
Abstract
The utility of human pluripotent stem cell–derived kidney organoids relies implicitly on the robustness and transferability of the protocol. Here we analyze the sources of transcriptional variation in a specific kidney organoid protocol. Although individual organoids within a differentiation batch showed strong transcriptional correlation, we noted significant variation between experimental batches, particularly in genes associated with temporal maturation. Single-cell profiling revealed shifts in nephron patterning and proportions of component cells. Distinct induced pluripotent stem cell clones showed congruent transcriptional programs, with interexperimental and interclonal variation also strongly associated with nephron patterning. Epithelial cells isolated from organoids aligned with total organoids at the same day of differentiation, again implicating relative maturation as a confounder. This understanding of experimental variation facilitated an optimized analysis of organoid-based disease modeling, thereby increasing the utility of kidney organoids for personalized medicine and functional genomics.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others


References
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007).
Bellin, M. et al. Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long-QT syndrome. EMBO J. 32, 3161–3175 (2013).
Kim, C. et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 494, 105–110 (2013).
Phelan, D. G. et al. ALPK3-deficient cardiomyocytes generated from patient-derived induced pluripotent stem cells and mutant human embryonic stem cells display abnormal calcium handling and establish that ALPK3 deficiency underlies familial cardiomyopathy. Eur. Heart J. 37, 2586–2590 (2016).
Ardhanareeswaran, K., Mariani, J., Coppola, G., Abyzov, A. & Vaccarino, F. M. Human induced pluripotent stem cells for modelling neurodevelopmental disorders. Nat. Rev. Neurol. 13, 265–278 (2017).
Aksoy, I. et al. Personalized genome sequencing coupled with iPSC technology identifies GTDC1 as a gene involved in neurodevelopmental disorders. Hum. Mol. Genet. 26, 367–382 (2017).
Jang, Y.-Y. & Ye, Z. Gene correction in patient-specific iPSCs for therapy development and disease modeling. Hum. Genet. 135, 1041–1058 (2016).
Paquet, D. et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533, 125–129 (2016).
Howden, S. E., Thomson, J. A. & Little, M. H. Simultaneous reprogramming and gene editing of human fibroblasts. Nat. Protoc. 13, 875–898 (2018).
Ader, M. & Tanaka, E. M. Modeling human development in 3D culture. Curr. Opin. Cell Biol. 31, 23–28 (2014).
Huch, M. & Koo, B.-K. Modeling mouse and human development using organoid cultures. Development 142, 3113–3125 (2015).
Suga, H. et al. Self-formation of functional adenohypophysis in three-dimensional culture. Nature 480, 57–62 (2011).
Eiraku, M. et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 472, 51–56 (2011).
Spence, J. R. et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470, 105–109 (2011).
Nakano, T. et al. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 10, 771–785 (2012).
Lancaster, M. A. et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379 (2013).
Kadoshima, T. et al. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl Acad. Sci. USA 110, 20284–20289 (2013).
McCracken, K. W. et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 516, 400–404 (2014).
Takasato, M. et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 526, 564–568 (2015).
Takasato, M., Er, P. X., Chiu, H. S. & Little, M. H. Generation of kidney organoids from human pluripotent stem cells. Nat. Protoc. 11, 1681–1692 (2016).
Pavenstädt, H., Kriz, W. & Kretzler, M. Cell biology of the glomerular podocyte. Physiol. Rev. 83, 253–307 (2003).
Brunskill, E. W., Georgas, K., Rumballe, B., Little, M. H. & Potter, S. S. Defining the molecular character of the developing and adult kidney podocyte. PLoS One 6, e24640 (2011).
Park, J. et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360, 758–763 (2018).
Lindström, N. O. et al. Conserved and divergent features of mesenchymal progenitor cell types within the cortical nephrogenic niche of the human and mouse kidney. J. Am. Soc. Nephrol. 29, 806–824 (2018).
Wu, D. et al. ROAST: rotation gene set tests for complex microarray experiments. Bioinformatics 26, 2176–2182 (2010).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Briggs, J. A. et al. Integration-free induced pluripotent stem cells model genetic and neural developmental features of Down syndrome etiology. Stem Cells 31, 467–478 (2013).
Yu, J. et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science 324, 797–801 (2009).
Forbes, T. A. et al. Patient-iPSC-derived kidney organoids show functional validation of a ciliopathic renal phenotype and reveal underlying pathogenetic mechanisms. Am. J. Hum. Genet. 102, 816–831 (2018).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Huber, W. et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121 (2015).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Risso, D., Ngai, J., Speed, T. P. & Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 32, 896–902 (2014).
Futschik, M. E. & Carlisle, B. Noise-robust soft clustering of gene expression time-course data. J. Bioinform. Comput. Biol. 3, 965–988 (2005).
Bates, D., Mächler, M., Bolker, B. & Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48 (2015).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
McCarthy, D. J. & Smyth, G. K. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics 25, 765–771 (2009).
Phipson, B., Lee, S., Majewski, I. J., Alexander, W. S. & Smyth, G. K. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann. Appl. Stat. 10, 946–963 (2016).
Young, M. D., Wakefield, M. J., Smyth, G. K. & Oshlack, A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, R14 (2010).
Chen, J., Bardes, E. E., Aronow, B. J. & Jegga, A. G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 37, W305–W311 (2009).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014).
Acknowledgements
We thank A. Christ and G. Baillie at the Institute for Molecular Bioscience, The University of Queensland, for sequencing services. We acknowledge A. Mallett and S. Alexander for assistance in ethics applications and patient recruitment. We thank D. Vukcevic and G.K. Smyth for valuable discussion regarding random effects modeling, and J. Maksimovic for initial analysis and mapping of the patient RNA-seq data. This study was funded by the National Institute of Diabetes and Digestive and Kidney Diseases (grant no. DK107344) and National Health and Medical Research Council of Australia (NHMRC) (grant nos. GNT1041277, GNT1100970, GNT1098654). The Murdoch Children’s Research Institute is supported by the Victorian Government’s Operational Infrastructure Support Program. M.H.L. is an NHMRC Senior Principal Research Fellow. A.O. is an NHMRC Career Development Fellow (grant no. GNT1126157). T.A.F. is an NHMRC Postgraduate Scholarship (grant no. GNT1114409) and Royal Australian College of Physicians Jacquot Award Recipient (grant no. APP1114409).
Author information
Authors and Affiliations
Contributions
B.P. advised on the experimental design, performed all the statistical analysis, and wrote the manuscript. P.X.E., M.T., L.J.H., J.S., T.A.F., and H.-J.Y. performed the differentiation experiments. P.X.E. prepared RNA and analyzed the qPCR data. H.-J.Y. and K.T.L. collected and presented the morphological immunofluorescence data. P.X.E. and A.N.C. performed the isolations and L.Z. performed the initial analysis for single-cell profiling. J.S., T.A.F., and L.J.H. performed the EpCAM+ and LTL+ MACS sorting, respectively. S.E.H., E.W., and J.S. generated the iPSC cell lines, and S.E.H. performed the CRISPR–Cas9 gene editing. A.O. advised on the experimental design and oversaw the statistical analysis. M.H.L. devised the study, designed and interpreted all the experimental data, and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
M.H.L. and M.T. hold intellectual property around the kidney organoid differentiation protocol. M.H.L. holds contract research agreements with Organovo Holdings. All other authors declare that they have no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 Differentially expressed genes and enriched pathways between consecutive time points in the kidney organoid differentiation protocol.
(a) Heat map of log2-normalized expression values for the top differentially expressed genes between consecutive time points in the kidney organoid differentiation protocol. Significant genes were determined with a TREAT test with an absolute log-fold-change cutoff of 1 and false discovery rate cutoff of 5% (two-sided). (b) Gene Ontology analysis of the top 100 upregulated genes between day 4 and day 0. (c) Gene Ontology analysis of the top 100 upregulated genes between day 7 and day 4. (d) Gene Ontology analysis of the top 100 upregulated genes between day 10 and day 7. (e) Gene Ontology analysis of the top 100 upregulated genes between day 18 and day 10. (f) Gene Ontology analysis of the top 100 upregulated genes between day 25 and day 18. The top 10 gene ontology categories for each comparison are shown. The top 100 upregulated genes between each comparison were tested for enrichment in the GO categories. A modified hypergeometric test was used to determine statistical significance, considering gene length bias. P values are one-sided.
Supplementary Figure 2 Expression patterns for 20 fuzzy clusters identified across the time points of the kidney organoid differentiation protocol.
Genes that displayed similar patterns of expression across the time course data were clustered using fuzzy c-means clustering, a soft clustering approach that assigns each gene gradual degrees of membership to each of the 20 clusters. We identified 7,682 genes to use as input for the Mfuzz algorithm. These genes were changing between at least one time point in the differentiation protocol.
Supplementary Figure 3 Analysis of the molecular program of human kidney differentiation.
(a) Selected cluster plots showing synexpression across the developmental time. Core genes with membership scores > 0.5 are shown in gray. Example genes of interest are shown in color for each cluster. Genes with an asterisk were validated with qPCR. Lists of core genes for each cluster are seen in Supplementary Table 3. (b) qPCR validation of selected genes from fuzzy clusters. At each time point qPCR was performed to measure expression of 10 genes in biologically independent samples. Each time point has n = 1 independent replicates (n = 4 in total).
Supplementary Figure 4 Pairwise correlation coefficients between all CRL1502-C32 organoids.
Spearman’s rho statistic was used to provide a rank-based measure of association. Correlations are estimated from 15,685 genes.
Supplementary Figure 6 Highly variable genes between CRL1502-C32 day 18 organoids.
(a) Unsupervised hierarchical clustering shows that organoids within batches cluster together. Expression values are log2-normalized. (b) Highly variable genes present in clusters 10 and 12. The overlap between the top 50 most variable genes and core genes in each cluster showed 7 and 16 genes in common in clusters 10 and 12, respectively. These genes are shown in color.
Supplementary Figure 7 Identification of key genes that predict relative maturation.
The 10 genes most significantly linearly related with the time series between days 7 and 25 were selected to build a multivariate linear model to predict organoid maturity of new samples.
Supplementary Figure 8 Evidence for nephron patterning and segmentation to capillary loop stage.
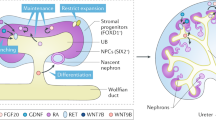
a. Diagram of anticipated morphological changes across nephron formation in mouse kidney. b. Immunofluorescence of nephrons forming within organoids across time using markers for proximal nephron (white; NPHS1), distal RV / medial nephron (red; JAG1) and distal nephron (green; CDH1). c. Serial single Z slices through confocal images of a single capillary-loop stage nephron showing segmentation along the length of the tubule and evidence for a lumen passing in and out of the plane of the image. Immunofluorescence performed using antibodies to markers of collecting duct (red; GATA3), epithelium (green; CDH1) and proximal nephron (white; NPHS1). Scale bar, 10 μm. d. QPCR of key nephrogenic genes between days 10 and 25 shows gradual loss of nephrogenic progenitors from day 10 to day 14 and formation of nephrons commencing at the same time. All images are representative images observed from within at least three organoids in this particular experiment. For the qPCR, each time point for each gene represents experimental triplicates.
Supplementary Figure 9 Heat map of log-normalized expression of selected kidney marker genes in single-cell data.
Expression values have been averaged across the cells per organoids per cluster.
Supplementary Figure 10 tSNE plots showing expression and distribution of selected kidney marker genes.
Each tSNE is made up of 8,361 cells from n = 4 biologically independent organoids.
Supplementary Figure 11 Stratified tSNE plot (by organoid) showing expression and distribution of three variable genes (NPHS2, PTPRO and MMP1) and two kidney marker genes (PAX2 and MAFB).
Each tSNE has 2,414 cells for organoid one, 2,202 cells for organoid two, 2,289 cells for organoid three and 1,418 cells for organoid four.
Supplementary Figure 12 Bright-field images of cell line RG_0019.0149.C6 displaying similar nephron formation and segmentation compared to the CRL1502-C32 line.
Scale bar is representative of 1 mm. There are three biologically independent organoids shown here for the RG_0019.0149.C6 line and one biologically independent organoid shown for CRL1502-C32.
Supplementary Figure 13 Heat map for the top 50 most highly variable genes between RG_0019.0149.C6 day 18 organoids.
Expression values are log2-normalized. Highly variable genes were identified using a random effects model.
Supplementary Figure 14 Top 10 enriched GO terms for genes contributing to vial-to-vial and residual variability between RG_0019.0149.C6 organoids.
The top 100 most variable genes were tested for enrichment in the GO categories. A modified hypergeometric test was used to determine statistical significance, taking into account gene length bias. P values are one-sided. GO categories with at least 10 genes are shown.
Supplementary Figure 15 Expression of key epithelial and interstitial genes across all organoids.
(a) Log-normalized expression of five epithelial-related genes for all total organoids and enriched nephron epithelium samples. (b) Log-normalized expression of five interstitial-related genes for all total organoids and enriched nephron epithelium samples. Day 7 and day 18 samples are stratified by batch.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–15
Supplementary Table 1
Differentially expressed genes between consecutive time points for the CRL1502-C32 time-series data
Supplementary Table 2
Top 20 enriched gene ontology and KEGG terms for top 100 upregulated and downregulated genes between consecutive time points
Supplementary Table 3
Core genes in 20 fuzzy clusters
Supplementary Table 4
Top 20 enriched gene ontology and KEGG for core genes in 20 fuzzy clusters
Supplementary Table 5
FANTOM5 transcription factors (hg19) present in each cluster
Supplementary Table 6
Random effects analysis identifying genes that contribute major sources of variation between CRL1502-C32 organoids
Supplementary Table 7
Marker gene analysis of single-cell clusters to identify cell types
Supplementary Table 8
Random effects analysis for cell line RG_0019.0149.C6, investigating contributions to vial-to-vial and residual variability
Supplementary Table 9
Top 20 enriched gene ontology terms for top 100 genes contributing to vial-to-vial and residual variability for RG_0019.0149.C6
Supplementary Table 10
Primer sequences for qRT–PCR
Rights and permissions
About this article

Cite this article
Phipson, B., Er, P.X., Combes, A.N. et al. Evaluation of variability in human kidney organoids. Nat Methods 16, 79–87 (2019). https://doi.org/10.1038/s41592-018-0253-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-018-0253-2
This article is cited by
-
Monocytes prevent apoptosis of iPSCs and promote differentiation of kidney organoids
Stem Cell Research & Therapy (2024)
-
Multiplexed bulk and single-cell RNA-seq hybrid enables cost-efficient disease modeling with chimeric organoids
Nature Communications (2024)
-
From cells to organs: progress and potential in cartilaginous organoids research
Journal of Translational Medicine (2023)
-
A live-cell image-based machine learning strategy for reducing variability in PSC differentiation systems
Cell Discovery (2023)
-
Integrated analysis of copy number variation-associated lncRNAs identifies candidates contributing to the etiologies of congenital kidney anomalies
Communications Biology (2023)
