
Abstract
The entry of SARS-CoV-2 into host cells depends on the refolding of the virus-encoded spike protein from a prefusion conformation, which is metastable after cleavage, to a lower-energy stable postfusion conformation1,2. This transition overcomes kinetic barriers for fusion of viral and target cell membranes3,4. Here we report a cryogenic electron microscopy (cryo-EM) structure of the intact postfusion spike in a lipid bilayer that represents the single-membrane product of the fusion reaction. The structure provides structural definition of the functionally critical membrane-interacting segments, including the fusion peptide and transmembrane anchor. The internal fusion peptide forms a hairpin-like wedge that spans almost the entire lipid bilayer and the transmembrane segment wraps around the fusion peptide at the last stage of membrane fusion. These results advance our understanding of the spike protein in a membrane environment and may guide development of intervention strategies.
Similar content being viewed by others

Main
The COVID-19 pandemic, caused by SARS-CoV-2, has resulted in the deaths of millions of people and has had devastating socioeconomic impacts worldwide. SARS-CoV-2 is an enveloped positive-stranded RNA virus that enters a host cell after fusion of the viral and cell membranes. Although membrane fusion is energetically favourable, there are high kinetic barriers when two membranes approach each other, primarily due to repulsive hydration forces3,4. Free energy, which is required for viral membrane fusion to overcome the kinetic barriers, comes from refolding of the virus-encoded fusion protein from a prefusion conformation, which is metastable after proteolytic cleavage to a lower-energy stable postfusion state1,2. The fusion protein of SARS-CoV-2 is its spike (S) protein, which is a type I, heavily glycosylated membrane protein with a transmembrane (TM) segment embedded in the viral membrane, and another membrane-interacting region, known as fusion peptide (FP), which can insert into the target cell membrane5. Like other class I viral fusion proteins, including HIV-1 envelope glycoprotein, influenza haemagglutinin and Ebola glycoprotein1,2, the S protein is synthesized as a single-chain precursor, trimerized and subsequently cleaved by a furin-like protease from the infected host cell into two fragments—the receptor-binding fragment S1 and the fusion fragment S26. To initiate the next round infection, S protein binds to the receptor angiotensin converting enzyme 2 (ACE2) on the surface of a new host cell and is further cleaved at a second site in S2 (S2′ site) by the host serine protease TMPRSS2 or the endosomal cysteine protease cathepsin L5,7,8. It then undergoes large conformational changes to insert the FP into the target cell membrane and then refold into a hairpin-like postfusion structure, placing the TM segment and FP at the same end of the molecule, thereby dragging two membranes close together to complete fusion9,10.
Soon after the release of the first SARS-CoV-2 genomic sequence11, the structures of the S protein fragments—such as its ectodomain stabilized in the prefusion conformation12,13, the receptor-binding domain (RBD) in complex with ACE214,15,16,17 and fragments of S2 in the postfusion conformation18—were reported. In the prefusion ectodomain structure, S1 folds into four different domains, the N-terminal domain (NTD), RBD, C-terminal domain 1 (CTD-1) and CTD-2, and they wrap around the prefusion conformation of S2. The structures of the purified full-length S protein in both the prefusion and postfusion conformations9, as well as those of the S proteins present on chemically inactivated SARS-CoV-2 virions19,20,21, were subsequently determined, revealing additional structural details. The fusion fragment S2 contains several segments that have important structural and functional roles, including the FP, heptad repeat 1 (HR1), central helix (CH), connector domain (CD), heptad repeat 2 (HR2), TM segment and cytoplasmic tail (CT) (Extended Data Fig. 1). In the postfusion trimer of the S2 ectodomain, HR1 and CH form a central three-stranded coiled coil, around 180 Å long9. Part of HR2 adopts a helical conformation and packs against the groove of the HR1 coiled-coil to form a six-helix bundle and stabilize the hairpin-like postfusion structure. It is consistent with a membrane fusion model, in which HR1 undergoes a ‘jack-knife’ transition to insert the FP into the target cell membrane and HR2 folds back to bring the FP and TM segments close together6, in turn causing the two membranes to fuse into a single lipid bilayer. However, in all previous structures, the regions of the S protein near the viral membrane, although structurally and functionally critical22,23,24,25, are either not present or disordered.
Among the membrane-interacting regions of coronavirus spike proteins, the FP is one of the most crucial structural elements for membrane fusion, but its exact location has been the source of various competing theories26. Three membranotropic regions in the SARS-CoV S2 have been suggested as putative fusion peptides (Extended Data Fig. 1), including a potentially glycosylated segment upstream of the S2′ cleavage site (residues 770–788, named the N-terminal FP; ref. 27,28); the segment immediately downstream of the S2′ cleavage site (residues 798–816 or 798–835, widely accepted as the bona fide FP29,30); and the segment immediately upstream of HR1 (residues 858–886, also known as the internal FP24,28,31). Early studies supported the assignment of the internal FP because it had strong membrane-perturbing capacities and mutations in this region led to inhibition of S-mediated cell–cell fusion24. Subsequent studies have shown that the bona fide FP, which is highly conserved among coronaviruses, has an even stronger calcium-dependent membrane ordering activity—a main characteristic of a viral FP when studied as a short peptide in solution—than the internal FP29,30. Recently, several broadly neutralizing antibodies have been identified, recognizing the bona fide FP region of spike proteins from all known human-infecting coronaviruses, and the FP has therefore been suggested to be a potential target for developing universal coronavirus vaccines32,33. A fourth hydrophobic region (residues 1190–1203, also called the pretransmembrane domain (pre-TM) or aromatic domain) adjacent to the TM domain has been shown to be important in SARS-CoV fusion, and may act in concert with the FP to support membrane fusion34,35.
To visualize the membrane-interacting regions of postfusion SARS-CoV-2 spike protein, we reconstituted the full-length protein in lipid-based nanodiscs, induced the conformational changes using soluble ACE2 to prepare a sample of the postfusion spike in lipid bilayer and determined its structure using cryo-EM to reveal structural details that are critical for a full understanding of the SARS-CoV-2 entry. This study shows that the true FP, as defined by target-membrane insertion, is the segment containing residues 867–909. We therefore use the designation FP to refer to that segment exclusively.
Postfusion S2 trimer in lipid nanodiscs
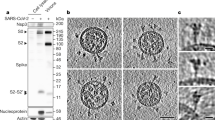
We reconstituted the purified, intact protein derived from the early variant G614 (B.1) in lipid nanodiscs using a circularized membrane scaffold protein (MSP)36,37. The reconstituted sample resolved by gel-filtration chromatography into three major peaks. SDS–PAGE analysis showed that peak I contained mainly the cleaved S protein in nanodiscs; peak II contained empty nanodiscs without the S protein; and peak III contained the free MSP (Fig. 1a). Negative-stain EM confirmed the identity of peak I as the nanodisc-associated prefusion S trimer, showing some flexibility between the ectodomain and the TM region. We induced the conformational changes of the prefusion S trimer by incubating the reconstitution reaction or the purified trimer in nanodiscs with soluble ACE2 (Fig. 1b and Extended Data Fig. 2). The ACE2-treated sample also resolved into three peaks on the basis of gel-filtration chromatography. Peak I contained primarily the dissociated S2 in nanodiscs; peak II contained empty nanodiscs co-eluting with dissociated S1 in complex with ACE2; and peak III contained unbound ACE2 co-eluting with free MSP (Fig. 1b and Extended Data Fig. 2). Negative-stain EM images of the peak I fraction showed a very rigid, postfusion S2 trimer projecting from the nanodisc. The S2′ site in the postfusion S2 trimer appeared to be uncleaved, as determined using SDS–PAGE analysis and N-terminal sequencing. These results demonstrate that ACE2 binding is sufficient to trigger the prefusion-to-postfusion conformational transition of the purified S trimer even in the context of the restricted bilayer membrane in a nanodisc.

a, The purified full-length SARS-CoV-2 S protein was reconstituted in lipid nanodiscs and resolved using gel-filtration chromatography on the Superose 6 column. The three major peaks (not at the void volume), as analysed by SDS–PAGE, are peak I, containing the prefusion S trimer in nanodiscs; peak II, containing empty nanodiscs; and peak III, containing free MSP. Representative 2D averages on the basis of negative-stain EM analysis of the peak I fractions are also shown. The box size of the 2D averages is around 880 Å. A280, absorbance at 280 nm. b, The purified full-length SARS-CoV-2 S protein was first reconstituted in nanodiscs, incubated with soluble ACE2 and then resolved using gel-filtration chromatography on the Superose 6 column. The three non-void-volume peaks, as analysed by SDS–PAGE, are peak I, containing the postfusion S2 trimer in nanodiscs; peak II, containing empty nanodiscs and dissociated S1 in complex with ACE2; and peak III, containing unbound ACE2 and free MSP. Representative 2D averages on the basis of negative-stain EM analysis of the peak I fractions are also shown. The box size of the 2D averages is around 880 Å. The experiments in a and b were repeated at least four times independently with similar results. Gel source data are provided in Supplementary Fig. 1.
Structure of postfusion S2 in membrane
We determined using cryo-EM the structure of the postfusion S2 trimer in nanodiscs, prepared as described above. The terminology for various segments of the S2 polypeptide chain are described in Fig. 2a. We recorded cryo-EM images on a Titan Krios electron microscope equipped with a Gatan K3 direct electron detector and used cryoSPARC38 for particle picking, two-dimensional (2D) classification, three-dimensional (3D) classification and refinement (Extended Data Fig. 3). One major class was obtained from 3D classification and refined to 2.9 Å resolution (Fig. 2b, Extended Data Figs. 3–5 and Extended Data Table 1). To improve the local resolution near the nanodisc, we performed additional masked local refinement, leading to a 3.3 Å map covering the TM and CT regions.

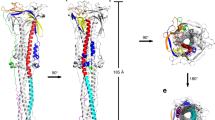
a, Schematic of the SARS-CoV-2 S2 fragment. Various segments include the S1/S2 cleavage site (S1/S2), the S2′ cleavage site (S2′), β718–729 in the S1/S2–S2′ fragment, the three-helix segment (3H), segment s816–834, the fusion peptide proximal region (FPPR), fusion peptide (FP), heptad repeat 1 (HR1), central helix (CH), connector domain (CD), β1127–1135, α1148–1155, heptad repeat 2 (HR2), the transmembrane anchor (TM), the cytoplasmic tail (CT) and glycans (tree-like symbols). b, The structure of the S2 trimer fits into a 2.9 Å density map. Three protomers (A, B and C) are coloured in green, blue and red, respectively. c, The overall structure of the S2 trimer in the postfusion conformation shown as a ribbon diagram. Various structural components, coloured as in a, include 3H, FPPR, FP, HR1, CH, CD, HR2, TM and CT. The S2′ cleavage site is in a disordered segment between Gln785 and Gly832, indicated by a dashed line. A top view of the transmembrane region in the nanodisc is also shown. d, One protomer each of the S2 trimer from the prefusion and the postfusion conformations superposed by the invariant CH and CD region. The locations of HR2, TM and CT disordered in the prefusion structure, as well as the b-FP in the postfusion structure are indicated by coloured rectangles. The N-terminal end of HR1, connected directly to the FP, translocates by about 185 Å during the prefusion-to-postfusion transition.
The overall structure of the postfusion S2 ectodomain in a nanodisc is nearly identical to that of the protein solubilized in detergent9 and that of SARS-CoV10. It also fits very well in the low-resolution map of the postfusion spike on the surface of SARS-CoV-2 virion, reconstructed by cryo-electron tomography20 (Extended Data Fig. 6). As reported previously, the core structure is a long central three-stranded coiled-coil made up by HR1 and CH (Fig. 2c). A three-stranded β-sheet formed by a β-hairpin from the CD and a segment (residues 718–729 (β718–729)) from the S1/S2-S2′ fragment, created by proteolytic cleavages, wraps around the C-terminal end of the CH coiled-coil. This connector β-sheet and CH form the invariant structure between the prefusion and postfusion structures. Another segment (residues 737–769 (s737–769)) in the S1/S2-S2′ fragment folds into three consecutive α-helices (3H), locked by two disulfide bonds, and also tightly packed against the groove of the CH coiled-coil. In our new structure, there is additional density for the segment immediately downstream 3H, which bends back towards the TM region and attaches to the surface of the postfusion structure, becoming disordered after Gln785. The S2′ site near Arg815, which is supposedly cleaved in this conformational state, remains invisible. In the C-terminal half of S2, a β-strand formed by residues 1127–1135 (β1127–1135) upstream of HR2 from another protomer joins the connector β-sheet to expand it into four strands, projecting HR2 towards the TM region and probably initiating the HR2 folding back. Furthermore, a two-turn helix formed by residues 1148–1155 (α1148–1155) wedges between two neighbouring 3Hs and a longer helix of HR2 makes up the six-helix bundle with the HR1 coiled-coil, together reinforcing the very rigid postfusion structure.
The membrane-interacting segments of S2, missing in all previous postfusion structures of any class I viral fusion proteins, are well resolved in our new structure. There are nine membrane-spanning helices (three per protomer) in the nanodisc region that were fully resolved in the refined maps. The FP region immediately upstream of HR1 forms a continuous α-helix extending the central coiled-coil to around 218 Å long and well into the lipid bilayer (Fig. 2c), possibly accounting for the rigidity of the entire postfusion structure including the transmembrane region. This first FP membrane-spanning helix is followed by a sharp U-turn within the lipid bilayer, and another helix that spans through the membrane once again and sends the adjacent FP proximal region (FPPR) back to the ectodomain side of the membrane. A total of six transmembrane helices of the FP from three protomers pack tightly together to form a blunted cone shape (Fig. 2c), probably required for effectively penetrating the target cell membrane. The third membrane-spanning helix is the TM segment, which tilts relative to the plane of the membrane, gently wrapping around the blunted cone. Part of the following CT appears to be embedded horizontally in the cytosolic headgroup region of the lipid bilayer, and three copies of it form a triangle that caps the tip of the transmembrane cone (Fig. 2c). Comparison of the prefusion and postfusion structures indicates that formation of the long postfusion central helix translocates the FP by 185 Å, and the FP reconfigures to become a hairpin-like wedge inserted into the target membrane (Fig. 2d). The FP not only inserts into the target membrane but also becomes a docking site for the TM helix in the final stages of membrane merger.
Definition of the SARS-CoV-2 FP and TM segments
The local refined maps with sidechain density for many residues in the transmembrane region enabled unambiguous modelling (Extended Data Fig. 5). It is clear that both s788–806 and s816–834 (Supplementary Fig. 2) are disordered but located outside the membrane (Fig. 2c), and they therefore do not function as the real FP. It is the FP that forms a hairpin-like wedge in a trimeric cone-shaped assembly to span almost the entire lipid bilayer (Fig. 3a). As expected, the membrane-inserted region of the wedge is largely hydrophobic with Trp886 and Phe888 at the blunted tip, while there are several charged residues (Asp867, Glu868 and Arg905) at the base of the wedge located in the headgroup region of lipid bilayer. Thus, a total of 43 residues (Asp867–Ile909) insert into membrane and make up the functional FP. Notably, Pro897 introduces a kink in the long central helix, making the region near the tip of the fusion wedge deviated from a canonical coiled-coil structure and also enabling the tight packing of the returning helix (residues 870–882) through an 876AXXXG880 motif to form the blunted cone shape (Fig. 3a). The interactions among the FPs from the three protomers are primarily mediated by polar residues (Thr881 and Gln895) before the Pro897-introduced kink, but by hydrophobic residues (Phe898, Met902, Phe906 and Ile909) after the kink (Extended Data Fig. 7).

a, The transmembrane region of the postfusion S2 fits in the EM density with one protomer (left). HR1 (yellow), FPPR (green), FP (red), HR2 (orange), TM segment (magenta) and CT (light green) are shown. Right, the atomic models for the FP and TM segment with key residues indicated. b, Interactions between the TM segment and FP near the ectodomain side of the membrane with key interface residues shown as a stick model. The elongated densities (grey) show potentially ordered phospholipids. c, Interactions between the TM segment and FP near the CT side of the membrane; key interface residues are shown as a stick model. d, Attachment of the FPPR and the segment of residues 857–866 to the lipid bilayer with the hydrophobic residues facing towards the hydrophobic core of the membrane highlighted as a stick model. e, Interactions between the FPPR and FP on the ectodomain side of the membrane; key interface residues are shown as a stick model.
For the transmembrane anchor, the TM helix begins at Tyr1215 and ends at Leu1234, and Trp1212 and Trp1217 from the pre-TM are embedded entirely in the lipid bilayer, indicating that the pre-TM is part of the TM domain, not a separate structural element. Furthermore, Cys1235 and Cys1236 are clearly not part of the TM helix and mark the beginning of the CT (Fig. 3a). These two cysteine residues, possibly palmitoylated in the virus39,40, are also very close to Cys1247 and Cys1248 from the CT of a neighbouring protomer, although formation of disulfide bonds between them on the cytosolic side is unlikely.
Interactions among the membrane regions
It is postulated that the FP of a class I viral fusion protein may interact with its TM domain in the final stage of the fusion process, but there has been no direct structural evidence for this interaction in an intact protein in the context of the membrane. The structure reported here shows an intimate FP–TM interaction in the SARS-CoV-2 postfusion spike. The TM segment packs against the FP from another protomer with two major contacts. First, Trp1212, Trp1217 and Phe1220 near the ectodomain side pack against the base of the fusion wedge; the contact may also involve some phospholipids, as suggested by strong elongated density in the vicinity, aligned perpendicular to the membrane plane (Fig. 3b). Second, Leu1234 near the CT side interacts with Trp886 and Phe888 from two different protomers at the tip of the fusion wedge (Fig. 3c). Three copies of the CT fragment come together to cap the tip of the fusion wedge.
The segment (residues 857–866) immediately upstream of the FP lies in the headgroup region of the lipid bilayer with several hydrophobic residues pointing towards the hydrophobic core of the membrane. This configuration probably helps orient the adjacent disulfide-locked FPPR (residues 835–856) in a manner with its C-terminal half embedded in the membrane as well (Fig. 3d,e). The N-terminal half of the FPPR, directly connected to the disordered s816–834, projects away from the membrane. Moreover, the FPPR packs against the FP wedge and Asp848 from the FPPR forms a salt bridge with Arg905 of the FP. Thus, the FPPR, which facilitates clamping down of the RBD in the prefusion conformation9, may also contribute to anchoring the membrane-interacting regions of the postfusion S2 structure.
Functional effect of designed mutations
To test the functional roles of the membrane-interacting structural elements, we assessed the impacts of several structure-guided mutations in the s816–834, FP, FPPR and CT regions. As shown in Fig. 4a, we substituted multiple hydrophobic residues in the s816–834, which would in principle be critical for membrane interaction, with charged residues (Mut-1); we mutated the key residues that participate in the FPPR interaction with the FP (Mut-2); we introduced multiple charged residues in the FP (Mut-3 and Mut-4) and mutated its AXXXG motif (Mut-5 and Mut-6); and we changed the two cysteine residues that mark the beginning of the CT (Mut-7).

a, Structure-guided design of mutations in the s816–834, FPPR, FP and CT regions. The wild-type sequences are shown in black and the mutated residues in red. aa, amino acids. b,c, Cell–cell fusion mediated by the G614 S and the designed mutants. HEK293T cells transfected with the full-length S protein expression plasmids were fused with ACE2-expressing cells. Cell–cell fusion led to reconstitution of the α and ω fragments of β-galactosidase to form an active enzyme, and the fusion activity was then quantified using a chemiluminescent assay. Cells not transfected with ACE2 were used as negative (neg.) controls. The experiment was repeated four times with similar results. Data are mean ± s.d. d, Cell–cell fusion mediated by the Delta-variant S and its mutants. The negative control conditions included no spike and no ACE2, no spike, no ω fragment of β-galactosidase and no α fragment of β-galactosidase. The experiment was repeated three times with similar results. Data are mean ± s.d.
When transiently transfected into HEK293T cells, these mutants expressed the same level of S, but the extent of cleavage between S1 and S2 was substantially reduced for Mut-3, Mut-4, Mut-5 and Mut-6, all of which are in the FP (Extended Data Fig. 8a). Like fusion peptides and fusion loops of many other viral fusion proteins41,42,43, the FP is buried in the prefusion conformation, in the present case by a segment near the N terminus of S2 from another promoter9,12,13. When detected by monoclonal antibodies using flow cytometry (Extended Data Fig. 8b), the recognition patterns by RBD-specific antibodies (C63C8, G32B6, C63C7, REGN10933 and REGN10987)44,45, NTD-specific antibodies (C12C9, 4A8 and C81D6)44,46, S2-specific antibodies (C163E6 and 3H3)44,46 and ACE2 for Mut-1, Mut-2 and Mut-7 were very similar to that of the wild-type S, except that the overall binding levels were substantially lower for Mut-1, suggesting that the mutations had some impact on protein secretion. The secretion level of Mut-3 was very low, as judged by binding to all antibodies and ACE2, indicating that this mutant protein might not be folded correctly. The patterns for binding to the antibodies and ACE2 were similar among Mut-4, Mut-5 and Mut-6, but they bound more weakly to RBD-specific antibodies and the neutralizing NTD-specific antibody 4A8 while much more strongly to S2-specific antibodies and to the non-neutralizing NTD-specific antibody C81D6 compared with the wild-type S, consistent with large structural rearrangements caused by the mutations in these proteins. These results suggest that the FP does not tolerate multiple mutations in the prefusion conformation, consistent with its buried position. In an S-mediated cell–cell fusion assay9, Mut-1 showed reduced fusion activity, whereas Mut-7 behaved like a wild-type spike; all of the other mutants showed no detectable fusion activity (Fig. 4b). Thus, as our structure shows, the s816–834 is not the FP and does not interact with the membrane, as introducing multiple charged residues does not abolish membrane fusion. Moreover, the two cysteine residues in the CT close to the TM domain, which have the potential to form interchain disulfide bonds, are dispensable for membrane fusion. Mut-3, Mut-4, Mut-5 and Mut-6 lost their membrane fusion activity, but their mutations also had a substantial impact on the prefusion structures of the mutant proteins. We next created additional constructs (Mut-8–Mut-15), each with a single point mutation in the FP, and identified three mutants (Mut-8, Mut-11 and Mut-14) that preserved the prefusion conformation but showed almost no detectable membrane fusion (Fig. 4c and Extended Data Fig. 8c,d). Similar results were obtained when the mutations of Mut-1, Mut-2, Mut-8, Mut-11 and Mut-14 were introduced in the Delta variant spike (Fig. 4d and Extended Data Fig. 8e,f). These results confirm that the FP is a key functional structural element in the SARS-CoV-2 spike.
Discussion
Our structure of the SARS-CoV-2 postfusion S2, with membrane-associated elements interacting with a lipid bilayer, has resolved a long-standing contentious issue in the coronavirus field regarding the functional identity of the FP. Although both the s788–806 and FP were suggested as candidates, the s816–834 subsequently became widely accepted as bona fide because it was more conserved among coronaviruses and showed a stronger activity than the other two in perturbing membrane structure as a synthetic peptide29,30. Our results show unambiguously that only the FP interacts with membrane. Our results underscore the risk of studying fusion peptides when taken out of the context of intact viral fusion proteins, because many hydrophobic peptides can induce structural changes of a lipid bilayer. For example, the FPPR, also identified as fusion peptide 2 for SARS-CoV in some studies30, projects part way into the membrane headgroup region and could therefore modify the local membrane structure, but it does not insert into the hydrophobic part of the lipid bilayer. From our structure, neither the s788–806 nor the s816–834 inserts into the membrane in the postfusion structure and therefore neither is likely to function as a real FP. The conclusion is further supported by the evidence that introducing multiple charged residues into s816–834 does not abolish the membrane fusion activity of the full-length S and by the presence of a conserved N-linked glycosylation site in s788–806. We cannot rule out the possibility that s788–806 and s816–834 may transiently interact with the membrane during the fusion process. Nevertheless, membrane insertion of the FP does not seem to depend on s788–806 or s816–834 as it is probably directly coupled with the refolding of adjacent HR1 into the coiled-coil. Moreover, a fusion peptide may not have to be fully conserved in its amino acid sequence provided that it maintains the ability to insert correctly into the target membrane. Indeed, there is a four-residue deletion at the tip of the FP in some coronaviruses, including HCoV-OC43 and HCoV-229E (Extended Data Fig. 6), but it is not likely to alter the overall shape of the fusion wedge and therefore its conserved function.
The substantial lipid-bilayer interaction surface of the fusion wedge observed here is consistent with the stability of the extended intermediate, which has been shown to persist for many minutes, even after release of S1 and its associated ACE2, when SARS-CoV-2 binds at the cell surface, unless the pH falls below neutral47. Fusion peptides with less extensive hydrophobic contacts, such as those postulated for the influenza virus fusion peptide48, would probably have shorter lifetimes.
Our structure also provides a more accurate structural definition of the TM domain, as well as the direct evidence for interactions between the FP and TM segment in the merged membrane for a class I viral fusion protein. The previously designated pre-TM segment, now shown to be part of the TM segment, contributes to the packing of the TM segment against the FP, explaining its functional role in membrane fusion. One study reported that a single-residue insertion into the TM segment of SARS-CoV S led to a complete loss in membrane fusion49, and the observation was difficult to interpret on the basis of the presumptive trimeric TM domain structure in the prefusion conformation50. The insertion was between Gly1201 and Phe1202 of SARS-CoV, which correspond to Gly1219 and Phe1220 in SARS-CoV-2. Such an insertion would cause a rotation of the C-terminal portion of the TM helix possibly together with the CT in the postfusion structure, thereby disrupting the interactions between the FP and the TM segment and CT and blocking the last step of membrane fusion. These results, together with our new structure, highlight the functional importance of the interactions among the membrane-interacting elements within the lipid bilayer, which have not been recognized previously. Nevertheless, it remains an open question how and to what extent these interactions functionally contribute to membrane fusion.
Finally, our structure suggests that the real FP of SARS-Cov-2 spike is probably not a useful vaccine target as it is well protected and is not accessible by antibodies in the prefusion conformation9,12,13, nor is it exposed after inserting into the target cell membrane in the prehairpin intermediate state. The s816–834, targeted by several broadly neutralizing antibodies32,33, may be conserved for other structural or functional reasons, which will require further investigation. Moreover, the interactions among the FP and FPPR, TM segment and CT shown in our structure may provide therapeutic targets for developing peptide-based or small-molecule fusion inhibitors. In particular, mutations in the FPPR, designed to disrupt its interaction with the FP, completely abolish fusion activity, raising the possibility of identifying therapeutic candidates that target this interaction and block viral infection.
Methods
Expression constructs
The expression constructs for the full-length S protein (residues 1–1273) of SARS-CoV-2 (G614) and monomeric soluble ACE2 protein were described previously51,52. To produce MSP, a modified gene encoding MSP2N236,37 fused to a 7×His tag and a TEV site at N-terminal end and a sortase eSrt site and a 6×His tag at C-terminal end, was synthesized and cloned into the pET28a vector between the NcoI and XhoI restriction sites to create the construct pET28a-lsMSP2N2 by GenScript. The expression constructs for the mutant spike proteins were generated according to standard PCR methods and were verified by DNA sequencing of the entire coding regions.
Expression and purification of recombinant proteins
A stable cell line was generated using HEK293T cells for large-scale production of the full-length G614 S protein according to a published protocol53. Purification of the full-length S protein was performed as previously described51. In brief, the stably transfected cells were grown in Expi293 expression medium (Thermo Fisher Scientific) containing 1% penicillin–streptomycin (Thermo Fisher Scientific) and 1.0 μg ml−1 puromycin (Thermo Fisher Scientific) to a density of around 4–5 × 106 per ml, then lysed in a lysis buffer containing buffer A (100 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA) and 1% (w/v) n-dodecyl-β-d-maltopyranoside (DDM) (Anatrace), EDTA-free complete protease inhibitor cocktail (Roche), and incubated at 4 °C for one hour. After centrifugation at 27,000g for 30 min, the supernatant was loaded onto a Strep-Tactin (IBA Lifesciences) column equilibrated with the lysis buffer. The column was washed with 50 column volumes of buffer A and 0.3% DDM, followed by additional washes with 50 column volumes of buffer A and 0.1% DDM, and with 50 column volumes of buffer A and 0.02% DDM. The S protein was eluted by buffer A containing 0.02% DDM and 5 mM desthiobiotin (IBA Lifesciences). The protein was further purified by gel-filtration chromatography on the Superose 6 10/300 column (GE Healthcare) in a buffer containing 25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.02% DDM.
Another stable cell line was also generated for large-scale production of monomeric soluble ACE2 protein. Purification of the ACE2 protein was performed as previously described52. In brief, the stably transfected cells were incubated in Expi293 expression medium containing 1% penicillin–streptomycin and 1.0 μg ml−1 puromycin to a density of around 4–5 × 106 per ml, the cell supernatant was collected by centrifugation at 3,000g for 30 min and loaded onto a column packed with Ni Sepharose excel resin (Cytiva Life Sciences). The column was washed with a buffer containing 20 mM Tris-HCl, pH 7.5, 300 mM NaCl and 10 mM imidazole. The ACE2 protein was eluted with a buffer containing 300 mM imidazole, and further purified by gel-filtration chromatography on the HiLoad 16/600 Superdex 200 pg column (GE Healthcare) in 25 mM Tris-HCl, pH 7.5 and 150 mM NaCl.
To produce the linear MSP2N2 (lsMSP2N2) protein, the expression construct was transformed into E. coli BL21 Star (DE3) (Thermo Fisher Scientific), and the cells were grown at 37 °C in LB medium with 50 µg ml−1 kanamycin to an optical density at 600 nm of 0.8 and then induced with 1 mM isopropyl-β-d-thiogalactopyranoside (Sigma-Aldrich) at 30 °C for 5 h, and collected by centrifugation. Purification and circularization of the lsMSP2N2 protein were performed as previously described37,54. In brief, the cells were completely resuspended in a buffer (40 mM Tris-HCl, pH 8.0, 300 mM NaCl) and lysed by sonication. The cell lysate was clarified by centrifugation at 30,000g for 30 min and the supernatant was loaded onto a column packed with Ni-NTA agarose (Qiagen). The column was sequentially washed with buffer I (40 mM Tris-HCl, pH 8.0, 300 mM NaCl, 1% Trion X-100), buffer II (40 mM Tris-HCl, pH 8.0, 300 mM NaCl, 50 mM sodium cholate, 20 mM imidazole) and buffer III (40 mM Tris-HCl, pH 8.0, 300 mM NaCl, 50 mM imidazole). The lsMSP2N2 protein was eluted with 40 mM Tris-HCl, pH 8.0, 300 mM NaCl, 400 mM imidazole. To remove the N-terminal His-tag, the protein was treated with TEV protease in the TEV buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 1 mM DTT) at room temperature for 3 h. To produce circularized protein (csMSP2N2) while removing its C-terminal His-tag, the sortase eSrt was mixed with the TEV-treated lsMSP2N2 with a molar ratio of 1:20 in a circularization buffer (30 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM CaCl2, 1 mM DTT), and incubated at 37 °C for 3 h. The circularization reaction was passed through a Ni-NTA agarose column to remove the His-tagged protein, and we collected the flow-through, which contained the csMSP2N2 protein. The protein was further purified by anion-exchange chromatography on the HiTrap Q HP column (Cytiva Life Sciences), dialysed against 20 mM Tris-HCl, pH 7.5 and 100 mM NaCl, then flash-frozen in liquid nitrogen and stored at −80 °C.
The trimeric ACE2 protein and S-specific monoclonal antibodies were described previously44,52
Reconstitution of the spike protein in nanodiscs
To reconstitute the spike protein in nanodiscs, soy extract polar lipid (Avanti) in chloroform was first dried under a nitrogen stream and dried further overnight in a vacuum desiccator. Lipid films were dissolved in a buffer containing 25 mM Tris pH 7.5, 150 mM NaCl, 2% DDM, 0.4% CHS, vortexed vigorously and sonicated until the solution became clear. The S protein and lipids were mixed and incubated on ice for 30 min. The csMSP2N2 was added to the mixture with a spike:csMSP2N2:lipid molar ratio of 1:8:700 and incubated on ice for another 30 min. Bio-beads SM2 (Bio-Rad) were added to remove detergents from the mixture and initiate the reconstitution. After gentle rotation at 4 °C overnight, the Bio-beads were removed through filtration. To induce the conformational transition of the S protein from the prefusion state to the postfusion state in the nanodiscs, the reconstitution mixture was added with soluble ACE2 protein at 6 μM and incubated at room temperature for 30 min. A control experiment confirmed that the prefusion S protein was stable at room temperature in the absence of ACE2. The ACE2-induced postfusion S-nanodisc sample was purified by gel-filtration chromatography on the Superose 6 Increase 10/300 column (GE Healthcare) in a buffer containing 25 mM Tris-HCl, pH 7.5, and 150 mM NaCl. Peak fractions containing the postfusion S-nanodisc sample were used for cryo-EM analysis.
Negative-stain EM
To prepare grids, 4 μl of freshly purified prefusion or postfusion S-nanodisc sample was adsorbed onto a glow-discharged carbon-coated copper grid (Electron Microscopy Sciences), washed with deionized water and stained with freshly prepared 1.5% uranyl formate. Images were recorded at room temperature on the Phillips CM10 transmission electron microscope with a nominal magnification of ×52,000. Particles were auto-picked and 2D class averages were generated using RELION (v.4.0.0)55.
Cryo-EM sample preparation and data collection
To prepare cryo-EM grids, the postfusion S-nanodisc sample at 1.5 mg ml−1 was applied to a 1.2/1.3 Quantifoil gold grid (Electron Microscopy Sciences) that had been glow-discharged with a PELCO easiGlow Glow Discharge Cleaning system (Ted Pella) for 60 s at 15 mA. The grids were immediately plunge-frozen into liquid ethane using the Vitrobot Mark IV (Thermo Fisher Scientific), and excess protein was blotted away using grade 595 filter paper (Ted Pella) with a blotting time of 4 s, a blotting force of −12 at 4 °C with 100% humidity. The grids were screened for ice thickness and particle distribution. Selected grids were used to acquire images with a Titan Krios transmission electron microscope (Thermo Fisher Scientific) operated at 300 keV and equipped with a BioQuantum GIF/K3 direct electron detector. Automated data collection was carried out using SerialEM (v.4.0.5)56 at a nominal magnification of ×105,000 and the K3 detector in counting mode (calibrated pixel size, 0.825 Å) at an exposure rate of ~13.8 electrons per pixel per second. Each video adds a total accumulated electron exposure of around 50.6/51.3 e− Å−2, fractionated in 50/51 frames. Datasets were acquired using a defocus range of 0.8–2.2 μm.
Image processing and 3D reconstructions
All data were processed using cryoSPARC (v.3.3.1)38 and RELION. Drift correction for cryo-EM images was performed using patch mode, and contrast transfer function was estimated by patch mode. Motion-corrected sums with dose weighting were used for all other image processing. Blob picking was performed for particle picking. In total, 18,339,830 particles were extracted from 14,299 images using a box size of 600 Å (downsizing to 128 Å) from dataset I, and 57,656,504 particles from 17,028 images from dataset II. The two sets of particles were processed separately and each was subjected to 8–10 rounds of 2D classification, giving 1,532,458 and 3,885,698 good particles, respectively. An initial model was produced using ab initio reconstruction in cryoSPARC on the basis of selected 2D class averages from dataset I.
For dataset I, good particles from 2D classification were used for one round of heterogeneous classification with six copies of the initial model as the reference in C1 symmetry. Three major classes with a total of 57.6% of the particles showing clear structural features were processed for two additional rounds of heterogeneous refinement with six copies of the initial model as the reference in C1 symmetry. The major class with a nanodisc shape at one end was re-extracted to an unbinned box size of 600 Å and processed for one round of 2D classification to remove bad particles, followed by one round of non-uniform refinement in C1 symmetry to produce a map at 3.18 Å resolution from 153,658 particles. Another round of non-uniform refinement in C3 symmetry was performed and improved the resolution to 3.0 Å. To further improve the local resolution in the nanodisc, these particles were transferred to RELION and subjected to one round of focus classification with a mask covering the entire protein, leading to a major class (78.9% particles) with continuous density at the transmembrane region. This class with 123,298 particles was imported back to cryoSPARC, and processed for another round of non-uniform refinement in C3 symmetry, giving a map at 2.96 Å resolution. One round of particle subtraction was performed to remove noises from the nanodiscs, followed by one round of local refinement with an overall mask in C3symmetry to produce a map at 2.84 Å resolution. Another round of local refinement in C3 symmetry focused on the transmembrane region was performed subsequently to improve its resolution, giving a map at 3.35 Å resolution. These overall and locally refined maps showed clear density for transmembrane helices, but the resolution was not high enough for unambiguously modelling the structure. Dataset II was therefore collected with the same batch of the S-nanodisc sample.
For dataset II, the selected particles from 2D classification were used for one round of heterogeneous classification with six copies of the initial model as the reference in C1 symmetry. Two major classes with total 37.8% of the particles showing clear structural features were processed for two additional rounds of heterogeneous refinement with six copies of the initial model as the reference in C1symmetry. The major class was re-extracted to an unbinned box size of 600 Å and processed for one round of 2D classification, followed by one round of duplicate removing and non-uniform refinement in C1symmetry, giving a map at 3.4 Å resolution from 131,604 particles. These particles were combined with the particle set from dataset I after the focus classification, and processed for one round of non-uniform refinement in C3 symmetry, contrast transfer function refinement and another round of non-uniform refinement in C3symmetry, giving a final overall map at 2.86 Å resolution from the combined 254,902 particles. Particle subtraction was also performed to reduce noises from the nanodiscs, followed by two rounds of local refinement with a different mask in size each time, giving a map at 3.26 Å resolution in the transmembrane region showing clear sidechain density for the transmembrane helices. The best maps from dataset I and the combined dataset were all used for model building.
All resolutions were reported from the gold-standard Fourier shell correlation using the 0.143 criterion. Density maps were corrected from the modulation transfer function of the K3 detector and sharpened by applying a temperature factor that was estimated using sharpening tools in cryoSPARC. The local resolution was also determined using cryoSPARC.
Model building
The initial template for model building was our postfusion S structure (Protein Data Bank: 6XRA). Several rounds of manual building were performed in Coot. The model around the transmembrane region was first refined in Phenix57 against the locally refined maps at 3.26/3.35 Å resolution, and further refined in Phenix against the 2.9 Å overall map. Iteratively, refinement was performed in both Phenix (real space refinement) and ISOLDE58, and the Phenix refinement strategy included minimization_global, local_grid_search, and adp, with rotamer, Ramachandran, and reference-model restraints, using 6XRA as the reference models. The local map from dataset I had stronger density for the CT region and was used for modelling the CT; the map from the combined dataset map had better sidechain density for the transmembrane helices and was therefore used for modelling them. The refinement statistics are summarized in Extended Data Table 1. Structural biology applications used in this project were compiled and configured by SBGrid59.
The intact S2 proteins from various coronaviruses were aligned by Clustal Omega60 and were displayed using the online server ESPript (v.3.0)61.
Western blot
Full-length S protein samples were resolved in 4–15% Mini-Protean TGX gel (Bio-Rad) and transferred onto PVDF membranes (Millipore) by an Iblot2 (Invitrogen by Thermo Fisher Scientific). Membranes were blocked with 5% skimmed milk in PBS for 1 h and incubated with a SARS-CoV-2 (2019-nCoV) Spike RBD Antibody (Sino Biological, 40592-T62) at a concentration of 1 μg ml−1 for another hour at room temperature. Alkaline-phosphatase-conjugated anti-rabbit IgG (1:5,000) (Sigma-Aldrich) was used as a secondary antibody. Proteins were visualized using one-step NBT/BCIP substrates (Promega).
Flow cytometry
Expi293F cells were grown in Expi293 expression medium. Cell surface display DNA constructs for the SARS-CoV-2 G614 or its mutants or S2 together with a plasmid expressing blue fluorescent protein (BFP) were transiently transfected into Expi293F cells using ExpiFectamine 293 reagent (Thermo Fisher Scientific) according to the manufacturer’s instruction. Then, 2 days after transfection, the cells were stained with primary antibodies at a concentration of 5 μg ml−1. An Alexa Fluor 647-conjugated donkey anti-human IgG Fc F(ab′)2 fragment (Jackson ImmunoResearch) was used as the secondary antibody at a concentration of 5 μg ml−1. Cells were run through an Intellicyt iQue Screener Plus flow cytometer. Cells gated for positive BFP expression were analysed for antibody binding.
Cell–cell fusion assay
The cell–cell fusion assay, based on the α-complementation of Escherichia coli β-galactosidase, was conducted to quantify the fusion activity mediated by SARS-CoV-2 S protein, as described previously9. In brief, the full-length G614 S or its mutants (10 μg) and the α fragment of E. coli β-galactosidase construct (10 μg), or the full-length ACE2 construct (5 μg) together with the ω fragment of E. coli β-galactosidase construct (10 μg), were transfected into HEK293T cells using polyethylenimine (80 μg). After incubation at 37 °C for 5 h, the medium was aspirated and replaced with complete DMEM (1% penicillin–streptomycin, 1% GlutaMax and 10% FBS), followed by incubation at 37 °C for additional 19 h. The cells were detached using PBS and resuspended in complete DMEM. S-expressing cells (50 μl, 1.0 × 106 cells per ml) were mixed with 50 μl ACE2-expressing cells (1.0 × 106 cells per ml) to allow cell–cell fusion to proceed at 37 °C for 4 h. Cell–cell fusion activity was quantified using a chemiluminescent assay system, Gal-Screen (Applied Biosystems), according to the standard protocol recommended by the manufacturer. The substrate was added to the mixture of the cells and allowed to react for 90 min in dark at room temperature. The luminescence signal was recorded using a Synergy Neo plate reader (Biotek).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The atomic structure coordinates and EM map have been deposited in the Protein Data Bank (8FDW) and Electron Microscopy Data Bank (EMD-29016, EMD-29017 and EMD-29018). All other related data generated during and/or analysed during this study, such as raw cryo-EM images, are available from the corresponding authors on reasonable request. Source data are provided with this paper.
References
Harrison, S. C. Viral membrane fusion. Virology 479-480, 498–507 (2015).
Kielian, M. Mechanisms of virus membrane fusion proteins. Annu. Rev. Virol. 1, 171–189 (2014).
Rand, R. P. & Parsegian, V. A. Physical force considerations in model and biological membranes. Can. J. Biochem. Cell Biol. 62, 752–759 (1984).
Parsegian, V. A., Fuller, N. & Rand, R. P. Measured work of deformation and repulsion of lecithin bilayers. Proc. Natl Acad. Sci. USA 76, 2750–2754 (1979).
Jackson, C. B., Farzan, M., Chen, B. & Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 23, 3–20 (2022).
Tortorici, M. A. & Veesler, D. Structural insights into coronavirus entry. Adv. Virus Res. 105, 93–116 (2019).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280 (2020).
Zhao, M. M. et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target. Ther. 6, 134 (2021).
Cai, Y. et al. Distinct conformational states of SARS-CoV-2 spike protein. Science 369, 1586–1592 (2020).
Fan, X., Cao, D., Kong, L. & Zhang, X. Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nat. Commun. 11, 3618 (2020).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020).
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292 (2020).
Lan, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220 (2020).
Yan, R. et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448 (2020).
Shang, J. et al. Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221–224 (2020).
Wang, Q. et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181, 894–904 (2020).
Xia, S. et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 30, 343–355 (2020).
Ke, Z. et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 588, 498–502 (2020).
Yao, H. et al. Molecular architecture of the SARS-CoV-2 virus. Cell 183, 730–738 (2020).
Liu, C. et al. The architecture of inactivated SARS-CoV-2 with postfusion spikes revealed by Cryo-EM and Cryo-ET. Structure 28, 1218–1224 (2020).
Bosch, B. J., de Haan, C. A., Smits, S. L. & Rottier, P. J. Spike protein assembly into the coronavirion: exploring the limits of its sequence requirements. Virology 334, 306–318 (2005).
Lontok, E., Corse, E. & Machamer, C. E. Intracellular targeting signals contribute to localization of coronavirus spike proteins near the virus assembly site. J. Virol. 78, 5913–5922 (2004).
Petit, C. M. et al. Genetic analysis of the SARS-coronavirus spike glycoprotein functional domains involved in cell-surface expression and cell-to-cell fusion. Virology 341, 215–230 (2005).
Ye, R., Montalto-Morrison, C. & Masters, P. S. Genetic analysis of determinants for spike glycoprotein assembly into murine coronavirus virions: distinct roles for charge-rich and cysteine-rich regions of the endodomain. J. Virol. 78, 9904–9917 (2004).
Belouzard, S., Millet, J. K., Licitra, B. N. & Whittaker, G. R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 4, 1011–1033 (2012).
Sainz, B. Jr, Rausch, J. M., Gallaher, W. R., Garry, R. F. & Wimley, W. C. Identification and characterization of the putative fusion peptide of the severe acute respiratory syndrome-associated coronavirus spike protein. J. Virol. 79, 7195–7206 (2005).
Basso, L. G., Vicente, E. F., Crusca, E. Jr, Cilli, E. M. & Costa-Filho, A. J. SARS-CoV fusion peptides induce membrane surface ordering and curvature. Sci. Rep. 6, 37131 (2016).
Madu, I. G., Roth, S. L., Belouzard, S. & Whittaker, G. R. Characterization of a highly conserved domain within the severe acute respiratory syndrome coronavirus spike protein S2 domain with characteristics of a viral fusion peptide. J. Virol. 83, 7411–7421 (2009).
Lai, A. L., Millet, J. K., Daniel, S., Freed, J. H. & Whittaker, G. R. The SARS-CoV fusion peptide forms an extended bipartite fusion platform that perturbs membrane order in a calcium-dependent manner. J. Mol. Biol. 429, 3875–3892 (2017).
Bosch, B. J. et al. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl Acad. Sci. USA 101, 8455–8460 (2004).
Dacon, C. et al. Broadly neutralizing antibodies target the coronavirus fusion peptide. Science 377, 728–735 (2022).
Low, J. S. et al. ACE2-binding exposes the SARS-CoV-2 fusion peptide to broadly neutralizing coronavirus antibodies. Science 377, 735–742 (2022).
Guillen, J., Perez-Berna, A. J., Moreno, M. R. & Villalain, J. Identification of the membrane-active regions of the severe acute respiratory syndrome coronavirus spike membrane glycoprotein using a 16/18-mer peptide scan: implications for the viral fusion mechanism. J. Virol. 79, 1743–1752 (2005).
Sainz, B. Jr, Rausch, J. M., Gallaher, W. R., Garry, R. F. & Wimley, W. C. The aromatic domain of the coronavirus class I viral fusion protein induces membrane permeabilization: putative role during viral entry. Biochemistry 44, 947–958 (2005).
Grinkova, Y. V., Denisov, I. G. & Sligar, S. G. Engineering extended membrane scaffold proteins for self-assembly of soluble nanoscale lipid bilayers. Protein Eng. Des. Sel. 23, 843–848 (2010).
Johansen, N. T. et al. Circularized and solubility-enhanced MSPs facilitate simple and high-yield production of stable nanodiscs for studies of membrane proteins in solution. FEBS J. 286, 1734–1751 (2019).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Ramadan, A. A. et al. Identification of SARS-CoV-2 spike palmitoylation inhibitors that results in release of attenuated virus with reduced infectivity. Viruses 14, 531 (2022).
Tien, C. F. et al. Glycosylation and S-palmitoylation regulate SARS-CoV-2 spike protein intracellular trafficking. iScience 25, 104709 (2022).
Modis, Y., Ogata, S., Clements, D. & Harrison, S. C. Structure of the dengue virus envelope protein after membrane fusion. Nature 427, 313–319 (2004).
Steinhauer, D. A., Wharton, S. A., Skehel, J. J. & Wiley, D. C. Studies of the membrane fusion activities of fusion peptide mutants of influenza virus hemagglutinin. J. Virol. 69, 6643–6651 (1995).
Wilson, I. A., Skehel, J. J. & Wiley, D. C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 289, 366–373 (1981).
Tong, P. et al. Memory B cell repertoire for recognition of evolving SARS-CoV-2 spike. Cell 184, 4969–4980 (2021).
Baum, A. et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 369, 1014–1018 (2020).
Chi, X. et al. A neutralizing human antibody binds to the N-terminal domain of the spike protein of SARS-CoV-2. Science 369, 650–655 (2020).
Kreutzberger, A. J. B. et al. SARS-CoV-2 requires acidic pH to infect cells. Proc. Natl Acad. Sci. USA 119, e2209514119 (2022).
Lorieau, J. L., Louis, J. M. & Bax, A. The complete influenza hemagglutinin fusion domain adopts a tight helical hairpin arrangement at the lipid:water interface. Proc. Natl Acad. Sci. USA 107, 11341–11346 (2010).
Corver, J., Broer, R., van Kasteren, P. & Spaan, W. Mutagenesis of the transmembrane domain of the SARS coronavirus spike glycoprotein: refinement of the requirements for SARS coronavirus cell entry. Virol. J. 6, 230 (2009).
Fu, Q. & Chou, J. J. A trimeric hydrophobic zipper mediates the intramembrane assembly of SARS-CoV-2 spike. J. Am. Chem. Soc. 143, 8543–8546 (2021).
Zhang, J. et al. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science 372, 525–530 (2021).
Xiao, T. et al. A trimeric human angiotensin-converting enzyme 2 as an anti-SARS-CoV-2 agent. Nat. Struct. Mol. Biol. 28, 202–209 (2021).
Chen, J. et al. Effect of the cytoplasmic domain on antigenic characteristics of HIV-1 envelope glycoprotein. Science 349, 191–195 (2015).
Ritchie, T. K. et al. Chapter 11—reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 464, 211–231 (2009).
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D 74, 519–530 (2018).
Morin, A. et al. Collaboration gets the most out of software. eLife 2, e01456 (2013).
Madeira, F. et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641 (2019).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Acknowledgements
We thank the members of the SBGrid team for computing support and Stephen Harrison for computing resources and for reading the manuscript. We acknowledge support for COVID-19-related structural biology research at Harvard from the Nancy Lurie Marks Family Foundation and the Massachusetts Consortium on Pathogen Readiness (MassCPR). This work was supported by COVID-19 Awards by MassCPR (to B.C.), a Fast grant by Emergent Ventures (to B.C.), and NIH grants AI127193 (to B.C. and J. Chou), AI147884 (to B.C.) and AI141002 (to B.C.).
Author information
Authors and Affiliations
Contributions
B.C., J.Z. and W.S. conceived the project. W.S. produced the full-length S protein and reconstituted it in nanodiscs with help from Y.C. and H.P. Y.C. also created the new MSP construct and established the initial conditions for nanodisc reconstitution. W.S. and J.Z. prepared cryo-grids and performed EM data collection with contributions from M.L.M., K.S. and C.X. J.Z. processed the cryo-EM data and built and refined the atomic models. H.C. and J.L. created the mutant constructs. H.Z. performed the flow cytometry experiment. J.Z. and W.S. designed the mutant constructs and performed the cell–cell fusion assay. J.V. and S.R.-V. contributed to plasmid preparation, cell culture and protein production. All of the authors analysed the data. B.C., J.Z. and W.S. wrote the manuscript with input from all of the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Steve Gamblin and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Schematic representations of the full-length SARS-CoV-2 and SARS-CoV spike proteins.
Segments of S1 and S2 include: NTD, N-terminal domain; RBD, receptor-binding domain; CTD1, C-terminal domain 1; CTD2, C-terminal domain 2; S1/S2, the furin cleavage site at the S1/S2 boundary; S2’, S2’ cleavage site; FP, fusion peptide; FPPR, fusion peptide proximal region; HR1, heptad repeat 1; CH, central helix region; CD, connector domain; HR2, heptad repeat 2; TM, transmembrane segment; CT, cytoplasmic tail; and tree-like symbols for glycans. Based on studies of the SARS-CoV S protein, three membranotropic regions in the SARS-CoV S2 have been suggested as putative fusion peptides, including a segment of residues 770–788, equivalent of the N-terminal FP in SARS-CoV S; a segment of residues 798–816 or 798–835, widely accepted as the “bona fide” FP; and a segment of residues 858–886, also equivalent of the internal FP in SARS-CoV S. A segment of residues 1190–1203 is called the pretransmembrane domain (pre-TM).
Extended Data Fig. 2 Additional details for preparation of the cryo-EM sample.
The prefusion S trimer was first reconstituted in nanodiscs (a) and further purified by gel filtration chromatography (b). Incubation of the stable prefusion S trimer in nanodiscs with soluble ACE2 led to S1 dissociation, and formation of the postfusion S2 trimer in nanodiscs (c). Samples from each step were analysed by SDS-PAGE and negative stain EM. The box size of 2D averages is ~880 Å. The experiment was repeated at least three times independently at different scales with similar results. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 3 Cryo-EM analysis of the postfusion S2 trimer.
Top, representative micrograph out of ~30,000 similar micrographs, and 2D averages (box dimension: 500 Å) of the cryo-EM particle images of the postfusion S2 trimer in nanodiscs. Bottom, data processing workflow for structure determination.
Extended Data Fig. 4 Analysis of the postfusion S2 trimer structures by cryoSPARC.
Gold standard FSC curves of the three refined 3D reconstructions of the postfusion S2 trimer and the corresponding cryoSPARC output for particle distribution of each reconstruction.
Extended Data Fig. 5 Additional analysis of the postfusion S2 trimer structure.
(a) 3D reconstructions of the postfusion S2 trimer from the overall refinement and local refinement are coloured according to local resolution estimated by ResMap with the FSC=0.143 criterion. (b) Representative density in grey surface representation from the EM maps of the postfusion S2.
Extended Data Fig. 6 Fit of the postfusion structure into the density map of the postfusion spike on the virion surface.
The postfusion structure was fit into the map of the viral postfusion spike reconstructed by cryo-ET (EMD-30428) at two different contour levels.
Extended Data Fig. 7 Interactions among three FP protomers in the postfusion SARS-CoV-2 spike.
Left, side view of packing of three FP protomer with each coloured differently shown in both ribbon diagram and stick model. Residues mediating the formation of the three-stranded coiled coil near the three-fold axis are indicated. Right, top views of representative residues that mediate the formation of the three-stranded coiled coil.
Extended Data Fig. 8 Expression and antigenic properties of the mutant spike proteins assessed by flow cytometry.
(a) Expression and processing of the full-length G614 S construct and its mutants (Mut-1 – Mut-7) in HEK293 cells. S protein samples prepared from HEK293 cells transiently transfected with 10 μg of the full-length S expression plasmids were detected by anti-RBD polyclonal antibodies. Bands for the uncleaved S and S1 fragment are indicated. The experiment was repeated four times with similar results. (b) Antibody binding to the full-length G614 S protein and its mutants (Mut-1 – Mut-7), as well as an S2 construct expressed on the cell surfaces analysed by flow cytometry. The antibodies and their targets are indicated. A designed ACE2-based fusion inhibitor ACE2615-foldon-T27W was used for detecting receptor binding52. MFI, mean fluorescent intensity. The error bars represent standard errors of mean from measurements using three independently transfected cell samples. The flow cytometry assays were repeated three times with essentially identical results. (c) Expression and processing of the full-length G614 S construct and its additional mutants (Mut-8 – Mut-15) in HEK293 cells. The experiment was repeated three times with similar results. (d) Antibody binding to the full-length G614 S protein and its additional mutants (Mut-8 – Mut-15), as well as an S2 construct expressed on the cell surfaces analysed by flow cytometry. The error bars represent standard errors of mean from measurements using three independently transfected cell samples. The flow cytometry assays were repeated three times with essentially identical results. (e) Expression and processing of the full-length Delta S construct and its mutants in HEK293 cells. The experiment was repeated four times with similar results. (f) Antibody binding to the full-length Delta S protein and its mutants, as well as an S2 construct expressed on the cell surfaces analysed by flow cytometry. The error bars represent standard errors of mean from measurements using three independently transfected cell samples. The flow cytometry assays were repeated three times with essentially identical results. For western blot source data, see Supplementary Fig. 1.
Supplementary information
Supplementary Figures
Supplementary Figs. 1–3. Supplementary Fig. 1: the uncropped blots. Supplementary Fig. 2: the sequence alignment of the S2 proteins from various coronaviruses. Supplementary Fig. 3: the gating graphs for the flow cytometry experiments.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shi, W., Cai, Y., Zhu, H. et al. Cryo-EM structure of SARS-CoV-2 postfusion spike in membrane. Nature 619, 403–409 (2023). https://doi.org/10.1038/s41586-023-06273-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-023-06273-4
This article is cited by
-
Functional and antigenic characterization of SARS-CoV-2 spike fusion peptide by deep mutational scanning
Nature Communications (2024)
-
SARS-CoV-2 proteins structural studies using synchrotron radiation
Biophysical Reviews (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
