significant interactions. This docking approach was validated by the successful pose-retrieval of the co-crystal ligand when docked into its corresponding binding site in the crystal. All graphical representations were rendered by MOE. 2016.0802. In silico investigation of Pharmacokinetic and toxicity properties of selected isolated compounds was carried out using the Graph-Based Signatures (pkCSM) program.
Statistical analysis
Data were analyzed with GraphPad Prism V 6 (GraphPad Software Inc., San Diego, CA, USA). Data were expressed as mean ± standard deviation (SD) and were analyzed using sample t-test using SPSS 26.0 software. Statistical differences yielding p < 0.05 were considered significant.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information file.
References
Walsh, N. C., Crotti, T. N., Goldring, S. R. & Gravallese, E. M. Rheumatic diseases: The effects of inflammation on bone. Immunol. Rev. 208, 228–251 (2005).
Ventura, E. et al. Homocysteine and inflammation as main determinants of oxidative stress in the elderly. Free Radical Biol. Med. 46, 737–744 (2009).
Ondua, M., Njoya, E. M., Abdalla, M. A. & McGaw, L. J. Anti-inflammatory and antioxidant properties of leaf extracts of eleven South African medicinal plants used traditionally to treat inflammation. J. Ethnopharmacol. 234, 27–35 (2019).
Yoon, J. H. & Baek, S. J. Molecular targets of dietary polyphenols with anti-inflammatory properties. Yonsei Med. J. 46, 585–596 (2005).
Chandra, S., Dey, P. & Bhattacharya, S. Preliminary in vitro assessment of anti-inflammatory property of Mikania scandens flower extract. J. Adv. Pharm. Edu. Res. 2, 25–31 (2012).
Rashid, M. A. Pharmacological and phytochemical screenings of ethanol extract of Sterculia villosa Roxb. J. Biomed. Pharm. Res. 2, 09–14 (2013).
Hill, K. D. & Johnson, L. A. Systematic studies in the eucalypts 7: A revision of the bloodwoods, genus Corymbia (Myrtaceae). Telopea 6, 185–504 (1995).
Rashwan, O. New phenylpropanoid glucosides from Eucalyptus maculata. Molecules 7, 75–80 (2002).
Abdel-Sattar, E., Kohiel, M., Shihata, I. & El-Askary, H. Phenolic compounds from Eucalyptus maculata. Pharmazie 55, 623–624 (2000).
El-Hawary, S. S., El-Kammar, H. A., Farag, M. A., Saleh, D. O. & El Dine, R. S. Metabolomic profiling of five Agave leaf taxa via UHPLC/PDA/ESI-MS inrelation to their anti-inflammatory, immunomodulatory and ulceroprotective activities. J. Steroids 160, 108648 (2020).
Mohamed, A.-F., Hasan, A. G. A., Hamamy, M. I. & Abdel-Sattar, E. Antioxidant and hepatoprotective effects of Eucalyptus maculata. Med. Sci. Monit. 11, 431 (2005).
Fang, R., Houghton, P. J. & Hylands, P. J. Cytotoxic effects of compounds from Iris tectorum on human cancer cell lines. J. Ethnopharmacol. 118, 257–263 (2008).
Zhang, W. Y. et al. 7-O-methylaromadendrin stimulates glucose uptake and improves insulin resistance in vitro. Biol. Pharm. Bull. 33, 1494–1499 (2010).
Stompor, M. A review on sources and pharmacological aspects of Sakuranetin. Nutrients 12, 513 (2020).
Zhang, X. et al. Anti-inflammatory activity of flavonoids fromPopulus davidiana. Arch. Pharmacal Res. 29, 1102–1108 (2006).
Ling, F., Jiang, C., Liu, G., Li, M. & Wang, G. Anthelmintic efficacy of cinnamaldehyde and cinnamic acid from cortex cinnamon essential oil against Dactylogyrus intermedius. Parasitology 142, 1744–1750 (2015).
Freitas, M. O., Ponte, F. A., Lima, M. A. S. & Silveira, E. R. Flavonoids and triterpenes from the nest of the stingless bee Trigona spinipes. J. Braz. Chem. Soc. 19, 532–535 (2008).
Abdelall, E. K. A., Lamie, P. F. & Ali, W. A. M. Cyclooxygenase-2 and 15-lipoxygenase inhibition, synthesis, anti-inflammatory activity and ulcer liability of new celecoxib analogues: Determination of region-specific pyrazole ring formation by NOESY. Bioorg. Med. Chem. Lett. 26, 2893–2899 (2016).
Jachak, S. M. et al. Anti-inflammatory, cyclooxygenase inhibitory and antioxidant activities of standardized extracts of Tridax procumbens L. Fitoterapia 82, 173–177 (2011).
Kim, K.-Y. & Kang, H. Sakuranetin inhibits inflammatory enzyme, cytokine, and costimulatory molecule expression in macrophages through modulation of JNK, p38, and STAT1. Evid. Based Complem. Altern. Med. 1, 1 (2016).
Lee, J.-W. et al. Aromadendrin inhibits lipopolysaccharide-induced nuclear translocation of NF-κB and phosphorylation of JNK in RAW 264.7 macrophage cells. Biomol. Ther. 21, 216 (2013).
Song, F., Li, H., Sun, J. & Wang, S. Protective effects of cinnamic acid and cinnamic aldehyde on isoproterenol-induced acute myocardial ischemia in rats. J. Ethnopharmacol. 150, 125–130 (2013).
Gaestel, M., Kotlyarov, A. & Kracht, M. Targeting innate immunity protein kinase signalling in inflammation. Nat. Rev. Drug Discovery 8, 480–499 (2009).
Zarghi, A. & Arfaei, S. Selective COX-2 inhibitors: A review of their structure-activity relationships. Iran. J. Pharm. Res. IJPR 10, 655–683 (2011).
El-Reedy, A. A. & Soliman, N. Synthesis, biological activity and molecular modeling study of novel 1, 2, 4-triazolo [4, 3-b][1, 2, 4, 5] tetrazines and 1, 2, 4-triazolo [4, 3-b][1, 2, 4] triazines. Sci. Rep. 10, 1–18 (2020).
Razak, S. et al. Molecular docking, pharmacokinetic studies, and in vivo pharmacological study of indole derivative 2-(5-methoxy-2-methyl-1H-indole-3-yl)-N′-[(E)-(3-nitrophenyl) methylidene] acetohydrazide as a promising chemoprotective agent against cisplatin induced organ damage. Sci. Rep. 11, 1–23 (2021).
Nguyen, H. T., Vu, T.-Y., Chandi, V., Polimati, H. & Tatipamula, V. B. Dual COX and 5-LOX inhibition by clerodane diterpenes from seeds of Polyalthia longifolia (Sonn.) Thwaites. Sci. Rep. 10, 1–10 (2020).
Singh, P., Kaur, J., Kaur, H., Kaur, A. & Bhatti, R. Synergy of physico-chemical and biological experiments for developing a cyclooxygenase-2 inhibitor. Sci. Rep. 8, 1–14 (2018).
Ritchie, T. J. & Macdonald, S. J. How drug-like are ‘ugly’drugs: do drug-likeness metrics predict ADME behaviour in humans?. Drug Discov. Today 19, 489–495 (2014).
Pires, D. E., Blundell, T. L. & Ascher, D. B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 58, 4066–4072 (2015).
Elgazar, A. A., Knany, H. R. & Ali, M. S. Insights on the molecular mechanism of anti-inflammatory effect of formula from Islamic traditional medicine: An in-silico study. J. Tradit. Complement. Med. 9, 353–363 (2019).
Sobeh, M. et al. Tannin-rich extracts from Lannea stuhlmannii and Lannea humilis (Anacardiaceae) exhibit hepatoprotective activities in vivo via enhancement of the anti-apoptotic protein Bcl-2. Sci. Rep. 8, 1–16 (2018).
Chakraborti, A. K., Garg, S. K., Kumar, R., Motiwala, H. F. & Jadhavar, P. S. Progress in COX-2 inhibitors: A journey so far. Curr. Med. Chem. 17, 1563–1593 (2010).
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
E.A.S., S.M.E. and M.R.M.: set the main idea of the manuscript; D.E.A. and E.A.E.G.: collecting and extraction of plant material, isolation and identification of pure compounds; all the authors: designing of the experiment; M.E.S.: carrying molecular docking of the isolated compounds; All authors: writing and revising the manuscript; E.A.S.: manuscript submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ali, D.E., Gedaily, R.A.E., Ezzat, S.M. et al. In silico and in vitro anti-inflammatory study of phenolic compounds isolated from Eucalyptus maculata resin. Sci Rep 13, 2093 (2023). https://doi.org/10.1038/s41598-023-28221-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-28221-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
