
Abstract
Sex differences in opioid dependence (OD) are genetically influenced. We conducted genomewide gene-by-sex interaction scans for the DSM-IV diagnosis of OD in 8,387 African-American (AA) or European-American subjects (43.6% women; 4,715 OD subjects). Among AAs, 9 SNPs were genome-wide significant at ADGRV1 (adhesion G-protein-coupled receptor V1, lead-SNP rs2366929*(C/T), p = 1.5 × 10−9) for sex-different risk of OD, with the rs2366929*C-allele increasing OD risk only for men. The top co-expressions in brain were between ADGRV1 and GRIK2 in substantia nigra and medullary inferior olivary nucleus, and between ADGRV1 and EFHC2 in frontal cortex and putamen. Significant sex-differential ADGRV1 expression from GTEx was detected in breast (Bonferroni-corrected-p < 0.002) and in heart (p < 0.0125), with nominal significance identified in brain, thyroid, lung, and stomach (p < 0.05). ADGRV1 co-expression and disease-enrichment analysis identifying the top 10 diseases showed strikingly sexually dimorphic risks. The enrichment and transcriptome analyses provided convergent support that ADGRV1 exerts a sex-different effect on OD risk. This is the first study to identify genetic variants contributing to sex differences in OD. It shows that ADGRV1 contributes to OD risk only in AA men, a finding that warrants further study.
Similar content being viewed by others

Introduction
Sex differences in opioid dependence (OD) have a strong biological basis1 with a genetic component2,3. Heritability estimates of heroin use are around 0.52. Although there is no report for sex-specific heritability estimates of OD specifically, heritability estimates of drug dependence are higher in men than women4. In animal models, genetic effects related to OD have differed by sex5,6,7. For example, a mouse model of the human OPRM1 (A118G) polymorphism found genotype-by-sex-specific reductions in the rewarding properties of morphine5. This variant does not, however, appear to affect OD risk in humans8. To our knowledge, the only systematic genome-wide search for the genes responsible for sex differences in human OD was a linkage study3.
Systematic search for the OD genetic risk variants (regardless of sex differences) using the genome-wide association study (GWAS) design has been reported. We published the first GWAS with genomewide significant findings9. The most compelling results in that study were the identification of genes involved in potassium signaling pathways (i.e., KCNC1 (Potassium Voltage-Gated Channel Subfamily C Member 1) and KCNG2 (Potassium Voltage-Gated Channel Modifier Subfamily G Member 2)) in the African American (AA) population, and genes involved in calcium signaling and long-term potentiation. Another GWAS was conducted in an Australian cohort, in which genetic data from opioid-dependent daily injectors were compared with that from opioid misusers who never progressed to daily injection, and identified several genomewide significant variants in CNIH3 (Cornichon Family AMPA Receptor Auxiliary Protein 3)10. In a recent study, we found that a variant on chromosome 15, rs12442183, near RGMA (Repulsive Guidance Molecule A), was genome-wide significantly associated with OD in the European American (EA) population11. RGMA encodes a central nervous system axon guidance protein called repulsive guidance molecule A. Risk allele rs12442183*T was related with higher expression of a specific RGMA transcript variant in frontal cortex. After chronic morphine injection, the homologous mouse gene, Rgma, was upregulated in the striatum of C57BL/6 J mice11.
We aimed to identify genetic variants exerting a sex difference in susceptibility to OD in a GWAS framework using the cohort of substance use disorder we have collected; characterize and annotate the identified genetic variants using publicly available databases of co-expressed genes and enrichment analysis; and use transcriptome analysis to identify biological mechanisms consistent with sex-specific effects on OD risk for the variants identified.
Materials and Methods
Subjects and the semi-structured assessment
This study analyzed data obtained from African-American (AA) and European-American (EA) participants in the Yale-Penn genetics of substance dependence cohort (total N = 8,387). The 4,944 AA and 3,443 EA samples were recruited from 2000 to 2013, as previously described9,12. These subjects represent two phases of collection: Yale-Penn-1 (N = 4,970) and Yale-Penn-2 (N = 3,417), which differed in their period of recruitment and the genotyping platforms used. The demographic characteristics of the study cohort are presented in Table 1. We assessed all subjects using the Semi-Structured Assessment for Drug Dependence and Alcoholism (SSADDA)13 and obtained certificates of confidentiality for all subjects from the National Institute on Drug Abuse (NIDA) and the National Institute on Alcohol Abuse and Alcoholism (NIAAA). The study protocol was approved by the Yale Institutional Review Board and the study was performed in accordance with the relevant guidelines and regulations. All subjects provided written informed consent
Genotype quality control, population stratification, and imputation
We genotyped the Yale-Penn-1 sample with the Illumina HumanOmni1-Quad array of approximately 988,000 single nucleotide polymorphisms (SNPs), and the Yale-Penn-2 sample with the Illumina HumanCore Exome array of approximately 266,000 exonic SNPs and 240,000 tagging SNPs. We excluded SNPs with a genotype call rate <98% or minor allele frequency (MAF) <1%.
We used the following measures to differentiate AA or EA subjects and control for population stratification. First, we conducted principal component (PC) analysis14 for the SNPs common among the genetic data for the Yale-Penn-1, Yale-Penn-2, and the 1000 Genomes phase 3 reference panel, which contains African, admixed American, European, East Asian and South Asian populations15. We then trimmed SNPs in LD (r2 > 0.2) using PLINK16. Using the remaining SNPs, we clustered the Yale-Penn subjects into different groups compared to the reference populations by the first three PCs in Euclidean space. Subjects were removed from the subsequent analyses if they were not clustered with African or European populations. Finally, we conducted a second PC analysis within each group to remove outliers greater than three standard deviations from the mean Euclidean distances. The resultant first 10 PCs were covariates in all subsequent association analyses to adjust for residual population stratification.
We imputed GWAS data to the 1000 Genomes phase 3 reference panel15, using Minimac3 implemented in the Michigan Imputation Server17. Post-imputation SNP exclusion metrics included: Hardy-Weinberg equilibrium p < 10−6, imputation accuracy < 0.8, or MAF < 5%. The final SNP counts for the subsequent association analyses were: Yale-Penn-1 sample, 8,775,706 SNPs in AAs and 6,417,418 in EAs; Yale-Penn-2 sample, 6,702,161 SNPs in AAs and 5,205,763 in EAs.
Genomewide gene-by-sex interaction scan for sex differences in OD
We performed genetic association tests for the DSM-IV diagnosis OD using a linear mixed model implemented in the program GEMMA18. A standard linear mixed-effect model was chosen to control for the relatedness among participants as our cohort contained a mixture of individuals ascertained using both an unrelated case-control design and a family design (11.9% of AAs and 7.9% of EAs came from the family design). We chose the standard linear mixed effect model implemented in GEMMA, where the effect estimates asymptotically converge to estimates of logistic regression for a binary outcome (such as OD affected versus OD unaffected here) when the sample size is large. We tested the gene-by-sex interaction effect and included the main effects of sex and SNP in addition to controlling for age, four other substance dependence diagnoses (alcohol, cocaine, nicotine, cannabis) and the first 10 PCs, to examine sex differences in genetic risk for OD. This model was examined separately for each of four datasets, i.e., both AA and EA subjects in the Yale-Penn-1 and Yale-Penn-2 samples. We then meta-analyzed the interactive effects of SNP-by-sex from Yale-Penn-1 and Yale-Penn-2 within each population using the inverse variance method that converted all the effects into a signed Z-score and tested for association using the Z-test, which was implemented in the program METAL19.
Co-expression, sex differences in gene expression, disease enrichment, and functional annotation
For the enrichment analysis, we first queried the target gene, i.e., the gene harboring the genomewide significant variants detected, to identify co-expressed genes using COXPRESdb20, a coexpression database of DNA-microarray and RNAseq-based expression data. After obtaining the top 100 co-expressed genes of the targeted gene (or genes) identified from the association analyses, we assessed functional enrichment using the WEB-based GEne SeT AnaLysis Toolkit (WebGestalt)21, a functional enrichment analysis web tool (see supplementary material for the setting of parameters). WebGestalt 2017 supports 12 organisms and 150,937 functional categories from public databases and computational analyses21. The enrichment analysis method ‘Disease Association Analysis’ of WebGestalt was used to test for enrichment of disease-associated genes among the top co-expressed genes with the targeted gene.
We studied sex differences in the expression of the targeted gene, i.e., ADGRV1, to investigate the functional roles of the top variants and the disease-enriched genes to provide insight into the molecular mechanism of sex differences in OD. To do this, we used the Genotype-Tissue Expression (GTEx) data and the tissue-specific gene expression database, the Brain eQTL Almanac, Braineac22. The RNA-Seq data of GTEx Analysis V7 (phs000424.v7.p2), included in the file, ‘GTEx_Analysis_2016-01-15_v7_RNASeQCv1.1.8_gene_tpm.gct.gz’, were downloaded from the GTEx Portal datasets23, and information about tissues and sex was retrieved from the files, ‘GTEx_v7_Annotations_SampleAttributesDS.txt’ and ‘GTEx_v7_Annotations_SubjectPhenotypesDS.txt’, respectively. We downloaded the gene expression (RNA-seq) data and the related phenotypes and analyzed the data using the general linear model, implemented in the SAS procedure “ proc glm,” where sex was modeled as the main effect in association with the levels of gene expression.
Results
Genomewide gene-by-sex interaction scan
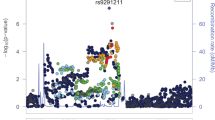
We summarized the result for each dataset and the meta-analysis within each population in the Manhattan plots (Supplementary Figure S1). The QQ plots show no systematic bias (Figure S1). A peak with the lead SNP rs2366929 and another eight SNPs in strong LD (r2 > 0.9) with the lead SNP (r2 > 0.8) reached genomewide significance (all p’s < 5 × 10−8) in the gene-by-sex interaction scans for OD in the meta-analyzed AA samples (Fig. 1a). For lead SNP rs2366929, each subsample also showed dose-response effects of nominal significance with p = 2.19 × 10−5 and p = 1.23 × 10−5 for Yale-Penn 1 and Yale-Penn 2, respectively. This peak of genomewide significance is located at ADGRV1 (Adhesion G protein-coupled receptor V1) on chromosome 5q14.3. The gene-by-sex interaction effects exhibit a gene dose-response relationship for the OD risk in AA men but not women (Fig. 1b). That is, the proportions of OD-affected to the total subjects (OD affected plus controls), within each of the three genotypes consistently show a gene dose-response trend relationship in men but not women (for lead SNP rs2366929*(C/T), chi-square test for trend in proportions, p = 1.1 × 10−14 in men and p = 0.15 in women). Figure 1b displays the increasing trend of the OD-affected proportions for men (CC > CT > TT) compared with women for the lead SNP rs2366929. This increasing trend in men versus women was observed for all nine SNPs identified in the association analyses (Supplementary Table S1 shows the trend tests for all nine SNPs). The MAF for the lead SNP increased 10.5% for the OD-affected versus the OD-unaffected men (0.353 vs. 0.248; comparison of two proportions, p < 0.0001); however, this increase was not present in women (0.241 vs. 0.265; p = 0.15). Table 2 displays the characteristics of the nine genome-wide significant SNPs.

Genomewide gene-by-sex interaction scans on opioid dependence (OD). (a) Regional association plot at the ADGRV1 (aka. GPR98) locus for the meta-analysis of the two phases of the African American (AA) sample. (b) Proportions of the OD affected for the AA sample for each genotype for the lead SNP rs2366929.
We identified no genomewide significant signals in the EA sample (Supplementary Figure S2) and the trans-ethnic meta-analysis of the AA and EA samples (Supplementary Figure S3).
Sex difference in ADGRV1 gene expression
We used GTEx data to identify tissues with differential ADGRV1 gene expression by sex for further annotating ADGRV1. We detected significant sex-differential ADGRV1 gene expression in breast (Bonferroni correction p < 0.002) and in heart (p < 0.0125) based on GTEx transcriptome data (dbGaP Accession phs000424.v7.p2) (Table 3). The ADGRV1 expression in heart supports a previous study of sexually dimorphic ADGRV1 expression in patients with non-ischemic human heart failure (Supplementary Table S2), in which ADGRV1 expression was lower in men than women, the same effect direction to what we found in the GTEx transcriptome. We identified four additional tissues with sex-differential ADGRV1 gene expression that were nominally significant, including brain (p = 0.018), thyroid (p = 0.021), lung (p = 0.038), and stomach (p = 0.045) (Table 3).
ADGRV1 co-expression and disease enrichment analysis
The top 100 genes co-expressed with ADGRV1 (Supplementary Table S3) identified by COEXPRESdb were subject to disease enrichment analysis. We found that ADGRV1 and 13 ADGRV1 co-expressed genes were significantly enriched within 10 diseases (all raw p’s < 0.01 and adjusted p’s < 0.05, Supplementary Table S4). The 13 genes are ABCC12, CFTR, CTAGE1, DAOA-AS1, EFHC2, FHL5, GRIK2, NAV3, SPO11, SYCP2, TAAR9, TPH2, and ZNF157. Among the 10 diseases, two are male-specific (male infertility and non-syndromic oligospermia), four affect males more than females (X-linked mental retardation, generalized epilepsy, Sezary syndrome, cutaneous T-cell lymphoma), three affect females more than males (chronic fatigue syndrome, panic disorder, and pseudoxanthoma elasticum), and one displays distinct disease characteristics between males and females (personality disorders).
We also correlated the expression of ADGRV1 with the expression of the 12 co-expressed disease risk genes (DAOA-AS1 expression was not available) across the 10 human brain tissues in the Braineac database. The correlation pattern of gene expression across the 10 brain tissues is displayed in Fig. 2. Robust correlations of gene expression between ADGRV1 and two genes, GRIK2 (Glutamate ionotropic receptor kainate type subunit 2) and EFHC2 (EF-hand domain containing 2), are ubiquitous across various human brain tissues. The highest correlations between the gene expressions of ADGRV1 and GRIK2 are in the substantia nigra and medulla inferior olivary nucleus and between ADGRV1 and EFHC2 in the frontal cortex and putamen (Fig. 2).

The mRNA expression of ADGRV1 was associated with 12 co-expressed genes across 10 human brain tissues in the Braineac database (Ramasamy et al.22). Pairwise Pearson correlation coefficients (r) for ADGRV1 and the 12 genes are labeled in each square of the heatmap. The color bar indicates the extent of significance, \(-{\log }10(p-{values})\), for the correlation coefficients.
Discussion
To our knowledge, this is the first systematic search for genetic variants contributing to sex differences in OD risk using GWAS. We identified a male-specific effect of ADGRV1 on risk of OD in the AA sample using genome-wide gene-by-sex interaction scans. Co-expressed genes, the enrichment analysis, and the transcriptome analysis provided mechanistic support for the finding that ADGRV1 exerts a sex-specific effect on OD risk.
ADGRV1 spans approximately 605.5 kb on chromosome 5q14.3 and encodes a member of the G-protein coupled receptor superfamily. In addition to the N- and C-terminal domains of ADGRV1, the encoded protein contains 7 putative Na( + )/Ca(2 + ) exchangers (defining the cation binding domain) and 21 N-linked glycosylation sites24. Its extracellular repeat domains bind Ca(2 + ) and are involved in signal transduction24. Members of the ADGR family play key roles in regulating cortical patterning, dendrite and synapse formation, and myelination25. Using GTEx transcriptome data and the Human Protein Atlas26, we found that ADGRV1 is also highly expressed in three endocrine glands (adrenal gland, thyroid, pituitary), lung, and multiple brain regions (Supplementary Figure S4). Opioid use disrupts hypothalamic-pituitary-adrenal (HPA) dynamics at the level of the pituitary or adrenal27. Chronic opioid use can cause pervasive endocrine dysfunction, which leads to hypogonadism, infertility, fatigue, anxiety, depression, menstrual irregularities, and so on28,29. Many of these disorders are among what we have identified for this study in the top ten diseases significantly enriched by the ADGRV1 co-expressed genes, such as fatigue, infertility, anxiety or panic disorder (Supplemental Table S4).
In the co-expressed genes and enrichment analyses, all of the top 10 diseases identified demonstrate sexually dimorphic risks or manifestations. We identified ADGRV1 as a male-specific risk factor in OD. The top 10 diseases also include two that are male-specific (i.e., male infertility and non-syndromic oligospermia) or four that predominantly affect males more than females (i.e., X-linked mental retardation, generalized epilepsy, Sezary syndrome, or cutaneous T-cell lymphoma). On the other hand, three of the 10 diseases affect more females than males (chronic fatigue syndrome, panic disorder, and pseudoxanthoma elasticum)30,31. Collectively, the top 10 diseases for ADGRV1 co-expressed genes are strikingly sexually dimorphic.
In disease-association studies, ADGRV1 has been well examined. Mutations in ADGRV1 are associated with familial febrile seizures32, and also contribute to focal and generalized epilepsy33,34, and epilepsy with myoclonic seizures35. Besides, ADGRV1 is associated with cardiac conduction disorder33 and ADGRV1 variants segregated in families with epilepsy co-occurring sudden death (due to cardiac conduction disorder), showing shared ADGRV1 risk variants between epilepsy and cardiac conduction disorder33. ADGRV1 expression in the heart has been reported for patients with non-ischemic heart failure (Supplementary Table S2), where expression was lower in men than women36. This sex difference in ADGRV1 expression in the heart was replicated in the GTEx transcriptome data (Table 3), although disease status was not controlled in this analysis because of the disease heterogeneity of GTEx donors. ADGRV1 has also been implicated in adverse metabolic effects of antipsychotic drugs37. Taken together, our novel discovery of ADGRV1 for contributing to OD risk in men add into this scientific literature on the disease-gene association of the strong candidate gene ADGRV1.
In the GTEx data, individual-level data do not include the diagnosis of OD. Thus our investigation into the association between sex differences in ADGRV1 expression and those in OD is not possible in that dataset. However, ADGRV1 is highly expressed in endocrine tissues (Supplementary Figure S4); opioid use could act on the tissues in which ADGRV1 is highly expressed (i.e., endocrine and brain), disrupting the normal G-protein coupled receptor signaling and hormone production and causing pathogenic cellular processes. The differences in ADGRV1 expression could affect stress responses or produce hormonal or behavioral effects that differ by sex in opioid users.
Regarding the co-expression patterns for the enriched genes expressed in brain, the identified brain regions show supportive evidence for neuropathology. The robust correlations in gene expression between ADGRV1 and GRIK2 and EFHC2 are ubiquitous in human brain. The top correlations in gene expressions between ADGRV1 and GRIK2 are in the substantia nigra and medulla inferior olivary nucleus (Fig. 2). The substantia nigra is enriched in dopaminergic neurons and plays an important role in reward38, while the medulla inferior olivary nucleus is implicated in motor learning and control39. Another set of top correlations in the expression are between ADGRV1 and EFHC2 in frontal cortex and putamen (Fig. 2). The frontal cortex contains most of the dopamine-sensitive neurons in the cerebrum and is associated with reward, attention, short-term memory, planning, and motivation40. The putamen is involved in reinforcement learning and various movements41. These results support a role for ADGRV1 in networks of co-expressed genes that regulate the neural activities involved in addiction-related functions, including reward, memory, and learning.
As for the other co-expressed genes involved in the top correlated brain regions, TPH2 encodes tryptophan hydroxylase 2, which catalyzes the rate-limiting step in the synthesis of serotonin. Chronic morphine and cocaine exert common effects on tyrosine hydroxylase in dopaminergic brain reward regions42. Mutations in TPH2 were associated with quality of life of patients in methadone maintenance treatment for heroin use disorder43, responses to cocaine treatment44, and a spectrum of psychiatric disorders (cf. a review and meta-analysis45). GRIK2 encodes a member of the kainate family of glutamate receptor subunits. In a mouse model, Grik2 mRNA levels were decreased after prolonged morphine treatment46. These results again support a role for ADGRV1 in networks of co-expressed genes that regulate the neural activities involved in opioid or drug addiction.
The variants at ADGRV1 reaching genomewide significance for a sex-difference in OD risk were identified in AAs, but not EAs. The minor allele “C” of lead SNP rs2366929 in the International Genome Sample Resource (IGSR)15 has MAF = 0.47 for Europeans, and MAF = 0.22 for Africans, consistent with the MAFs (0.248 male; 0.265 female) we observed in our admixed AA population (Table 2). Local or population-specific variation is important in mapping disease risk47,48. Although the current information is not enough to determine why the genetic effects in ADGRV1 were not seen in EAs, there are several plausible explanations. For example, effect sizes might be smaller in EA ancestries such that larger sample size may be necessary for discovery. Further studies with larger sample sizes and more power are warranted to investigate sex-differences in OD risk.
The GEMMA model we used for the current study includes 17 main effects (i.e., age, sex, SNP, and four other substance dependence on alcohol, cocaine, marijuana, and nicotine, and ten PCs) and one two-way interaction (i.e., SNP-by-sex). In theory, a saturated model that incorporates all of the two-way interaction effects would be ideal but would involve a total of 153 terms in the model, comprised of the 17 main effects and 136 pairwise interaction effects. We chose not to apply the saturated model because in previous simulations (unpublished data) we obtained effect estimates for the targeted interactions that were generally close to those estimated by the saturated model. However, the power was greatly reduced by the inclusion of a large number of interaction terms. We opted to use the more limited model.
The strengths of the present study include a moderately large cohort of substance dependence subjects who were carefully phenotyped and whose data were rigorously analyzed. However, the study findings are limited by the absence of a suitable cohort of AAs with OD in which to replicate the findings after we made extensive efforts to locate an existing dataset sufficiently powered for replication. Despite this limitation, we believe that the findings from this first study of sex- differences in genetic risk for OD can aid in understanding the underlying differences and facilitate better sex-specific prevention and treatment efforts for OD.
In summary, we identified ADGRV1 as a risk locus contributing to increased risk of OD in AA men by examining genetic variants systematically on a genome-wide scale. Functional annotation of this finding corroborated a substantial role for ADGRV1 in increasing OD risk, especially the potential pathogenic effects of variation in AGDRV1 on cardio-cerebral mechanisms, which could contribute to the risk of fatal opioid overdose or respiratory depression that has been observed following high-dose opioid exposure. Further study of this finding is warranted.
Data availability
The datasets analyzed for the current study are not all publicly available. The Yale-Penn-1 subsample can be requested from the NCBI dbGaP repository: A Genome-Wide Association Study of Heroin Dependence (dbGaP Study Accession: phs000277.v1.p1). The Yale-Penn-2 subsample has not deposited to the dbGaP, but will be available from the corresponding author on reasonable request.
References
Craft, R. M. Sex differences in analgesic, reinforcing, discriminative, and motoric effects of opioids. Experimental and clinical psychopharmacology 16, 376–385, https://doi.org/10.1037/a0012931 (2008).
Mistry, C. J., Bawor, M., Desai, D., Marsh, D. C. & Samaan, Z. Genetics of Opioid Dependence: A Review of the Genetic Contribution to Opioid Dependence. Current psychiatry reviews 10, 156–167, https://doi.org/10.2174/1573400510666140320000928 (2014).
Yang, B. Z., Han, S., Kranzler, H. R., Palmer, A. A. & Gelernter, J. Sex-specific linkage scans in opioid dependence. American journal of medical genetics. Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics 174, 261–268, https://doi.org/10.1002/ajmg.b.32507 (2017).
Kendler, K. S., Maes, H. H., Sundquist, K., Ohlsson, H. & Sundquist, J. Genetic and family and community environmental effects on drug abuse in adolescence: a Swedish national twin and sibling study. The American journal of psychiatry 171, 209–217, https://doi.org/10.1176/appi.ajp.2013.12101300 (2014).
Mague, S. D. et al. Mouse model of OPRM1 (A118G) polymorphism has sex-specific effects on drug-mediated behavior. Proceedings of the National Academy of Sciences of the United States of America 106, 10847–10852, https://doi.org/10.1073/pnas.0901800106 (2009).
Mogil, J. S., Chesler, E. J., Wilson, S. G., Juraska, J. M. & Sternberg, W. F. Sex differences in thermal nociception and morphine antinociception in rodents depend on genotype. Neuroscience and biobehavioral reviews 24, 375–389 (2000).
Verzillo, V., Madia, P. A., Liu, N. J., Chakrabarti, S. & Gintzler, A. R. Mu-opioid receptor splice variants: sex-dependent regulation by chronic morphine. Journal of neurochemistry 130, 790–796, https://doi.org/10.1111/jnc.12768 (2014).
Schwantes-An, T. H. et al. Association of the OPRM1 Variant rs1799971 (A118G) with Non-Specific Liability to Substance Dependence in a Collaborative de novo Meta-Analysis of European-Ancestry Cohorts. Behavior genetics 46, 151–169, https://doi.org/10.1007/s10519-015-9737-3 (2016).
Gelernter, J. et al. Genome-wide association study of opioid dependence: multiple associations mapped to calcium and potassium pathways. Biological psychiatry 76, 66–74, https://doi.org/10.1016/j.biopsych.2013.08.034 (2014).
Nelson, E. C. et al. Evidence of CNIH3 involvement in opioid dependence. Molecular psychiatry 21, 608–614, https://doi.org/10.1038/mp.2015.102 (2016).
Cheng, Z. et al. Genome-wide Association Study Identifies a Regulatory Variant of RGMA Associated With Opioid Dependence in European Americans. Biological psychiatry. https://doi.org/10.1016/j.biopsych.2017.12.016 (2018).
Sherva, R. et al. Genome-wide Association Study of Cannabis Dependence Severity, Novel Risk Variants, and Shared Genetic Risks. JAMA psychiatry 73, 472–480, https://doi.org/10.1001/jamapsychiatry.2016.0036 (2016).
Pierucci-Lagha, A. et al. Reliability of DSM-IV diagnostic criteria using the semi-structured assessment for drug dependence and alcoholism (SSADDA). Drug and alcohol dependence 91, 85–90, https://doi.org/10.1016/j.drugalcdep.2007.04.014 (2007).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nature genetics 38, 904–909, https://doi.org/10.1038/ng1847 (2006).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74, https://doi.org/10.1038/nature15393 (2015).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics 81, 559–575, https://doi.org/10.1086/519795 (2007).
Das, S. et al. Next-generation genotype imputation service and methods. Nature genetics 48, 1284–1287, https://doi.org/10.1038/ng.3656 (2016).
Zhou, X. & Stephens, M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nature methods 11, 407–409, https://doi.org/10.1038/nmeth.2848 (2014).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics (Oxford, England) 26, 2190–2191, https://doi.org/10.1093/bioinformatics/btq340 (2010).
Okamura, Y. et al. COXPRESdb in 2015: coexpression database for animal species by DNA-microarray and RNAseq-based expression data with multiple quality assessment systems. Nucleic Acids Res 43, D82–86, https://doi.org/10.1093/nar/gku1163 (2015).
Wang, J., Vasaikar, S., Shi, Z., Greer, M. & Zhang, B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res 45, W130–w137, https://doi.org/10.1093/nar/gkx356 (2017).
Ramasamy, A. et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nature neuroscience 17, 1418–1428, https://doi.org/10.1038/nn.3801 (2014).
GTEx-Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science (New York, N.Y.) 348, 648–660, https://doi.org/10.1126/science.1262110 (2015).
Nikkila, H. et al. Sequence similarities between a novel putative G protein-coupled receptor and Na+/Ca2+ exchangers define a cation binding domain. Molecular endocrinology (Baltimore, Md.) 14, 1351–1364, https://doi.org/10.1210/mend.14.9.0511 (2000).
Langenhan, T., Piao, X. & Monk, K. R. Adhesion G protein-coupled receptors in nervous system development and disease. Nature reviews. Neuroscience 17, 550–561, https://doi.org/10.1038/nrn.2016.86 (2016).
Thul, P. J. et al. A subcellular map of the human proteome. Science (New York, N.Y.) 356, https://doi.org/10.1126/science.aal3321 (2017).
Brown, T. T., Wisniewski, A. B. & Dobs, A. S. Gonadal and Adrenal Abnormalities in Drug Users: Cause or Consequence of Drug Use Behavior and Poor Health Outcomes. American journal of infectious diseases 2, 130–135 (2006).
Das, G. Chronic heroin dependence leading to adrenal insufficiency. Case reports in endocrinology 2014, 461816, https://doi.org/10.1155/2014/461816 (2014).
Rhodin, A., Stridsberg, M. & Gordh, T. Opioid endocrinopathy: a clinical problem in patients with chronic pain and long-term oral opioid treatment. The Clinical journal of pain 26, 374–380, https://doi.org/10.1097/AJP.0b013e3181d1059d (2010).
Faro, M. et al. Gender differences in chronic fatigue syndrome. Reumatologia clinica 12, 72–77, https://doi.org/10.1016/j.reuma.2015.05.007 (2016).
Germain, D. P. Pseudoxanthoma elasticum. Orphanet journal of rare diseases 12, 85, https://doi.org/10.1186/s13023-017-0639-8 (2017).
Nakayama, J. et al. A nonsense mutation of the MASS1 gene in a family with febrile and afebrile seizures. Annals of neurology 52, 654–657, https://doi.org/10.1002/ana.10347 (2002).
Coll, M. et al. Targeted next-generation sequencing provides novel clues for associated epilepsy and cardiac conduction disorder/SUDEP. PloS one 12, e0189618, https://doi.org/10.1371/journal.pone.0189618 (2017).
Scheel, H., Tomiuk, S. & Hofmann, K. A common protein interaction domain links two recently identified epilepsy genes. Human molecular genetics 11, 1757–1762 (2002).
Myers, K. A. et al. ADGRV1 is implicated in myoclonic epilepsy. Epilepsia, https://doi.org/10.1111/epi.13980 (2017).
Fermin, D. R. et al. Sex and age dimorphism of myocardial gene expression in nonischemic human heart failure. Circulation. Cardiovascular genetics 1, 117–125, https://doi.org/10.1161/circgenetics.108.802652 (2008).
Adkins, D. E. et al. Genomewide pharmacogenomic study of metabolic side effects to antipsychotic drugs. Molecular psychiatry 16, 321–332, https://doi.org/10.1038/mp.2010.14 (2011).
Ilango, A. et al. Similar roles of substantia nigra and ventral tegmental dopamine neurons in reward and aversion. The Journal of neuroscience: the official journal of the Society for Neuroscience 34, 817–822, https://doi.org/10.1523/jneurosci.1703-13.2014 (2014).
Schweighofer, N., Lang, E. J. & Kawato, M. Role of the olivo-cerebellar complex in motor learning and control. Frontiers in neural circuits 7, 94, https://doi.org/10.3389/fncir.2013.00094 (2013).
Kandel, E. R., Schwartz, J. H. & Jessell, T. M. Principles of Neural Science (Fourth ed.). 324 (McGraw-Hill, 2000).
Packard, M. G. & Knowlton, B. J. Learning and memory functions of the Basal Ganglia. Annual review of neuroscience 25, 563–593, https://doi.org/10.1146/annurev.neuro.25.112701.142937 (2002).
Beitner-Johnson, D. & Nestler, E. J. Morphine and cocaine exert common chronic actions on tyrosine hydroxylase in dopaminergic brain reward regions. Journal of neurochemistry 57, 344–347 (1991).
Wang, R. Y. et al. Impacts of GRIN3A, GRM6 and TPH2 genetic polymorphisms on quality of life in methadone maintenance therapy population. PloS one 13, e0201408, https://doi.org/10.1371/journal.pone.0201408 (2018).
Nielsen, D. A., Harding, M. J., Hamon, S. C., Huang, W. & Kosten, T. R. Modifying the role of serotonergic 5-HTTLPR and TPH2 variants on disulfiram treatment of cocaine addiction: a preliminary study. Genes, brain, and behavior 11, 1001–1008, https://doi.org/10.1111/j.1601-183X.2012.00839.x (2012).
Ottenhof, K. W., Sild, M., Levesque, M. L., Ruhe, H. G. & Booij, L. TPH2 polymorphisms across the spectrum of psychiatric morbidity: A systematic review and meta-analysis. Neuroscience and biobehavioral reviews 92, 29–42, https://doi.org/10.1016/j.neubiorev.2018.05.018 (2018).
Korostynski, M., Piechota, M., Kaminska, D., Solecki, W. & Przewlocki, R. Morphine effects on striatal transcriptome in mice. Genome biology 8, R128, https://doi.org/10.1186/gb-2007-8-6-r128 (2007).
Bustamante, C. D., Burchard, E. G. & De la Vega, F. M. Genomics for the world. Nature 475, 163–165, https://doi.org/10.1038/475163a (2011).
Kim, M. S., Patel, K. P., Teng, A. K., Berens, A. J. & Lachance, J. Genetic disease risks can be misestimated across global populations. Genome biology 19, 179, https://doi.org/10.1186/s13059-018-1561-7 (2018).
Acknowledgements
The Genotype-Tissue Expression Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from: ‘GTEx_Analysis_2016-01-15_v7_RNASeQCv1.1.8_gene_tpm.gct.gz’ in the GTEx Portal and the dbGaP accession number phs000424.v7.p2 on 07/31/2018. This work was supported by the National Institutes of Health Grant Nos R03DA047562 (to B.Z.Y.), and RC2 DA028909, R01 DA12690, R01 DA12690S1, R01 DA12849, R01 DA18432, R01 AA11330, and R01 AA017535 (to J.G.) and VA Connecticut Healthcare Center and Philadelphia VA Medical Center Mental Illness Research, Education and Clinical Centers. Genotyping services for a part of our genome-wide association study were provided by the Center for Inherited Disease Research and the Yale University Center for Genome Analysis, which is fully funded by Federal Contract No. N01-HG-65403 from the National Institutes of Health to the Johns Hopkins University. We thank James Poling, Ph.D., Yale University School of Medicine and APT Foundation, Roger Weiss, M.D., McLean Hospital, Kathleen Brady, M.D., Ph.D., and Raymond Anton, M.D., Medical University of South Carolina, and David Oslin, M.D., University of Pennsylvania, for their work in recruitment and assessment. We thank Ann Marie Lacobelle, M.S., and Christa Robinson, B.S., who provided technical assistance; Yaira Nunez and Michelle Slivinsky, who led the quality control effort for the Semi-Structured Assessment for Drug Dependence and Alcoholism interviews and phenotyping for the study sample; and John Farrell, Ph.D., Section of Biomedical Genetics, Boston University School of Medicine, who provided database management assistance.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study design, data acquisition, or data analysis and interpretation. B.Z.Y. conceived the study, contributed to analytical modeling, data analysis, and interpretation, and drafted the manuscript. Both H.Z. and Z.C. performed data analysis and interpretation. H.R.K. contributed to data acquisition and interpretation. J.G. contributed to the study design as well as data acquisition and interpretation. All authors participated in revising the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
H.R.K. has been a consultant or advisory board member for Alkermes, Indivior, and Lundbeck, and he is a member of the American Society of Clinical Psychopharmacology Alcohol Clinical Trials Initiative, which was supported in the past 3 years by AbbVie, Alkermes, Ethypharm, Indivior, Eli Lilly, Lundbeck, Otsuka, Pfizer, Arbor Pharmaceuticals, and Amygdala Neurosciences. Drs Kranzler and Gelernter are named as inventors on PCT patent application #15/878,640 entitled: “Genotype-guided dosing of opioid agonists”, filed January 24, 2018.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, BZ., Zhou, H., Cheng, Z. et al. Genomewide Gene-by-Sex Interaction Scans Identify ADGRV1 for Sex Differences in Opioid Dependent African Americans. Sci Rep 9, 18070 (2019). https://doi.org/10.1038/s41598-019-53560-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53560-0
This article is cited by
-
Tyrosine Hydroxylase Gene Polymorphisms Contribute to Opioid Dependence and Addiction by Affecting Promoter Region Function
NeuroMolecular Medicine (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
