
Abstract
Fibroblast growth factor receptor (FGFR) alterations drive oncogenesis in multiple tumor types. Here we studied pemigatinib, a selective, potent, oral FGFR1–FGFR3 inhibitor, in the phase 2 FIGHT-207 basket study of FGFR-altered advanced solid tumors. Primary end points were objective response rate (ORR) in cohorts A (fusions/rearrangements) and B (activating non-kinase domain mutations). Secondary end points were progression-free survival, duration of response and overall survival in cohorts A and B, and safety. Exploratory end points included ORR of cohort C (kinase domain mutations, potentially pathogenic variants of unknown significance) and analysis of co-alterations associated with resistance and response. ORRs for cohorts A, B and C were 26.5%, 9.4% and 3.8%, respectively. Tumors with no approved FGFR inhibitors or those with alterations not previously confirmed to be sensitive to FGFR inhibition had objective responses. In cohorts A and B, the median progression-free survival was 4.5 and 3.7 months, median duration of response was 7.8 and 6.9 months and median overall survival was 17.5 and 11.4 months, respectively. Safety was consistent with previous reports. The most common any-grade treatment-emergent adverse events were hyperphosphatemia (84%) and stomatitis (53%). TP53 co-mutations were associated with lack of response and BAP1 alterations with higher response rates. FGFR1–FGFR3 gatekeeper and molecular brake mutations led to acquired resistance. New therapeutic areas for FGFR inhibition and drug failure mechanisms were identified across tumor types. ClinicalTrials.gov identifier: NCT03822117.
Similar content being viewed by others


Main
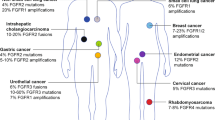
FGFR genes harbor pathogenic variants in an array of cancers1. Mutations, fusions and amplifications involving FGFR1–FGFR3 collectively occur in up to 7% of cancers1,2,3. As a key regulator of physiological functions, including cell migration, proliferation and survival, FGFR can drive oncogenesis when its signaling is altered by mutation1,4. Thus, FGFR is an attractive drug target, with selective FGFR inhibitors gaining regulatory approval in disease-specific contexts5,6,7,8.
The FGFR-altered tumor types with approved FGFR inhibitors are urothelial cancer, cholangiocarcinoma and myeloid and lymphoid neoplasms (MLNs). In advanced refractory urothelial tract and bladder cancers, where FGFR3 mutations are frequent2, the reversible FGFR1–FGFR4 inhibitor erdafitinib is approved for tumors harboring FGFR2 or FGFR3 point mutations or fusions6. In advanced refractory cholangiocarcinoma, where FGFR2 fusions predominate2, the reversible FGFR1–FGFR3 inhibitor pemigatinib5 and the irreversible FGFR1–FGFR4 inhibitor futibatinib are approved for tumors with FGFR2 fusions or other rearrangements7. In relapsed or refractory MLNs, pemigatinib gained approval for patients with FGFR1 rearrangements5.
Evidence of other potentially oncogenic and actionable FGFR alterations and potentially responsive tumors are emerging, providing compelling rationale for evaluating FGFR inhibition in a tumor-agnostic trial. FGFR1–FGFR3 fusions and point mutations in tumors of different histologies have demonstrated sensitivity to FGFR inhibition in early phase studies, including FIGHT-101, the first-in-human, phase 1 study of pemigatinib8,9,10,11,12,13,14,15,16. Moreover, FGFR alterations, including in-frame insertions and truncating deletions, have been described as potential oncogenic drivers but have not been clinically established as actionable17. Essential questions remain about the sensitivity of these rarer gene alterations to FGFR inhibition, the sensitivity of different FGFR-altered tumor histologies, the impact of specific gene co-alterations on response to FGFR inhibitors and mechanisms of drug failure across histologies.
Given the diversity of FGFR alterations and the variety of histologic contexts in which they appear, we sought to evaluate the therapeutic importance of FGFR alterations in multiple tumor types. Building on preclinical and phase 1 data9,13, the phase 2 FIGHT-207 basket study was designed to evaluate pemigatinib in patients with previously treated unresectable or metastatic solid tumors with FGFR1–FGFR3 fusions/rearrangements or mutations (NCT03822117; EudraCT, 2018-004768-69). Here we report the clinical outcomes of the study and the biological correlates of intrinsic and acquired resistance from analysis of tissue and circulating tumor DNA (ctDNA) samples.
Results
End points
The primary end points were ORR (percentage of patients with complete responses or partial responses) confirmed by independent review committee (IRC) per Response Evaluation in Solid Tumors (RECIST) v.1.1 criteria or Response Assessment in Neuro-Oncology (RANO) in cohorts A and B. Secondary end points were duration of response (DOR), IRC-assessed progression-free survival (PFS), overall survival (OS) and safety and tolerability as assessed by the incidence, type, and severity of adverse events (AEs) in cohorts A and B. Selected exploratory end points were ORR, DOR, PFS and OS in cohort C and genomic analysis of baseline and on-treatment tumor and plasma samples for markers of response and pemigatinib resistance. IRC-assessed clinical benefit rate (CBR) in all cohorts was conducted as a post hoc analysis.
Patients
Between 17 October 2019 and 12 July 2021, 111 patients enrolled. Of these, 107 patients were divided into three cohorts: A (FGFR1–FGFR3 fusions/rearrangements; n = 49), B (activating FGFR1–FGFR3 non-kinase domain single-nucleotide variants (SNVs); n = 32) or C (FGFR1–FGFR3 kinase domain mutations or variants of unknown significance (VUS) with potential pathogenicity; n = 26; Fig. 1a). Four remaining patients were included in the safety analysis but were excluded from the efficacy analysis per protocol because their FGFR alterations were not centrally confirmed (Supplementary Table 1). All patients received pemigatinib 13.5 mg orally once daily (QD) continuously. Of the patients in the efficacy-evaluable cohorts, 89 had ctDNA analysis for plasma collected at baseline and, among these, 73 had both baseline and progression samples (Fig. 1b).

a, Patient disposition. b, Samples for genomic analysis. The primary reason for treatment discontinuation is shown for each patient. *FoundationOne, FMI. †The four patients originally misassigned to cohort C based on local test uncertainty were analyzed here with the relevant set of gene alterations in cohorts A and B. EOT, end of treatment; FMI, Foundation Medicine, Inc.
Median age among efficacy-evaluable patients was 62 (range, 25–84) years. Overall, 57% of patients were women, 69% were white and 23% were Asian (Table 1). Cholangiocarcinoma (16%), urothelial tract/bladder cancer (11%) and glioblastoma (9.3%) were the most common tumors. Duration of treatment was longest in cohort A (median [range], 4.1 months [0.3–20.2]), followed by cohort B (3.2 months [0.2–15.4]) and cohort C (2.1 months [0.2–18.6]). The most common primary reason for treatment discontinuation was disease progression (77%) and the least common primary reason was AEs (5.4%).
Efficacy
The primary end points were ORRs in cohorts A and B. ORR (95% confidence interval (CI)) in cohort A was 27% (15%, 41%; n = 13) and 9.4% (2%, 25%; n = 3) in cohort B. ORR (95% CI) in cohort C, which was an exploratory end point, was 3.8% (0.1%, 20%; n = 1; Fig. 2 and Table 2). One patient in cohort A had a complete response. Secondary end points were DOR, PFS and OS in cohorts A and B. Median DOR was 7.8 months in cohort A and 6.9 months in cohort B. Median PFS and OS in cohort A were 4.5 and 17.5 months, respectively, and 3.7 and 11.4 months in cohort B, respectively. Efficacy outcomes are summarized in Table 2 and Extended Data Fig. 1.

Best percent change from baseline by RECIST or RANO for all evaluable patients with tissue NGS report and reported best change in lesion size: FGFR fusions/rearrangements (n = 48); FGFR actionable SNVs (n = 32); FGFR kinase domain mutations or VUS (n = 20). Best OR and PFS by IRC indicated where evaluable. Patients are arranged by FGFR alteration type. Bars are colored by major tumor histologies. Dashed lines indicate a criterion for partial response (change from baseline in target lesion size ≥30%). Tumors are grouped into the following histologies based on ≥5 patients: Cholangiocarcinoma, gynecologic cancers (cervical, endometrial and uterine), CNS (glioblastoma, low-grade pediatric glioma and astrocytoma), pancreatic cancer, breast cancer, urothelial tract/bladder cancer, non-small cell lung cancer and other (adrenal cancer, anal cancer, cancer of unknown primary origin, colorectal cancer, gastric/gastroesophageal cancer, gallbladder cancer, giant cell bone tumor, head and neck cancer, lung neuroendocrine cancer, nasopharyngeal cancer, ovarian cancer, prostate cancer, renal cell cancer, sarcoma and solitary fibrous tumor). Genomic analysis is included for all reportable samples and included NGS analysis of tumor tissues and ctDNA at baseline, and of ctDNA at time of progression (gray boxes indicate no report).
Objective responses were observed in multiple tumor types, including histologies for which no FGFR inhibitors are approved (Fig. 3 and Supplementary Table 2). Histologies of particular note included central nervous system (CNS) tumors, pancreatic tumors (all KRAS wild-type), cervical tumors and urothelial carcinomas harboring FGFR fusions or mutations.

Best percent change from baseline by RECIST or RANO (denoted by +) for all evaluable patients with tissue NGS report and reported best change in lesion size; BOR and PFS by IRC indicated where evaluable. Patients are arranged by major tumor histologies as previously described. Bars are colored by FGFR alteration type. Dashed lines indicate a criterion for partial response (change from baseline in target lesion size ≥30%; top) and clinical benefit (PFS ≥ 6 months; bottom). Tumors are grouped into the following histologies based on ≥5 patients: Cholangiocarcinoma, gynecologic cancers (cervical, endometrial and uterine), CNS (glioblastoma, low-grade pediatric glioma and astrocytoma), pancreatic cancer, breast cancer, urothelial tract/bladder cancer, non-small cell lung cancer and other (adrenal cancer, anal cancer, cancer of unknown primary origin, colorectal cancer, gastric/gastroesophageal cancer, gallbladder cancer, giant cell bone tumor, head and neck cancer, lung neuroendocrine cancer, nasopharyngeal cancer, ovarian cancer, prostate cancer, renal cell cancer, sarcoma and solitary fibrous tumor).
Safety
Among 111 patients who received ≥1 dose of pemigatinib, no new safety signals were seen. A full list of treatment-emergent AEs (TEAEs) is provided in Supplementary Table 3. The rate of grade ≥3 TEAEs was 68% (Extended Data Table 1). Fatal TEAEs occurred in six patients and included general physical health deterioration (n = 3; 2.7%), acute respiratory failure (n = 1; 0.9%), confusional state (n = 1; 0.9%) and sepsis (n = 1; 0.9%). None of the fatal TEAEs was considered by investigators to be related to pemigatinib. TEAEs leading to dose interruption and reduction occurred in 79 (71%) and 48 (43%) patients, respectively. Eight (7.2%) patients discontinued pemigatinib due to TEAEs. The most common any-grade TEAEs were hyperphosphatemia (84%) and stomatitis (53%). Nail toxicities and serous retinal detachment occurred in 45% and 14% of patients.
Genomic analysis of putative primary driver FGFR alterations
Clinical genomic analysis was performed on tissue and plasma samples collected from patients in cohorts A, B and C. Four patients from cohort C, initially determined with local testing to have VUS, were reassigned for this translational analysis to the other cohorts based on central review and reconsideration of their gene alterations. DMBT1-FGFR2 (patient 16) and FGFR1 rearrangements with indeterminate partner (patient 26 and patient 48) were assigned to cohort A and FGFR3 G370C (patient 57) was assigned to cohort B.
Among the FGFR gene alterations, fusions were most sensitive to FGFR inhibition (Fig. 2). The majority of patients in this cohort had type II FGFR fusions (n = 49; 94%), wherein FGFR was the 5′ fusion gene and the breakpoint occurred after the kinase domain in the region spanning intron 17 to exon 18 (ref. 18). Three additional rearrangements (BAG4-FGFR1, RGS12-FGFR3 and DMBT1-FGFR2) were considered putative type I fusions, a less-common oncogenic FGFR rearrangement observed primarily in MLNs, wherein a 5′ partner gene fuses with FGFR at a breakpoint after the transmembrane domain18. Both type I and II fusions are typically oncogenic and can be sensitive to FGFR inhibition. Although FGFR fusions and rearrangements were the most responsive gene alterations across tumor histologies, response was not uniform across histologies; differential rates of objective response and clinical benefit may indicate differential dependencies on FGFR across histologies with common gene alterations subgroups; however, given the relatively small populations evaluated for each histology, analysis of larger populations will likely be required for a more definitive assessment of FGFR pathway dependencies.
FGFR non-kinase domain SNVs that were considered actionable based on publicly available alterations databases or clinical study data (cohort B) were localized in extracellular and transmembrane domains. Among these FGFR SNVs, clinical benefit was observed for patients with urothelial carcinoma (n = 4), cholangiocarcinoma (n = 3) and squamous cell carcinoma (n = 1). Among five patients with intrahepatic cholangiocarcinoma that had FGFR2 SNVs, two (C382R (patient 79) and extracellular domain in-frame deletion I291_Y308D del (patient 78)) experienced partial response and two (W290C (patient 75) and Y375C (patient 77)) had stable disease with PFS of 10.5 and 3.7 months, respectively. While cholangiocarcinomas harboring these actionable mutations are less prevalent than FGFR2 rearrangements, they seem to represent an additional population that may benefit from FGFR inhibition.
FGFR kinase domain mutations (cohort C) were considered to be of uncertain actionability given that some kinase domain mutations demonstrate reduced sensitivity to FGFR inhibitors, including pemigatinib in preclinical models19. Notably, 2 of 12 patients with FGFR kinase domain mutations experienced clinical benefit. One patient with FGFR1 K656E grade II diffuse astrocytoma had a partial response (patient 100) and one patient with an FGFR1 N546K low-grade pediatric type glioma had stable disease and a 6.2-month PFS. Notably, activating mutations in K656 in the FGFR1 activation loop and N546, a controlling residue in the ‘molecular brake’ function, represent the two most common sites of activating FGFR1 SNVs in gliomas and other CNS tumors; however, among the remaining ten patients with kinase domain mutations without clinical benefit, eight had mutations in molecular brake residues (Extended Data Table 2; FGFR1 N546K/D (n = 5); FGFR2 N549K (n = 3)). Four additional patients in cohort C had mutations downstream of the FGFR2 kinase domain (patients 82, 89, 98 and 99). These mutations produce truncations before exon 18 and were recently described to be potentially pathogenic17. Among these, two patients (Q774* (patient 99) and E769fs (patient 98)) had stable disease ≥6 months, suggesting a modest but real clinical benefit.
Tissue next-generation sequencing (NGS) analysis also identified instances of FGFR amplification (defined as FGFR copy number ≥6). Concurrent FGFR gene amplifications were detected in nine patients (Supplementary Table 4), including concurrent amplifications with the corresponding FGFR mutation (n = 4) or FGFR fusion/rearrangement (n = 1) as well as FGFR amplifications occurring in an alternative FGFR to the enrollable FGFR gene alteration (n = 4). There were not enough patients in FIGHT-207 with concurrent FGFR gene amplification to conclude whether it had a meaningful impact on response to pemigatinib.
Correlation of co-alterations with patient outcomes
This FIGHT-207 basket study provided the opportunity to assess possible patterns of intrinsic resistance associated with co-alterations across multiple histologies and multiple FGFR alterations using combined genomic analysis of tumor tissue and ctDNA. Among patients with FGFR fusions/rearrangements and actionable SNVs (cohorts A and B, respectively), 79 evaluable patients had baseline tissue sequencing and 55 of these additionally had baseline ctDNA sequencing. Baseline ctDNA analysis had limited concordance with tissue NGS analysis for detection of FGFR variants and some co-alterations across all study samples (Supplementary Fig. 1), likely explained by multiple technical (for example, assay sensitivity, analytical thresholds for variant reporting and variable variant annotations) and biological (for example, age of samples and variable ctDNA shedding) factors. This correlation analysis is therefore focused on the complementary value of combining the gene alterations detectable by the two methods. Tumors were categorized as having a specific co-mutation if this mutation was seen by tissue or ctDNA analysis or both. Based on baseline tissue NGS analysis alone, patterns seen in patients with FGFR2 fusion-positive cholangiocarcinoma in FIGHT-202 were recapitulated here across multiple histologies harboring a variety of FGFR1–FGFR3 fusions and mutations. Specifically, none of 27 patients with tumors harboring alterations in TP53 had an objective response. Moreover, patients with tumors with TP53 alterations or one of several other tumor-suppressor genes had a lower PFS than those with wild-type copies of these genes (Extended Data Table 3). New correlations seen in FIGHT-207 included the associations with oncogenic alterations in the MAPK pathway or inactivating alterations in ARID1A with low PFS and between alterations in BAP1 and high clinical benefit. Notably, by baseline ctDNA analysis alone, these associations with ARID1A, MAPK pathway and BAP1 alterations held, but the association seen with TP53 and tumor-suppressor gene alterations did not (Extended Data Tables 4–6).
Acquired resistance in multiple histologies
All 73 patients who had post-progression ctDNA samples with matched baseline ctDNA also had baseline tumor biopsy molecular profiling. Fourteen (19%) patients acquired one or more secondary FGFR mutation in the kinase domain, in residues known or likely to confer resistance (Extended Data Table 7)20,21,22,23,24,25. For patients with cholangiocarcinoma, kinase domain mutations emerged exclusively in patients with clinical benefit from pemigatinib, supporting the case for acquired-resistance mechanisms. While diverse FGFR1–FGFR3 alterations and multiple tumor types were represented, the common pattern across histologies was the emergence of mutations in the gatekeeper residues (FGFR2 V564F/I/L; FGFR3 V555L/M) or closely neighboring residues (FGFR1 V559L/M) and molecular brake residues (FGFR1 N546K; FGFR2 N549D/H/K, E565A and K641R). Other emergent FGFR2 mutations included M537I, L617V and K659M. Ten of 14 (71%) patients developed polyclonal FGFR resistance mutations, with most patients developing concurrent gatekeeper and molecular brake residue mutations and many developing co-occurring mutations at the same codon (N549K and N549D). No mutations in an FGFR gene other than the originally altered FGFR gene were detected in post-progression plasma samples (for example, FGFR2 mutations were not detected in FGFR1-altered tumors).
In addition to secondary FGFR variants, new mutations in co-altered genes emerged in end-of-treatment but not baseline plasma ctDNA samples that may be associated with resistance as they involved TP53, PIK3CA and/or RAS (Extended Data Fig. 2)26,27. A larger set of additional emergent variants is presented in Extended Data Fig. 3.
Pooled co-alteration data from pemigatinib studies
To increase the power of our analysis, we investigated pooling the FIGHT-207 data with datasets from previous pemigatinib clinical studies, including FIGHT-101 (ref. 9) (phase 1/2; multiple histologies), FIGHT-201 (ref. 28) (phase 2; urothelial tract/bladder cancer) and FIGHT-202 (ref. 26) (phase 2; cholangiocarcinoma) in which co-alteration analysis has been previously reported. This analysis included patients with available tissue NGS analysis, FGFR fusions/rearrangements or actionable FGFR SNVs, centrally determined best overall response and treatment with pemigatinib at or above the recommended dose. Combined FIGHT-101 (n = 20) and FIGHT-207 (n = 72) data increased the power of the analysis for various solid tumors, but did not result in any change to the identification of co-altered genes significantly correlated with best overall response to pemigatinib. The tumor suppressors BAP1 and TP53 remained the genes whose alteration correlated significantly with objective response (Supplementary Table 5). Similarly, analysis of combined FIGHT-202 (n = 104) and FIGHT-207 (n = 11) data for patients with cholangiocarcinoma (Supplementary Table 6) did not result in any change to the identification of co-altered genes significantly correlated with best overall response to pemigatinib, and only TP53 was found to be nominally significant (significance was not maintained following stringency correction for multiple testing). Combined FIGHT-201 (n = 149) and FIGHT-207 (n = 13) data for patients with urothelial carcinoma (Supplementary Table 7) identified TSC1, which was reported in earlier analysis and CDKN1A, which was now found to be correlated nominally significantly with objective response. Notably, a combined analysis including samples from all four studies was not considered to be valid due to skewing resulting from the inclusion of larger sample sets for cholangiocarcinoma and urothelial carcinoma. This imbalance precludes inference of global correlations of co-alterations with response to pemigatinib.
Discussion
Oncogenic FGFR1–FGFR3 alterations are diverse in genomic structural changes, localization and functional consequences1. Although clinically validated only in cholangiocarcinoma and bladder cancer, FGFR alterations are present in multiple histologies2. Basket trials such as FIGHT-207 and the recently completed phase 1 basket study of futibatinib and the phase 2 RAGNAR basket study of erdafitinib offer growing evidence for expanding indications that seem to be actionable with FGFR inhibitors8,10. We report not only the safety and efficacy of pemigatinib in this exploratory phase 2 basket study, but leverage the depth of translational data collected in FIGHT-207 to provide five key insights into the biology of FGFR inhibition and the clinical utility of FGFR inhibitors.
First, we observed antitumor activity in cancers beyond cholangiocarcinoma and bladder cancer. Pemigatinib demonstrated activity in patients with CNS tumors, pancreatic cancer and cervical cancer. Similarly, clinical activity in multiple tumor types has been previously reported in other FGFR inhibitor studies8,10,12,29,30. While actionable FGFR alterations in these cancers are rare (<6%)2,3, the benefit of FGFR inhibition seen in this study highlights the value of routine comprehensive molecular screening in solid tumors.
Second, in addition to confirming previous reports that FGFR2 fusions and other rearrangements in cholangiocarcinoma are sensitive to FGFR inhibition10,12,30,31, this study showed in a dedicated cohort of FGFR-mutated tumors that specific FGFR2 SNVs, namely C382R and in-frame deletions, are associated with response to pemigatinib, suggesting that FGFR inhibitors may be effective in cholangiocarcinoma with FGFR2 alterations other than fusions and rearrangements.
Third, the dedicated cohort for activating FGFR2 mutations allowed us to explore the sensitivity of previously clinically unvalidated classes of mutations. In-frame deletions are consistently associated with objective responses. Exon 18 truncating mutations are associated with prolonged stable disease in some instances32. In general, de novo FGFR kinase domain mutations showed low response to pemigatinib, which was not unexpected as secondary mutations in the kinase domain represent a mechanism of acquired resistance21,22,24,33,34,35,36; however, we note that exceptional cases of clinical benefit did occur, including one patient with FGFR1 K656E and one patient with molecular brake mutation FGFR1 N546K. To systematically characterize the sensitivity of a diverse array of FGFR1–FGFR3 SNVs to FGFR inhibition in the clinic, we compiled available data from these patients from multiple FGFR inhibitor trials. We reviewed response data for 254 patients with FGFR1–FGFR3 SNVs treated with at least one of five FGFR inhibitors: pemigatinib (FIGHT-101 (ref. 9), FIGHT-201 (ref. 28), FIGHT-202 (ref. 31) and FIGHT-207), futibatinib10, infigratinib37,38, Debio1347 (refs. 32,39) or RLY-4008 (ref. 16) (Fig. 4). The resulting maps indicate that certain activating FGFR1–FGFR3 SNVs show repeated evidence of clinical benefit in response to FGFR inhibition, providing a rationale for clinical development for these patients.

Clinical response data for patients with alternative FGFR1–FGFR3 SNVs treated with pemigatinib (FIGHT-101 (n = 9)9, FIGHT-201 (n = 154)28, FIGHT-202 (n = 5)31, FIGHT-207 (n = 53)), futibatinib (n = 6)10, infigratinib (n = 5)37,38, Debio1347 (n = 5)32,39 or RLY-4008 (n = 14)16 are compiled by site of mutation with indicated rates of BOR. For cases with multiple FGFR co-mutations, additional mutations are noted in parentheses. Ig, immunoglobulin.
Fourth, study of potential mechanisms of primary resistance to pemigatinib revealed that baseline co-alterations in tumor suppressors, particularly TP53 and ARID1A, and oncogenic co-alterations in the MAPK pathway were associated with shorter PFS compared to those without alterations. Notably, consistent with data seen in FIGHT-202 where none of nine patients with cholangiocarcinoma and concurrent TP53 mutations showed an objective response26, in FIGHT-207 none of 27 FGFR-altered tumors of various histologies with concurrent TP53 mutations detected in tumor tissue showed an objective response to pemigatinib. Similarly, TP53 co-alterations were associated with lower ORRs in a cohort of patients with urothelial carcinoma and FGFR3 alterations treated with erdafitinib under real-world conditions40; however, in the FIGHT-201 study in FGFR-altered bladder cancer28, baseline concurrent TP53 alterations did not correlate with response or nonresponse to pemigatinib, cautioning against overgeneralization of subgroup analyses. A positive correlation was seen between alterations in BAP1 and both clinical benefit from and response to pemigatinib. FGFR2 and BAP1 alterations commonly co-occur in intrahepatic cholangiocarcinoma41, suggesting that the FGFR2 and BAP1 co-alteration may represent a distinct cooperative molecular etiology for some cancers. Overall, further prospective studies are needed to validate the correlations seen in this study to assess whether co-mutation status can inform patient selection.
Fifth, serial ctDNA analysis revealed mechanisms of acquired resistance to pemigatinib in a variety of tumor types. To date, our knowledge of acquired resistance to FGFR inhibitors has largely been restricted to FGFR2 fusion-positive cholangiocarcinoma22,24,33,34,35,36 and FGFR3-altered urothelial cancer21,25,40. In our study, patient 16 with advanced pancreatic cancer harboring a FGFR1–PDE4DIP fusion developed newly detected mutations in a residue near the gatekeeper (FGFR1 V559L/M) and in a molecular brake residue (FGFR1 N546K), standing as the first report of clinical on-target resistance to an FGFR inhibitor in an FGFR1-altered tumor or in pancreatic cancer to our knowledge. Consistent with laboratory characterization of acquired FGFR2 and FGFR3 resistance mutations in patients with cholangiocarcinoma and urothelial carcinoma, respectively21,24, our study also revealed that across FGFR1–FGFR3, the most common sites for progression-emergent kinase domain mutations are the gatekeeper residues and the molecular brake residues. Mutations in the gatekeeper residue sterically hinder pemigatinib from binding the receptor23, and mutations in the molecular brake residues result in functional gain and conformational shifts that disfavor inhibitor binding20,23. Polyclonal resistance with multiple mutations emerging at progression in the same patient was common in our study, as has previously been observed in cholangiocarcinoma but less commonly in urothelial carcinoma21,22,25. In addition to patients with cholangiocarcinoma, we saw polyclonal acquired resistance in patients with FGFR2-altered gastroesophageal/gastroesophageal junction cancer and cancer of unknown primary origin, FGFR3-altered non-small cell lung cancer and FGFR1-altered pancreatic cancer. Notably, several next-generation FGFR inhibitors have shown preclinical activity and preliminary clinical activity in patients with cholangiocarcinoma harboring FGFR2 kinase domain mutations and urothelial cancer harboring FGFR3 kinase domain mutations following previous FGFR inhibitor treatment16,21,32,42,43,44.
Besides the observed secondary mutations in FGFRs, molecular analysis of ctDNA at the time of progression identified other emergent gene variants that may contribute to acquired resistance (on-pathway resistance mutations). Genes with emergent variants were PIK3CA and RAS family genes (KRAS, NRAS and HRAS), presumably conferring alternatives for downstream pathway activation. In cholangiocarcinoma, FGFR2 fusions are generally mutually exclusive with alterations in MAPK pathway (KRAS, NRAS and BRAF) in baseline samples26, reflecting their roles as alternative oncogenic drivers. Notably, among the eight evaluable patients with pancreatic tumors in FIGHT-207, seven patients had FGFR fusions in the context of the KRAS wild-type background, highlighting the importance of testing for FGFR2 fusions in this population with few therapeutic options. Emergent PIK3CA and RAS family mutations were also found to co-occur with acquired FGFR2 resistance mutations in some patients with cholangiocarcinoma24. Co-alterations in PI3K and RAS pathways have similarly been described as conferring bypass resistance in nonclinical models for other FGFR inhibitors21,24. The interplay between oncogenic FGFR1–FGFR3 alterations, acquired on-target resistance mutations and emergent co-alterations compensating for FGFR inhibition requires further study and clinical validation.
One inherent limitation of the basket study design is that heterogeneous tumors and genetic alterations were included, some of which were not well represented. While tumor heterogeneity was intentional by design and a strength for signal finding, the study was terminated early by the sponsor for business reasons and some tumor and molecular cohorts, cohorts A and B, specifically, were therefore underpowered to definitively conclude questions of FGFR dependency for specific alterations and tumor types. The observations of response in this study are nevertheless valuable as indicators for potentially actionable FGFR alterations and tumors that warrant deeper investigation. Additionally, heavily pretreated patients enrolled in FIGHT-207 may have had more co-alterations that impacted response. Our study was not designed to evaluate whether the co-alterations we found to be associated with response and PFS were predictive of tumor response to pemigatinib. Interpreting these findings should be carried out with caution, as the association between co-alterations and outcomes may only be prognostic in nature. Finally, it should be noted that safety in this basket study is consistent with what was previously reported in patients with either cholangiocarcinoma or urothelial carcinoma treated with pemigatinib in the FIGHT-202 (ref. 31) and FIGHT-201 (ref. 28) studies.
In conclusion, we evaluated the clinical activity of pemigatinib in this phase 2 basket study comprising multiple tumor types and including previously untested FGFR1–FGFR3 alterations. We identified new therapeutic areas for FGFR inhibition in this study and ascertained the highest-sensitivity FGFR mutations from a compilation of studies, such that this curated list of mutations can be considered for eligibility in future FGFR inhibitor trials. We also discovered aspects of FGFR biology that transcend observations in cholangiocarcinoma and urothelial cancers and highlight the value of testing for FGFR alterations in multiple tumor types. Future work to predict response to pemigatinib is needed to better identify patients with cancer who might benefit from FGFR inhibitor therapy.
Methods
Study design
This open-label, single-arm, multicenter phase 2 study consisted of three cohorts defined by FGFR alteration category. Patients with in-frame FGFR1–FGFR3 fusions and rearrangements, including intact kinase domains, were assigned to cohort A. Cohort B consisted of patients with FGFR actionable SNVs, excluding kinase domain SNVs, considered known or likely to be activating and actionable. This set included specific somatic missense mutations, insertions or deletions of FGFR1–FGFR3 that were known or likely activating (based on clinical trial data and public alterations annotations by OncoKB, ClinVar and Omim)45,46,47. Cohort C included the remaining patients with FGFR1–FGFR3 mutations in the kinase domain or FGFR1–3 VUS with potential pathogenicity (Fig. 1). Patient enrollment and initial cohort assignment based on genomic or fluorescence in situ hybridization testing results from a local laboratory were permitted. Most patients had local testing using the FoundationOne CDx assay (Foundation Medicine), which detects genomic alterations in 324 genes (>500× median coverage for target genes)48. Additional local tests were performed by Caris, Tempus, Guardant360, Oncomine, Riken Genesis Oncoguard and Sophia Genetics laboratories.
Sex and/or gender were not considered in the study design or statistical analysis plan because FGFR alterations across histologies have not been shown consistently to predominate in one sex2. Moreover, the sex distribution in our study is similar to that of other basket studies of FGFR inhibitors8,10. Patients were recruited into FIGHT-207 irrespective of sex or gender. The sex of patients was self-reported, and gender was not collected.
The study was performed in accordance with the International Council for Harmonisation Good Clinical Practice, the principles embodied by the Declaration of Helsinki and local regulatory requirements. The study protocol was approved by the institutional review board of each study site before patient enrollment. All patients provided written informed consent before screening. The sponsor provided medical monitoring of the study, but no data safety monitoring board was established. A full list of investigators and study sites is provided in Supplementary Table 8. The study was terminated by the sponsor for business reasons.
Patients
Eligible patients were ≥18 years old with a histologically or cytologically confirmed advanced/metastatic or surgically unresectable solid tumor and radiographically measurable disease per RECIST v.1.1 or RANO criteria. Patients were required to have a documented FGFR1–FGFR3 mutation or fusion/rearrangement, disease progression after ≥1 line of previous systemic therapy, no therapy available likely to provide clinical benefit, ECOG PS ≤2, a baseline tumor specimen and willingness to avoid pregnancy or fathering children.
Exclusion criteria were previous receipt of a selective FGFR inhibitor; concurrent administration or receipt of anticancer medications ≤28 days before first pemigatinib dose; candidacy for potentially curative surgery; clinically notable corneal or retinal disorder confirmed by ophthalmologic examination; current evidence of ectopic mineralization or calcification; radiation administered ≤2 weeks before the first dose of pemigatinib or inadequate recovery from radiation-related toxicities; untreated CNS metastases or CNS metastases that have progressed; additional malignancy requiring active treatment or that is progressing, except for basal cell carcinoma of the skin, squamous cell carcinoma of the skin or in situ cervical cancer that has undergone potentially curative therapy; gastrointestinal disorders that could interfere with the absorption, metabolism or excretion of pemigatinib; inability to swallow and retain oral medication; clinically notable or uncontrolled cardiac disease, except for patients with a pacemaker or well-controlled atrial fibrillation; history or presence of clinically meaningful abnormal electrocardiogram; active chronic or current infectious disease requiring systemic antibiotic, antifungal or antiviral treatment ≤2 weeks before enrollment; active hepatitis B or hepatitis C infections; HIV infection; use of potent cytochrome P450 3A4 (CYP3A4) inhibitors or inducers or moderate CYP3A4 inducers ≤14 days or ≤5 half-lives, whichever is longer, before the first dose of pemigatinib; known hypersensitivity or severe reaction to pemigatinib or its excipients; inadequate recovery from toxicity or complications from major surgery; pregnancy or breastfeeding; receipt of an investigational drug for any indication; history of hypovitaminosis D requiring supraphysiologic doses to correct the deficiency; inability or unlikeliness of the patient to comply with the dose schedule and evaluations; any condition that in the investigator’s opinion may interfere with the full participation in the study, pose a notable risk to the patient or interfere with data interpretation; and inability of the patient to provide informed consent. Patients with laboratory values outside of normal ranges were also excluded. Nonpermitted hematology values were platelets ≤75 × 109 l−1, hemoglobin ≤9.0 g dl−1 or absolute neutrophil count ≤1.5 × 109 l−1. Transfusions were allowed with a 2-week washout period. Laboratory values suggesting hepatic dysfunction were alanine aminotransferase ≥3 × upper limit of normal (ULN; >5 × ULN for liver metastasis), aspartate aminotransferase ≥3 × ULN (>5 × ULN for liver metastasis), total bilirubin ≥1.5 × ULN (≥2.5 × ULN if Gilbert’s syndrome or liver metastasis) or alkaline phosphatase ≥3 × ULN. Prohibited renal values were serum creatinine clearance ≤30 ml min−1 based on the Cockcroft–Gault formula. Patients with serum phosphate >ULN or serum calcium outside of normal range or serum albumin-corrected calcium outside of the normal range when serum albumin is outside of the normal range were also excluded.
Treatment
Patients self-administered pemigatinib on a continuous basis at a starting oral dose of 13.5 mg QD in 21-day cycles until documented radiological disease progression, unacceptable toxicity, withdrawal of consent or physician decision.
End points and assessments
The primary end points were ORRs in cohorts A and B as determined by IRC. ORR was defined as the percentage of patients who achieved complete response or partial response per RECIST v.1.1 or RANO criteria. Disease was assessed by computed tomography or magnetic resonance imaging at baseline, every three cycles and at the end of treatment.
Secondary end points were IRC-assessed PFS (time from first dose to progressive disease or death, whichever is first) in cohorts A and B, respectively, DOR (time from the first assessment of complete response or partial response until progressive disease or death, whichever is first) in cohorts A and B, respectively, OS (time from first dose to death) in cohorts A and B, respectively, and safety and tolerability as assessed by the incidence and severity of TEAEs and treatment-related AEs according to the National Cancer Institute Common Terminology Criteria for Adverse Events v.5.0.
Selected exploratory end points included ORR, PFS, OS and DOR in cohort C, and baseline and on-treatment tumor and plasma genomic analysis associated with response and resistance.
IRC-assessed CBR (percentage of patients with CR, PR or SD ≥6 months) was also calculated for all cohorts as a post hoc analysis.
Statistical analyses
Approximately 60 and 90 patients were planned for cohorts A and B, respectively. Assuming ORRs of 35% in cohort A and 30% in cohort B, respectively, 60 and 90 patients were needed to ensure ≥90% power to reject the null hypothesis of ORR ≤ 15% with a one-sided test at the overall 0.025 level of significance. In cohort C, ≈20 patients were enrolled to provide ≥80% chance of observing at least four responders if the underlying ORR was 30%.
The efficacy population included all enrolled patients (n = 107) in cohorts A, B and C with FGFR alterations confirmed based on genomic testing results from the Foundation Medicine central laboratory who received ≥1 pemigatinib dose. The safety population included all enrolled patients who received ≥1 pemigatinib dose. The primary analysis of ORR in efficacy-evaluable patients in cohorts A and B was based on IRC-confirmed tumor responses, with 95% CI for ORR in all cohorts estimated using the Clopper–Pearson method. PFS, DOR and OS in efficacy-evaluable patients in all cohorts were analyzed with the Kaplan–Meier method; 95% CI for median PFS, DOR and OS were calculated using the generalization of Brookmeyer and Crowley’s method with log–log transformation. The exact 95% CI for the CBR in all cohorts was calculated. Data analyses were performed according to the statistical analysis plan using SAS v.9.4.
Translational analyses
Genomic data for baseline tissue included all evaluable patients (n = 107). Genomic data for plasma ctDNA data from baseline (n = 89) and paired at disease progression (n = 73) included all available samples from efficacy-evaluable patients. For available samples, PredicineCARE49 (Predicine) NGS analysis of plasma cell-free DNA was conducted for 152 genes (approximately 20,000× coverage for target genes) at baseline and at disease progression. Analysis focused on gene alterations, including SNVs, copy-number variants or rearrangements considered to be known or likely pathogenic based on the Foundation Medicine database and incorporating COSMIC status. Analysis of the gene co-alterations correlation with ORR or CBR used Fisher’s exact test, two-sided and correlation with PFS used a log-rank test. Analysis of genes with emergent pathogenic variants at progression included all genes with variants detected in ctDNA exclusively at progression. Translational data analyses were performed in R v.4.1.1.
Key protocol amendments
Amendment 3 (current version): February 2021
In the current version of the protocol, cohort definitions were further refined based on evolving terminology and to clarify which alterations were accepted for cohorts A and C. The current version includes other updates regarding tumor biopsy timing, COVID-19 pandemic mitigation strategies and regulatory requirements in Japan. This version of the full study protocol with confidential information redacted is included in the Supplementary Information supporting the article.
Amendment 2: January 2020
Cohort definitions were updated and details of the efficacy analysis were clarified. Other changes were made to incorporate US Food and Drug Administration review feedback received for other pemigatinib study protocols.
Amendment 1: February 2019
The protocol was amended to clarify the cohort assignment for patients with unknown fusion partners. Cohort A alterations were updated to include FGFR2 intron 17 rearrangements and cohort C to include FGFR1 and FGFR3 rearrangements with unknown fusion partners. Other revisions were made to incorporate updated safety information and Voluntary Harmonisation Procedure review feedback received for other pemigatinib study protocols.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Incyte Corporation is committed to data sharing that advances science and medicine while protecting patient privacy. The study protocol with confidential information redacted is provided in the Supplementary Information. Qualified external scientific researchers may request anonymized datasets owned by Incyte for the purpose of conducting legitimate scientific research. Researchers may request anonymized datasets from any interventional study (except phase 1 studies) for which the product and indication have been approved on or after 1 January 2020 in at least one major market (for example, United States, EU and Japan). Data will be available for request after the primary publication or 2 years after the study has ended. Information on Incyte’s clinical trial data-sharing policy and instructions for submitting clinical trial data requests are available at https://www.incyte.com/Portals/0/Assets/Compliance%20and%20Transparency/clinical-trial-data-sharing.pdf?ver=2020-05-21-132838-960. Anonymized gene variant analyses are available through controlled access at dbGaP, accession number: phs003590.v1.p1.
References
Babina, I. S. & Turner, N. C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 17, 318–332 (2017).
Murugesan, K. et al. Pan-tumor landscape of fibroblast growth factor receptor 1-4 genomic alterations. ESMO Open 7, 100641 (2022).
Helsten, T. et al. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin. Cancer Res. 22, 259–267 (2016).
Xie, Y. et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target Ther. 5, 181 (2020).
Incyte. PEMAZYRE (pemigatinib). Full prescribing information. (2022).
Janssen Biotech. BALVERSA (erdafitinib). Full prescribing information. (2022).
Taiho Oncology. LYTGOBI (futibatinib). Full prescribing information. (2023).
Pant, S. et al. Erdafitinib in patients with advanced solid tumours with FGFR alterations (RAGNAR): an international, single-arm, phase 2 study. Lancet Oncol. 24, 925–935 (2023).
Subbiah, V. et al. FIGHT-101, a first-in-human study of potent and selective FGFR 1-3 inhibitor pemigatinib in pan-cancer patients with FGF/FGFR alterations and advanced malignancies. Ann. Oncol. 33, 522–533 (2022).
Meric-Bernstam, F. et al. Futibatinib, an irreversible FGFR1-4 inhibitor, in patients with advanced solid tumors harboring FGF/FGFR aberrations: a phase I dose-expansion study. Cancer Discov. 12, 402–415 (2022).
Schram, A. M. et al. First-in-human study of highly selective FGFR2 inhibitor, RLY-4008, in patients with intrahepatic cholangiocarcinoma and other advanced solid tumors. J. Clin. Oncol. 39, TPS4165–TPS4165 (2021).
Nogova, L. et al. Evaluation of BGJ398, a fibroblast growth factor receptor 1-3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: results of a global phase I, dose-escalation and dose-expansion study. J. Clin. Oncol. 35, 157–165 (2017).
Liu, P. C. C. et al. INCB054828 (pemigatinib), a potent and selective inhibitor of fibroblast growth factor receptors 1, 2, and 3, displays activity against genetically defined tumor models. PLoS ONE 15, e0231877 (2020).
Sootome, H. et al. Futibatinib is a novel irreversible FGFR 1-4 inhibitor that shows selective antitumor activity against FGFR-deregulated tumors. Cancer Res. 80, 4986–4997 (2020).
Karkera, J. D. et al. Oncogenic characterization and pharmacologic sensitivity of activating fibroblast growth factor receptor (FGFR) genetic alterations to the selective FGFR inhibitor erdafitinib. Mol. Cancer Ther. 16, 1717–1726 (2017).
Subbiah, V. et al. RLY-4008, the first highly selective FGFR2 inhibitor with activity across FGFR2 alterations and resistance mutations. Cancer Discov. 13, 2012–2031 (2023).
Zingg, D. et al. Truncated FGFR2 is a clinically actionable oncogene in multiple cancers. Nature 608, 609–617 (2022).
De Luca, A. et al. FGFR fusions in cancer: from diagnostic approaches to therapeutic intervention. Int. J. Mol. Sci. 21, 6856 (2020).
Nakamura, I. T. et al. Comprehensive functional evaluation of variants of fibroblast growth factor receptor genes in cancer. NPJ Precis. Oncol. 5, 66 (2021).
Chen, H. et al. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol. Cell 27, 717–730 (2007).
Facchinetti, F. et al. Resistance to selective FGFR inhibitors in FGFR-driven urothelial cancer. Cancer Discov. 13, 1998–2011 (2023).
Goyal, L. et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov. 7, 252–263 (2017).
Lin, Q. et al. Characterization of the cholangiocarcinoma drug pemigatinib against FGFR gatekeeper mutants. Commun. Chem. 5, 100 (2022).
Wu, Q. et al. Landscape of clinical resistance mechanisms to FGFR inhibitors in FGFR2-altered cholangiocarcinoma. Clin. Cancer Res. 30, 198–208 (2023).
Pal, S. K. et al. Efficacy of BGJ398, a fibroblast growth factor receptor 1-3 inhibitor, in patients with previously treated advanced urothelial carcinoma with FGFR3 alterations. Cancer Discov. 8, 812–821 (2018).
Silverman, I. M. et al. Clinicogenomic analysis of FGFR2-rearranged cholangiocarcinoma identifies correlates of response and mechanisms of resistance to pemigatinib. Cancer Discov. 11, 326–339 (2021).
Yue, S. et al. FGFR-TKI resistance in cancer: current status and perspectives. J. Hematol. Oncol. 14, 23 (2021).
Necchi, A. et al. Pemigatinib for metastatic or surgically unresectable urothelial carcinoma with FGF/FGFR genomic alterations: final results from FIGHT-201. Ann. Oncol. 35, 200–210 (2024).
Chae, Y. K. et al. Phase II study of AZD4547 in patients with tumors harboring aberrations in the FGFR pathway: results from the NCI-MATCH trial (EAY131) subprotocol W. J. Clin. Oncol. 38, 2407–2417 (2020).
Papadopoulos, K. P. et al. A phase 1 study of ARQ 087, an oral pan-FGFR inhibitor in patients with advanced solid tumours. Br. J. Cancer 117, 1592–1599 (2017).
Abou-Alfa, G. K. et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 21, 671–684 (2020).
Cleary, J. M. et al. FGFR2 extracellular domain in-frame deletions are therapeutically targetable genomic alterations that function as oncogenic drivers in cholangiocarcinoma. Cancer Discov. 11, 2488–2505 (2021).
Goyal, L., Kongpetch, S., Crolley, V. E. & Bridgewater, J. Targeting FGFR inhibition in cholangiocarcinoma. Cancer Treat. Rev. 95, 102170 (2021).
Krook, M. A. et al. Efficacy of FGFR inhibitors and combination therapies for acquired resistance in FGFR2-fusion cholangiocarcinoma. Mol. Cancer Ther. 19, 847–857 (2020).
Krook, M. A. et al. Tumor heterogeneity and acquired drug resistance in FGFR2-fusion-positive cholangiocarcinoma through rapid research autopsy. Cold Spring Harb. Mol. Case Study 5, a004002 (2019).
Varghese, A. M. et al. Noninvasive detection of polyclonal acquired resistance to FGFR inhibition in patients with cholangiocarcinoma harboring FGFR2 alterations. JCO Precis. Oncol. 5, PO.20.00178 (2021).
Lassman, A. B. et al. Infigratinib in patients with recurrent gliomas and FGFR alterations: a multicenter phase II study. Clin. Cancer Res. 28, 2270–2277 (2022).
Gile, J. J. et al. FGFR inhibitor toxicity and efficacy in cholangiocarcinoma: multicenter single-institution cohort experience. JCO Precis. Oncol. 5, PO.21.00064 (2021).
Farouk Sait, S. et al. Debio1347, an oral FGFR inhibitor: results from a single-center study in pediatric patients with recurrent or refractory FGFR-altered gliomas. JCO Precis. Oncol. 5, PO.20.00444 (2021).
Guercio, B. J. et al. Clinical and genomic landscape of FGFR3-altered urothelial carcinoma and treatment outcomes with erdafitinib: a real-world experience. Clin. Cancer Res. 29, 4586–4595 (2023).
Mody, K. et al. Clinical, genomic, and transcriptomic data profiling of biliary tract cancer reveals subtype-specific immune signatures. JCO Precis. Oncol. 6, e2100510 (2022).
Rengan, A. K. & Denlinger, C. S. Robust response to futibatinib in a patient with metastatic FGFR-addicted cholangiocarcinoma previously treated using pemigatinib. J. Natl Compr. Cancer Netw. 20, 430–435 (2022).
Goyal, L. et al. TAS-120 overcomes resistance to ATP-competitive FGFR inhibitors in patients with FGFR2 fusion-positive intrahepatic cholangiocarcinoma. Cancer Discov. 9, 1064–1079 (2019).
Javle, M. M. et al. Phase II study of FGFR1-3 inhibitor tinengotinib as monotherapy in patients with advanced or metastatic cholangiocarcinoma: interim analysis. J. Clin. Oncol. 41, 539–539 (2023).
Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 (2018).
Chakravarty, D. et al. OncoKB: a precision oncology knowledge base. JCO Prec. Oncol. https://doi.org/10.1200/po.17.00011 (2017).
An Online Catalog of Human Genes and Genetic Disorders (OMIM, 2024); https://omim.org/
FoundationOne CDx. Technical Information (Foundation Medicine, 2023).
Predicine Inc. PredicineCARE (Predicine, 2023).
Acknowledgements
This study was funded by Incyte. We thank the investigators, staff and patients who participated in the FIGHT-207 study. We also thank J. Li, an employee of MD Anderson. Writing assistance was provided by E. McClure, an employee of ICON, and was funded by Incyte.
Author information
Authors and Affiliations
Contributions
S.D., M.F., J.G.-D., H.I., A.I., I.S., M.U. and T.Y. made substantial contributions to the acquisition and interpretation of the data, revised the paper critically for important intellectual content and approved the final version. X.L. made substantial contributions to the conception and design of the study and analysis and interpretation of the data, revised the paper critically for important intellectual content and approved the final version. J.R., M.L.V., N.O., A.G. and L.G. made substantial contributions to the conception and design of the study and the analysis and interpretation of the data, revised the paper critically for important intellectual content and approved the final version. M.S. made substantial contributions to the analysis and interpretation of the data, revised the paper critically for important intellectual content and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
J.R. served as a consultant or advisor for AADi, Avoro Capital Advisors, Boxer Capital, Chinese University of Hong Kong, Clarion Healthcare, Columbus Venture Partners, Cullgen, Debiopharm Group, Ellipses Pharma, Envision Pharma Group, Incyte, iOnctura, Macrogenics, Merus, Monte Rosa Therapeutics, Oncology One, Pfizer, Sardona Therapeutics, Vall d’Hebron Institute of Oncology/Ministerio de Empleo y Seguridad Social and Tang Advisors; received travel support from ESMO; received research funding paid directly to the institution from AADi, Amgen, Bayer, Bicycle Therapeutics, BioAtla, BioMed Valley Discoveries, Black Diamond Therapeutics, Blueprint Medicines, Cellestia Biotech, Curis, CytomX Therapeutics, Deciphera, Fore Biotherapeutics, Genmab, GlaxoSmithKline, Hummingbird, Hutchison MediPharma, IDEAYA Biosciences, Incyte, Kelun, Linnaeus Therapeutics, Loxo, Merck Sharp & Dohme, Merus, Mirati Therapeutics, Novartis, Nuvation Bio, Pfizer, Roche, Spectrum Pharmaceuticals, Symphogen, Taiho Pharmaceutical, Takeda/Millennium, Tango Therapeutics, Vall d’Hebron Institute of Oncology/Cancer Core Europe and Yingli Pharma; and reported a relationship with Vall d’Hebron Institute of Oncology/Ministerio de Empleo y Seguridad Social. S.D. received research funding paid directly to the institution from Basilea Pharmaceutica, Incyte, Nerviano Medical Science, Pfizer and Roche. M.F. received institutional research grants from AbbVie, Amgen, Aprea, AstraZeneca, Beigene, BMS, Checkmate, Elicio, Genmab, Gilead, GSK, Incyte, Jacobio, Lilly, Merck, Mirati and Novartis; served on advisory boards for AbbVie, AstraZeneca, Jazz Pharma, Beigene and Mirati; and is a consultant for Omega Therapeutics and Novartis. J.G.-D. received research funding from Astellas, BMS, GSK, Ipsen, Janssen, Pfizer, Roche and Sanofi and honoraria for serving as a speaker for AstraZeneca, BMS, Janssen and Roche. A.I. served on advisory boards for AstraZeneca, Bayer, Chugai, Daiichi Sankyo, GSK, Merck, MSD, Parthenon and Roche and received research grants from AstraZeneca, Bayer, BMS, GSK, Merck, MSD, Novartis, Parthenon, Pfizer and Roche. I.S. received institutional research grants from Alligator Bioscience, AstraZeneca, BMS, Cantargia AB, Genentech, Genmab, Incyte, Loxo/Bayer, Loxo/Lilly, MSD, Novartis, Orion, Roche, Pfizer, Puma Biotechnology and Symphogen and support for attending meetings and/or travel expenses from AstraZeneca, Incyte, Merck and Pfizer. M.U. received research grants from Astellas Pharma, AstraZeneca, Boehringer Ingelheim, CHUGAI Pharmaceutical, DFP, Eisai, Eli Lilly, Incyte, J-Pharma, Merck Biopharma, MSD, Novartis, Ono Pharmaceutical and Taiho Pharmaceutical and honoraria from AstraZeneca, CHUGAI Pharmaceutical, Eisai, Incyte, MSD, Novartis, Ono Pharmaceutical and Taiho Pharmaceutical. T.Y. received research grants from AbbVie, AMED, Ascent, AstraZeneca, GlaxoSmithKlineINBC, Incyte, Lilly, Merck Biopharma, MSD, Nanobiotix, Novartis, Ono Pharmaceutical, Pfizer, Roche and Syneos Health and lecture fees from AstraZeneca, Bristol-Meyers Squibb, Chugai, Eisai, Merck Biopharma, MSD, Ono Pharmaceutical and Rakuten Medical. M.L.V., N.O., X.L., A.G. and M.S. are employees and shareholders of Incyte. L.G. served on a data safety and monitoring committee for AstraZeneca and on advisory boards or as a consultant for Alentis Therapeutics AG, Black Diamond, Blueprint Medicine, Compass Therapeutics, Eisai/H3Biomedicine, Exelixis, Genentech, Kinnate, Incyte Corporation, Merck, QED Therapeutics, Servier, Sirtex Medical, Surface Oncology, Taiho Oncology, TranstheraBio, Tyra Biosciences, AbbVie, AstraZeneca and Cogent Biosciences.
Peer review
Peer review information
Nature Medicine thanks Alison Schram, Hui Zhang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Saheli Sadanand, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 (A) DOR and (B) PFS Based on IRC Assessment per RECIST v1.1 or RANO and (C) OS (Efficacy-Evaluable Population).
DOR, duration of response; IRC, independent review committee; NE, not estimable; OS, overall survival; PFS, progression-free survival; RANO, Response Assessment in Neuro-Oncology; RECIST, Response Evaluation Criteria in Solid Tumors.
Extended Data Fig. 2 Genes With Most Frequent Emergent Pathogenic or Resistance Variants at Progression by ctDNA.
Genes with pathogenic or known resistance variants detected by ctDNA at progression but not at baseline are plotted by number of emergent variants. ctDNA, circulating tumor DNA.
Extended Data Fig. 3 Across-Indication Analysis of Baseline Co-alterations.
Analysis of tumor tissue samples includes all evaluable patients from cohorts A, B, and C and central tissue next-generation sequencing (Foundation Medicine, Inc.) reporting. Known or likely pathogenic somatic gene alterations occurring in ≥2% of patients are shown. Patients are arranged by best percent change from baseline per RECIST or RANO. BOR, best overall response; cnv, copy number variation; CR, complete response; FGFR, fibroblast growth factor receptor; IRC, independent review committee; NA, not applicable; NE, not evaluable; PChg, percent change from baseline; PD, progressive disease; PR, partial response; SD, stable disease; snv, single-nucleotide variant.
Supplementary information
Supplementary Information
Supplementary Tables 1–8 and Fig. 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rodón, J., Damian, S., Furqan, M. et al. Pemigatinib in previously treated solid tumors with activating FGFR1–FGFR3 alterations: phase 2 FIGHT-207 basket trial. Nat Med (2024). https://doi.org/10.1038/s41591-024-02934-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41591-024-02934-7
