
Abstract
‘Glomerulonephritis’ (GN) is a term used to describe a group of heterogeneous immune-mediated disorders characterized by inflammation of the filtration units of the kidney (the glomeruli). These disorders are currently classified largely on the basis of histopathological lesion patterns, but these patterns do not align well with their diverse pathological mechanisms and hence do not inform optimal therapy. Instead, we propose grouping GN disorders into five categories according to their immunopathogenesis: infection-related GN, autoimmune GN, alloimmune GN, autoinflammatory GN and monoclonal gammopathy-related GN. This categorization can inform the appropriate treatment; for example, infection control for infection-related GN, suppression of adaptive immunity for autoimmune GN and alloimmune GN, inhibition of single cytokines or complement factors for autoinflammatory GN arising from inborn errors in innate immunity, and plasma cell clone-directed or B cell clone-directed therapy for monoclonal gammopathies. Here we present the immunopathogenesis of GN and immunotherapies in use and in development and discuss how an immunopathogenesis-based GN classification can focus research, and improve patient management and teaching.
Similar content being viewed by others
Introduction
Glomerulonephritis (GN) describes a variety of relatively rare immune-mediated diseases characterized by damage to the glomerular compartment of the nephrons of the kidney1. If not properly treated, acute GN can lead to chronic kidney disease and irreversible kidney failure2,3, with patients requiring kidney replacement therapy (dialysis or kidney transplantation). Data from a US Medicare cohort (with average age of 75 years) indicated that up to 1.2% of individuals are affected by GN4. Moreover, GN accounts for 18.7% of Germans with chronic kidney disease and 30–36% of end-stage kidney disease in US children and adolescents5,6. In addition, GN is more prevalent and may be more severe in certain ethnic groups, such as African American, Hispanic, Asian, and Australian and Canadian First Nations populations.
Traditionally, GN has been classified by histopathological lesion patterns. However, the growing understanding of immunopathogenesis of the wide spectrum of GN and the increasing numbers of immunomodulatory drugs require a categorization that better connects with effective treatments. Here we propose a new classification for GN, present the latest insights on their respective immunopathogenesis and discuss how these imply specific immunotherapies.
Key features of glomerular anatomy and function
The glomerular filtration barrier is responsible for creating an ultrafiltrate of water and low molecular weight solutes, while retaining most high molecular weight proteins and blood cells within the vasculature7. The glomerular microvasculature is particularly vulnerable to immune-mediated injury because the filtration process involves delicate anatomical structures that are exposed to substantial shear stress and perfusion pressure. Highly specialized glomerular endothelial cells build a size- and charge-dependent barrier to serum proteins, while in a similar manner the glomerular basement membrane (GBM) itself is also highly specialized. The outer aspect of the filter membrane harbours postmitotic epithelial cells with octopus-like primary, secondary and interdigitating tertiary foot processes, cells known as podocytes8 (Fig. 1). A zipper-like slit membrane between these foot processes comprising nephrin and other podocyte proteins forms the ultimate barrier to small serum proteins9. The glomerular tuft is surrounded by parietal epithelial cells along the Bowman capsule, which directs the ultrafiltrate towards the draining tubule. Immune-mediated injury to any of these structures can cause loss of serum proteins into the urine or even stop filtration, the key function of the kidney needed to maintain internal homeostasis1,10.

The glomerulus is the blood-filtering unit of the kidney. Each glomerulus drains the filtrate into its own tubule, and the glomerulus and its tubules together constitute the functional unit of the kidney, the nephron. The vascular part of each glomerulus includes an afferent arteriole, an efferent arteriole and a capillary network inside the glomerulus, where the filtration occurs under conditions of high perfusion pressure and shear stress. The capillary network is held together by mesenchymal cells, known as mesangial cells, and a matrix, which regulate capillary tension. Parts of the glomerular filtrate pass through the mesangium; hence, circulating antigens and immunoglobulins can get trapped there. Glomerular capillaries are characterized by a fenestrated endothelium covered with glycocalyx and attached to the glomerular basement membrane. At the outer aspect of the glomerular capillaries, podocytes attach to the glomerular basement membrane. Podocytes are specialized epithelial cells with neuron-like primary and secondary foot processes interdigitated with the respective secondary foot processes of neighbouring podocytes. Between podocyte foot processes is the slit diaphragm, which covers a large area of the filtration barrier and is essential for preventing the passage of serum proteins such as albumin into the filtrate. Water, ions and other small solutes cross the filtration barrier through pores in the slit diaphragm. Inflammatory processes in the glomerulus typically alter the barrier function and cause leakage of serum proteins and frequently also of intact blood cells into the urine.
Infectious organisms, their immunostimulatory or toxic components, immunoglobulins activating complement, spontaneous activation of the complement system, neutrophil extracellular traps (NETs), infiltrating myeloid cells, helper or cytotoxic T cells and other innate and adaptive immune effectors can cause glomerular injury, detectable as leakage of albumin or other serum proteins, erythrocytes and leukocytes into the urine1. Depending on the acuity of the immune process, GN presents as a chronic, a subacute or even a peracute illness1. For example, certain infectious or autoimmune stimuli can lead to massive activation of complement or NET release inside glomeruli, causing diffuse capillary loop necrosis and necroinflammation and impairing glomerular filtration, leading to a rapid decline in the excretory function of the kidneys11,12. Because the filtration process occurs under high perfusion pressure across a delicate barrier, serum proteins can be easily trapped within the mesangium or along the GBM. Diabetes mellitus, obesity, a high-salt diet and hypertension further increase filtration pressure and accelerate glomerular injury (glomerular barotrauma) and promote barrier dysfunction even in less severe inflammatory states13. Finally, leukocytes pass through tight glomerular capillaries at a higher pressure and flow velocity compared with other capillary beds, which promotes the release of NETs14 followed by focal capillary necrosis in the glomerulus10,14.
Clinical presentation and diagnosis of GN
Acute GN most frequently presents with high blood pressure (hypertension), proteinuria (excessive protein in the urine) and haematuria (blood in the urine), whereas GN with predominant podocyte injury causes nephrotic syndrome presenting with a massive proteinuria causing leg oedema (Box 1). In general, proteinuria with a predominant component of albumin indicates podocyte injury, whereas haematuria implies ruptures of the GBM. As these unspecific clinical signs rarely allow a precise diagnosis of the GN subtype, only kidney biopsy can confirm GN, and histopathological lesion patterns visualized by immunostaining for immunoglobulin and complement components are used to define GN subcategories15,16.
Lesion patterns and histological signs
A diagnostic kidney biopsy can distinguish GN from other kidney disorders and define the injured glomerular compartment. This has led to descriptive terms such as ‘mesangioproliferative glomerulonephritis’, ‘membranoproliferative glomerulonephritis’, ‘membranous glomerulonephritis’, ‘crescentic glomerulonephritis’, ‘minimal change disease’ and ‘focal segmental glomerulosclerosis’ (Table 1 and Fig. 2). Histological signs of immunological activity, such as the presence of deposits of complement factors and immune complexes, their isotypes and their clonality, as well as ultrastructural changes observed with electron microscopy, provide further clues to the underlying cause of GN. For example, expansion of the cisternae of the endoplasmic reticulum, named ‘tubuloreticular structures’, in glomerular cells is considered an ultrastructural hallmark of type I interferon signalling, although the molecular mechanisms remain unknown17. However, lesion patterns such as immune complex GN, complement factor C3 GN (C3GN) and pauci-immune GN are nonspecific regarding the underlying pathophysiology. Frequently, additional immunophenotyping is needed to precisely define the type of GN (Box 1).

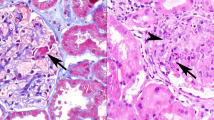
Common histopathological lesion patterns characteristic of glomerulonephritis (GN) are shown; for example, endocapillary lesions (part a), mesangioproliferative lesions (part b) and membranoproliferative lesions (part c). Immunostaining is routinely performed as one element of immunophenotyping and shows, for example, granular IgG4 positivity along the filtration barrier in a membranous GN (part d), diffuse fibrinogen positivity in necrotizing and crescentic GN (part e) and a relative lack of immunoglobulins and complement deposition in pauci-immune GN (part f). Mesangial IgA positivity is typical of IgA nephropathy (part g) and complement factor C3 positivity in absence of IgG deposits defines complement factor C3 GN (C3GN) (part h). Staining for κ and λ immunoglobulin chains (part i) helps to distinguish monotypic and polytypic immune deposits. For example, monotypic λ deposits occur in proliferative GN with monoclonal immunoglobulin deposits. HE, hematoxylin–eosin.
Kidney biopsy also allows active and potentially reversible lesions to be distinguished from inactive disease or chronic and irreversible lesions (Table 1), and this informs immunotherapy. Active lesions include intravascular neutrophil karyorrhexis or NETosis, immunothrombosis and fibrin deposition, endothelial and mesangial cell proliferation, glomerular leukocytic infiltrates, vascular loop necrosis, cellular crescents (that is, massive hyperplasia of parietal epithelial cells in the Bowman space obstructing glomerular outflow)10 and periglomerular lymphocyte infiltrates18. Glomerular deposits of IgM, IgG, C1q, C3c and C4d or a combination of these support complement activation. Only GN with such evidence of immunological activity may benefit from immunotherapy. By contrast, any fibrous transformation of glomerular structures and atrophy of kidney tubules or fibrosis within the interstitium indicate irreversible loss of kidney parenchyma. However, pathology lesion-based GN categories often do not dissect the diversity of underlying immunological disorders, which require different treatments19,20,21; hence, treatment of lesion-based GN categories results in many ‘non-responders’22.
Pathophysiology-based classification
With use of modern immunophenotyping, it is possible to define five major GN categories that connect directly with their respective immunotherapies (Table 2). The first category is GN due to infection, which involves glomerular injury via humoral and cellular mechanisms of host defence. This category of GN responds to infection control with antibiotics or antivirals. The second category is autoimmune GN, which involves adaptive immune responses directed against distinct autoantigens, and thus therapeutic targets involve suppression of autoantigen presentation and clones of autoreactive lymphocytes. Third, alloimmune GN can occur in the context of kidney transplantation and requires suppression of donor-specific immunity. Fourth, autoinflammatory GN originates from inborn errors of innate immunity and responds to inhibitors of single cytokines or complement factors. Finally, monoclonal gammopathy-related GN involves direct glomerular deposition of monoclonal immunoglobulins and requires therapies directed against the pathogenic plasma cell clone or B cell clone. Here we discuss recent developments in immunophenotyping, immune mechanisms of glomerular injury, and recent and upcoming immunotherapies for each of these GN subtypes.
Infection-related GN
Risk factors and epidemiology
Infection-related GN is caused by an immune response to pathogens (Table 3). For example, poststreptococcal GN, which occurs following infection with group A streptococci, has an estimated 470,000 new annual cases worldwide, mostly in low-income and middle-income countries owing to social factors and the absence of early antibiotic treatment23,24. GN affects 30% of patients with endocarditis (a rare and potentially fatal infection of the inner lining of the heart)25 and 0.7–2% of patients with infected ventriculoatrial shunts25,26. HIV-associated nephropathy is more common in individuals of sub-Saharan descent due in part to the high prevalence of apolipoprotein L1 (APOL1) risk alleles in this population27,28. GN occurs in more than 50% of patients with hepatitis C virus (HCV) infection29. GN due to hepatitis B virus (HBV) infection is common in HBV-endemic areas but has become rare in HBV-vaccinated populations30. Like other helminthic diseases, schistosomiasis can cause GN31,32. Limited data exist in developing countries, but in sub-Saharan Africa, eosinophil-mediated glomerular injury is common in children, likely due to the infections prevalent in this region33. Indeed, Epstein–Barr virus (EBV), parvovirus B19, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), syphilis, tuberculosis and malaria have all been linked to the development of GN34.
In situ immune complex formation
Poststreptococcal GN occurs after an acute infection with certain nephrogenic strains of group A β-haemolytic streptococci, because their antigens can attach to glomerular endothelial cells24. IgG, IgM and sometimes IgA bind to streptococcal antigens trapped in glomeruli, and the in situ formation of immune complexes triggers endothelial damage by activating the classical complement pathway. However, chemokine-binding evasins secreted by streptococci and other proteins of the bacterial surface suppress activation of the classical complement pathway35. The leading candidate antigens are nephritis-associated plasmin receptor (NAPlr; identified as glyceraldehyde 3-phosphate dehydrogenase) and streptococcal pyrogenic exotoxin B (SpeB; a cationic cysteine proteinase)36. These antigens also induce the release of IL-6, tumour necrosis factor (TNF), IL-8 and transforming growth factor-β from peripheral blood leukocytes, promoting glomerular inflammation36. NAPlr activates C3, and SpeB evades innate host defence by degrading lytic complement factors35,36,37. Both induce adhesion molecule expression, increase leukocyte recruitment and directly activate the alternative complement pathway. The alternative complement pathway may also be activated by transiently produced autoantibodies targeting factor B, a regulator of the alternative complement pathway38. Sometimes, infections also induce a transient production of antineutrophil cytoplasmic antibodies (ANCAs), antinuclear antibodies or cryoglobulins24 as an additional autoimmune component to infection-related GN.
Deposition of circulating immune complexes
The most important mechanism precipitating kidney injury during active infection involves the deposition of circulating immune complexes, as observed in endocarditis, skin ulcers and cellulitis, osteomyelitis, pneumonia, visceral abscess and urinary tract infection39, a process that implies persistent presence of antigens in the blood40. Indeed, impaired clearance of pathogens in patients with primary or secondary immunodeficiencies may contribute to their enhanced susceptibility to infection-related GN and the deposition of pathogen antigens in the glomeruli. Antigens can also persist in the blood in infections with pathogens that integrate into the host genome, such as HCV, HIV, EBV, intracellular Staphylococcus aureus or Mycobacterium tuberculosis, leading to deposition of antigen–antibody immune complexes in the glomeruli. Similarly, parasites that encode strategies of immune evasion, such as members of the genus Schistosoma, which cover themselves with host proteins, can persist in the blood. Some body compartments are difficult to reach for immune effectors or antibiotics, including biofilms on implants, heart valves or bones, allowing pathogens to escape elimination and establish chronic infection.
Direct cytotoxic effects of pathogens
Some pathogens have direct effects on kidney cells, precipitating glomerular filtration barrier impairments (Fig. 3). HIV, EBV, arbovirus, parvovirus B19 or SARS-CoV-2 can infect and injure glomerular epithelial cells41,42,43. Moreover, viral nucleic acids promote intraglomerular production of type I interferons leading to podocyte detachment or death, reduced production of podocyte-derived vascular endothelial growth factor needed for maintaining glomerular endothelial cells and collapse of glomerular capillaries8,44. Podocyte loss is associated with proteinuria and irreversible glomerulosclerosis45. Type I interferons induce podocyte death by catastrophic mitosis and block the differentiation of local podocyte progenitors into new podocytes17,46,47. By contrast, activation of immature progenitors among the parietal epithelial cells can result in the formation of hyperplastic lesions that obliterate the Bowman space and block glomerular filtration loss48.

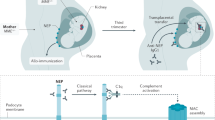
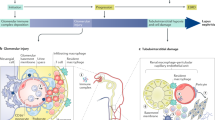
Glomerulonephritis (GN) comprises a group of immune-mediated disorders with the involvement of different innate and adaptive immune pathways in glomerular injury. a, Infection-related GN is triggered by pathogens or pathogen-associated molecular patterns (PAMPs) that elicit host defence mechanisms, which may affect the kidney in various ways as indicated. Circulating immune complexes (ICs) can become trapped in the mesangium or they can form in situ in the subendothelial space, where they can trigger complement activation. In addition, some pathogens have direct cytopathic effects, precipitating glomerular filtration barrier impairments. b, Autoimmune GN involves loss of tolerance to self-antigens in glomerular cells but frequently also to extrarenal antigens, which localize to the kidney or affect the kidney in other ways. Memory T cells in lymphoid tissues and long-lived plasma cells in the bone marrow maintain chronic autoimmunity. Infections can trigger flares of autoimmune GN via a nonspecific activation of autoreactive lymphocyte clones. c, Alloimmune GN can occur following transplantation and is associated with the development of donor-specific antibodies to HLA and non-HLA antigens of the graft and can lead to thrombotic microangiopathy and endothelial damage. d, Genetic variants in genes encoding cytokine pathways or regulatory elements of the complement cascade cause autoinflammatory disorders, some of which cause GN. Mechanisms of kidney pathology include spontaneous complement activation in complementopathies and AA amyloid deposits in periodic fever syndromes. e, Monoclonal gammopathy-related GN develops from somatic mutations in B cell clones or plasma cell clones that produce immunoglobulins or immunoglobulin components with nephrotoxic properties. These reach the glomerulus via the circulation. ANCA, antineutrophil cytoplasmic antibody; GBM, glomerular basement membrane; NET, neutrophil extracellular trap.
Podocyte loss in viral infections also relates to their ability to promote the release of type I interferons through activation of the NLR family pyrin domain-containing protein 3 (NLRP3) inflammasome and the cytosolic nucleotide sensor stimulator of interferon genes (STING). This pathway is confounded by the presence of APOL1 risk kidney alleles27, which have been selected in individuals of sub-Saharan ancestry, including 30% of African Americans, as they confer protection against Trypanosoma brucei rhodesiense infection (sleeping sickness)27. However, they predispose to progressive disease in a variety of glomerular diseases27. Expression of high-risk APOL1 genotypes, called ‘G1’ and ‘G2’, induces activation of STING, leading to the production of type I interferons, with consequent podocyte loss. High-risk APOL1 alleles also activate the NLRP3 inflammasome, with consequent release of IL-1β49 that promotes the death of podocytes by pyroptosis50. In turn, APOL1 expression itself is upregulated by type I interferons and this further enhances podocyte loss8, in an autoactivation loop21,50. This may explain why Schistosoma infection-related GN progresses quickly to kidney failure in APOL1 carriers or in patients co-infected with HIV, HCV or HBV8. Type I interferons, IL-1β, IL-6 and TNF induced by SARS-CoV-2 trigger the expression of pathogenic APOL1 via JAK–STAT signalling, resulting in podocyte loss and COVID-19-associated nephropathy51.
Targets for immunotherapy
Prevention or treatment of infection-related GN has focused on addressing the underlying infection with antivirals or antibiotics and/or by removing an infected device52. For example, combination antiretroviral therapy has greatly reduced the incidence of HIV-associated nephropathy53. Similarly, treatment of HCV infection has largely abolished HCV-related GN54. By contrast, the benefit of corticosteroids or other immunosuppressive drugs to limit irreversible damage in infection-related GN remains uncertain55. A recent randomized controlled trial of corticosteroid treatment showed no significant increase in kidney recovery rates in infection-related GN and was associated with a sixfold increase in adverse events56. Targeted therapies with the potential to interrupt the interferon–APOL1 loop are now being explored in carriers of high-risk APOL1 alleles. A preliminary communication on a phase II clinical trial reported that treatment with the APOL1 inhibitor inaxaplin (NCT05312879) reduced the degree of proteinuria in 47.6% of patients with diverse proteinuric kidney diseases and was well tolerated. A JAK/STAT inhibitor, baricitinib, is also being explored in a phase II clinical trial (NCT05237388) for the treatment of patients with APOL1-mediated glomerular disorders51.
Autoimmune GN
Autoimmune GN is characterized by an adaptive immune response directed against a series of different self-antigens (Table 3), some of which exclusively localize to the kidney (podocyte antigens and GBM antigens), whereas others are expressed systemically, such as IgA (IgA nephropathy), IgG (cryoglobulinaemic GN), neutrophil antigens (ANCA GN) and chromatin components (lupus nephritis), and often lead to extrarenal manifestations. Circulating extrarenal antigens can cause GN upon entrapment in the filtration barrier. Finally, autoantibodies to complement factors can induce GN by inducing unnecessary complement activation. Autoimmune GN associated with high levels of nephritogenic autoantibodies produces active lesions followed by immediate atrophy and scarring (necrotizing and crescentic GN), whereas low antibody titres and a low nephritogenic potential produce less active lesions and result in chronic and smouldering GN with damage accruing over longer periods.
Risk factors and epidemiology
The exact prevalence of autoimmune GN is unclear, but collectively the different subtypes account for a significant proportion of all GN. Autoantibodies directed against IgA cause IgA nephropathy, the most prevalent autoimmune GN, especially in Asia. Its autoimmune nature is most obvious when presenting as an acute small vessel vasculitis with IgA deposits affecting the skin, intestinal tract, joints and kidneys. GN involving ANCAs, anti-podocyte antibodies or anti-chromatin antibodies is also relatively common57,58,59 (Table 3). The higher prevalence and disease severity of IgA nephropathy in Asia or of lupus nephritis in people of African descent may be explained by gene variants that affect checkpoints of immune tolerance, including monogenic primary immunodeficiencies60 or variants in HLA-DRB1, HLA-DQA1, HLA-DQB1 and Fc receptors61,62. Alternatively, it has been proposed that somatic mutation-driven expansion of autoreactive B cell clones resembling features of clonal haematopoiesis may explain the pathogenesis of autoimmune GN63,64. Somatic mutations may allow proliferating self-reactive lymphocytes to bypass regulatory checkpoints and to account for a high percentage of the overall lymphocyte population64,65. However, this recent concept awaits exploration in the context of autoimmune GN.
Immunopathogenesis
In all autoimmune GN disorders, the central pathogenic elements are loss of tolerance and an adaptive immune response to a self-antigen (Fig. 3). The reasons for loss of tolerance differ, and are usually difficult to determine. Primary immunodeficiencies and impaired regulatory T cell function are rarely clinically obvious65. Despite the shared involvement of innate and adaptive immunity, the nature and distribution of the self-antigen within and outside the kidney explains the spectrum of different subtypes of autoimmune GN. For example, loss of tolerance to IgA or IgG can cause IgA vasculitis (Henoch–Schönlein purpura) or cryoglobulinaemic vasculitis, respectively, with highly active polyclonal immune complex GN as a typical classical lesion pattern66,67,68. For reasons still unknown, some individuals release hypoglycosylated IgA from intestinal B cell reservoirs into the blood, where it is handled as an autoantigen, resulting in circulating anti-IgA–IgG immune complexes and IgA nephropathy66.
Another form of autoimmune GN involving ubiquitous antigens is lupus nephritis, as commonly occurs in patients with systemic lupus erythematosus, and is characterized by loss of tolerance to chromatin components and other ubiquitous self-antigens69. The various lesion patterns involve polytypic deposits of IgA, IgM, IgG and complement factors C1q and C3, indicating involvement of the classical complement pathway70. Autoimmune vasculitis characterized by ANCAs, specific for the neutrophil antigen myeloperoxidase or proteinase 3, frequently results in severe GN. ANCAs bind to and prime neutrophils71,72. When migrating through the high shear stress glomerular capillary network, neutrophils degranulate and release NETs, which induces complement-driven microvascular endothelial injury12,58,73. Moreover, the C5 cleavage product C5a acts as an anaphylatoxin in ANCA GN and drives local neutrophil recruitment and inflammation72; hence, C5aR blockade is efficacious in active ANCA GN74. Despite its autoimmune nature, ANCA GN is misleadingly described as pauci-immune due to minimal antibody and complement deposition in the glomerulus upon standard kidney biopsy assessment58,75.
C3GN, a GN lesion pattern defined by complement deposits without concomitant immunoglobulin deposits, can be categorized as autoimmune in the presence of circulating autoantibodies known as nephritic factors to C3, C5 or factors B and H that lower the physiological threshold for activation of the alternative complement pathway, leading to complement-mediated glomerular injury76,77,78. Autoimmune C3GN, involving all elements of the adaptive immune system in the production of the causative nephritogenic autoantibody within secondary lymphoid organs, is distinguishable from autoinflammatory C3GN that lacks any involvement of the adaptive immune system and from monoclonal gammopathy-related C3GN caused by an aberrant plasma cell clone or B cell clone in the bone marrow.
Autoimmune GN involving high-affinity antibodies to the non-collagenous domain of the α3 chain of type IV collagen (α3(IV)NC1) presents as rapidly progressive GN with or without pulmonary manifestations (historically known as Goodpasture syndrome), due to the restricted expression of α3(IV) in the GBM and the alveolar basement membrane79. However, anti-α3(IV)NC1 antibodies can recognize α3(IV)NC1 within structural collagen networks only upon dysfunction of peroxidasin, a unique extracellular peroxidase that catalyses the formation of sulfilimine crosslinks in collagen IV (refs. 80,81). Indeed, peroxidasin antibodies reduce peroxidasin activity before anti-GBM IgG production and the onset of GN, probably by exposing the cryptic B cell α3(IV)NC1 epitope81. In addition, individuals expressing HLA-DR15 serotypes present the immunodominant α3(IV)NC1 peptide in a way that thymic selection results in the generation of conventional α3(IV)NC1 peptide-specific CD4+ T cells that can potentially target self-tissues. However, the same peptide adopts a different structural confirmation when bound to HLA-DR1 and preferentially selects thymus-derived antigen-specific regulatory T cells that dominantly protect from loss of tolerance in mice and in human in vitro assays65.
Finally, autoimmune GN involving podocytes as the sole target structure induces lesion patterns of either membranous nephropathy (targeting antigens localized at podocyte surfaces) or minimal change disease and focal segmental glomerulosclerosis (targeting filtration slit antigen) and has no extrarenal manifestations. Podocytes localize to the outside of glomerular capillaries, a site that is not accessible to intravascular immune cells under normal conditions (Fig. 3), which may limit the effectiveness of peripheral tolerogenic mechanisms. Indeed, a surprising spectrum of antigens are involved in autoimmune podocytopathies, including the podocyte proteins M-type PLA2R82 and thrombospondin type 1 domain-containing protein 7A83, as well as the extrarenal proteins semaphorin 3B19, protocatherin 7 (ref. 84), human high temperature requirement A1 (ref. 85), contactin 1 (ref. 86), netrin G1 (ref. 87) and NEL-like protein 1 (ref. 88) (Table 3). Antibody binding to these antigens at the podocyte–GBM interface is followed by in situ immune complex formation and complement-driven podocyte injury along the outer aspect of the GBM. Autoimmunity to PLA2R is the most common autoimmune podocytopathy, and genome-wide association studies document unusual variants at a locus in proximity to the gene encoding PLA2R as a potential factor in the loss of tolerance to this podocyte antigen62. By contrast, autoantibodies binding to components of the slit diaphragm between podocyte foot processes appear as tiny dust-like IgG deposits that are detectable only on frozen sections by confocal microscopy89. As the resulting podocyte foot process effacement is not visible by standard microscopy, this variant of autoimmune GN has been referred to as ‘minimal change disease’8. Indeed, autoimmune podocytopathies are not characterized by immune cell infiltrates due to their antigens being in a relatively immune privileged location to which leukocytes do not have ready access.
Immunopathology
Glomerular injury in immune complex GN occurs upon activation of Fc receptors on resident cells or infiltrating immune cells, as well as upon activation of the classical complement pathway90. In addition, the lectin complement pathways can be involved91; for example, in membranous GN associated with glomerular deposits of anti-PLA2R IgG4 (ref. 92). In patients with anti-PLA2R GN and genetic deficiency of mannan-binding lectin, activation of the alternative pathway predominates93. Subendothelial immune complex deposits first activate endothelial cells, triggering endocapillary lesions. Entrapment of immune complexes in the mesangium leads to mesangioproliferative or membranoproliferative lesions. These glomerular compartments are also accessible to leukocytes; hence, antagonists of pro-inflammatory chemokines, such as CC-chemokine ligand 2, and their chemokine receptors can attenuate such lesions94.
The specialized nature of the glomerular capillaries influences effector T cell functions within the glomerulus. CD4+ T cells migrate within capillaries95, but in healthy conditions or in the early stages of disease they do not extravasate inside the glomerulus. Studies using model antigens have demonstrated that the distribution of the antigen within the glomerulus is an important determinant of the pattern of injury. When the antigen is present on the endothelial side of the GBM (or in the GBM itself), both T helper 1 (TH1) cells and TH17 cells induce antigen-specific endocapillary injury95,96,97. Unusually, in this setting antigens can be presented within glomerular capillaries by intravascular monocytes to effector CD4+ T cells95. Thus, in autoimmune GN and infection-related GN, endocapillary antigens result in proliferative GN that can progress rapidly. When T cells accumulate around the Bowman capsule, the capsule itself serves as a niche for the cells of the glomerular tuft that protects them from cytotoxic CD8+ T cells, at least until there is concurrent significant glomerular endothelial injury98. Kidney-resident memory TH17 cells, generated following infection of the kidney, can be reactivated during subsequent GN by local pro-inflammatory cytokines and can exacerbate GN through the production of IL-17A in an antigen-independent manner99. Incident infections also exacerbate pre-existing GN, for example via circulating pathogen-associated molecular patterns that activate Toll-like receptors and other pattern recognition receptors in macrophage infiltrates, which accelerates glomerular inflammation and injury100,101. Bacterial lipopeptides specifically injure the glomerular filtration apparatus by activating Toll-like receptors 2 and 4 on glomerular endothelial cells and podocytes102.
Targets for immunotherapy
In types of autoimmune GN with acute and irreversible kidney injury caused by circulating antibodies, plasma exchange or treatment with the endopeptidase imlifidase is used to quickly remove nephritogenic antibodies103,104 (Fig. 4). Glucocorticoids are still in use to control autoimmune GN activity, but their nonspecific mechanism of action and metabolic side effects compromise their safety profile; hence, glucocorticoid replacement is a key goal for GN immunotherapy (Table 4 and Fig. 4). For example, intestinal-release capsules of the glucocorticoid budesonide limits the release of aberrantly glycosylated IgA from Peyer patches in the intestinal wall of patients with IgA nephropathy and shows less systemic steroid toxicity105.

Most immunosuppressive drugs primarily act inside lymphoid organs, whereas complement inhibitors and calcineurin inhibitors directly act at the site of glomerular injury. Steroids (glucocorticoids) suppress local inflammation in the kidney as well as adaptive immunity in lymphoid organs. Imlifidase degrades IgG in the circulatory system but possibly also IgG deposits inside the kidney. Drugs in use are shown in bold. APOL1, apolipoprotein L1; APRIL, a proliferation-inducing ligand; BAFF, B cell activating factor; BTK, Bruton’s tyrosine kinase; C5aR, C5a receptor; CAR, chimeric antigen receptor; CTLA4, cytotoxic T lymphocyte-associated protein 4; DC, dendritic cell; FB, factor B; FcR, Fc receptor; FD, factor D; IFNAR1, type I interferon receptor; JAK, Janus kinase; MASP2, mannan-binding lectin serine protease 2; MC1R, melanocortin 1 receptor; MC3R, melanocortin 3 receptor; siRNA, small interfering RNA; SLIT2, Slit homologue 2 protein; STAT, signal transducer and activator of transcription; TACI, transmembrane activator and CAML interactor; TRPC5, transient receptor potential cation channel subfamily C member 5.
Potential targets of immunotherapy for autoimmune GN include cells of the adaptive immune system and molecules involved in antigen presentation and recognition, for example using B cell-directed therapies and co-stimulation blockers, acting primarily inside lymphoid organs (Fig. 4). Belimumab, a monoclonal antibody to B cell activating factor, is approved for the treatment of lupus nephritis, and rituximab, a monoclonal antibody to CD20, is approved for the treatment of ANCA GN and is also broadly used as a steroid-sparing agent in autoimmune podocytopathies. Other B cell targets are under evaluation (for example, APRIL and TACI). Cathepsin S inhibition can attenuate autoimmune GN by suppressing MHC class II-mediated CD4+ T cell and B cell priming106. Another group of drugs targets the maturation, activation, proliferation and survival of autoreactive lymphocyte clones; for example, mycophenolate mofetil, azathioprine, cyclophosphamide, calcineurin inhibitors, anti-IL-23, anti-IL-17A and anti-CD40 ligand. Calcineurin inhibitors combine the T cell-directed immunosuppressive effect with a stabilization of the podocyte actin cytoskeleton, which generates strong antiproteinuric effects in addition to suppressing T cell function, for example in lupus nephritis107,108 (Fig. 4). The first reports support the efficacy of chimeric antigen receptor T cell therapy directed against CD19+ B cells and of a monoclonal antibody to CD38 (daratumumab) for targeting long-lived plasma cells in lupus nephritis109,110.
In addition, overwhelming preclinical evidence supports the role of innate immune pathways in autoimmune GN. For example, mice deficient in Toll-like receptors, complement factors C3, C5 and C5aR, adhesion molecules, pro-inflammatory chemokines and their receptors, or TH1-type, TH2-type and TH17-type cytokines are generally protected from immunopathology in GN models111. Therefore, complement inhibitors could possibly replace glucocorticoids for the control of glomerular inflammation. Indeed, the C5aR inhibitor avacopan is now used to minimize steroid use in active ANCA GN74, because this anaphylatoxin antagonist can quickly suppress glomerular inflammation and injury. Numerous other antibody-based or small interfering RNA-based complement inhibitors are currently under study (Table 4). By contrast, despite robust preclinical evidence, IFNAR1 blockade failed to control proteinuria in active lupus nephritis112.
Alloimmune GN
Risk factors and epidemiology
Glomerular injury can occur in recipients of any type of transplant, but it is most common in kidney transplant recipients. HLA mismatch and the presence of anti-HLA alloantibodies (donor-specific antibodies) are risk factors for alloimmune GN113. Chronic rejection mediated by alloimmunity resulting in chronic transplant glomerulopathy is a significant cause of graft loss, although acute antibody-mediated rejection can also involve the glomerulus.
A less common type of alloimmune response develops in those who have received a kidney transplant for a genetic deficiency of a specific protein (for example, one of the chains of type IV collagen or the podocyte-specific protein nephrin) that has led to kidney failure. The subsequent transplantation of a genetically intact kidney results in alloimmunity to the ‘non-self’ protein present in the graft114. Autoimmune glomerular disease can recur in the transplanted kidney (for example, autoimmune IgA nephropathy) despite immunosuppression prescribed to limit allogeneic responses114. Lastly, de novo glomerular disease can occur in kidney transplants and in allogeneic stem cell recipients who develop graft-versus-host disease (GVHD)115.
Immunopathogenesis and immunopathology
In kidney transplantation, donor reactivity to recipient HLA molecules of the graft is detectable as donor-specific antibodies in the blood that initially induce microvascular inflammation, often with the participation of complement and innate immune cells, in the form of acute glomerulitis113 (Fig. 3). The kidney graft is selectively impacted as it is only the transplanted tissue that expresses the allogeneic HLA molecules. Glomerular endothelial cells are most commonly affected by anti-HLA antibodies, although mesangial cells and to a lesser extent podocytes can be targeted. Active acute antibody-mediated rejection can exhibit various features, including immune cell infiltrates and leukocytes within glomerular capillaries, thrombotic microangiopathy, necrosis, endothelial and mesangial cell swelling, capillary occlusion and C4d deposition116. Acute glomerulitis due to antibody-mediated rejection can progress to transplant glomerulopathy, which is characterized by persistent or recurrent glomerular endothelial cell injury and GBM duplication usually in the absence of immune complex deposits despite the involvement of anti-HLA alloantibodies or other antibodies113,116. As in other glomerular diseases, chronic inflammation leads to glomerulosclerosis. In some patients, donor-specific antibodies targeting HLA class II may be more important than anti-HLA class I antibodies117. Pre-existing donor-specific antibodies at transplantation are likely to induce antibody-mediated rejection in glomeruli, but de novo donor-specific antibodies are important in many patients.
Besides anti-HLA antibodies, antibodies to self-antigens can develop, including antibodies to glomerular matrix proteins and endothelial cell antigens, in patients with transplant glomerulopathy118. Some of these autoantigens are cryptic and are released by the obligate ischaemia–reperfusion injury occurring in kidney transplantation. Antibody-mediated mechanisms of injury are often critical in transplant glomerulopathy, but in some circumstances, particularly in the absence of detectable anti-HLA antibodies and glomerular C4d deposition, other cellular effectors (for example, natural killer cells119) participate in injury independently of humoral effectors120.
Other immunological processes can result in glomerular injury in the transplanted kidney. Uncommonly, immunosuppressive agents, specifically calcineurin inhibitors, trigger glomerular endothelial injury and thrombotic microangiopathy in the graft121. In patients with genetic kidney disease who lack tolerance to an absent or aberrant glomerular protein, implantation of a non-mutated kidney results in processing and presentation of a normal protein as an apparent foreign antigen, followed by an adaptive immune response (for example, alloantibody deposition in the glomerulus via in situ immune complex formation). Although the immunopathogenesis of GVHD affecting the native kidneys remains unclear, observations in humans and rodent models of GVHD suggest several different patterns of injury and implicate a variety of immune mediators115. Glomerular lesions reported in GVHD include subepithelial deposits representing immune complex deposition or in situ immune complex formation122. However, patterns of injury are variable, reflecting not only the variable participation of immune responses but also the involvement of non-immune mediators or endothelial and mesangial injury. At a molecular level, data from animal models early in GVHD implicate antigen-presenting genes and the T cell chemoattractants CXC-chemokine ligands 9 and 11, with corresponding T cell infiltrates123. In humans, urinary levels of IL-6, IL-15 and CC-chemokine ligand 2 were associated with the risk of developing proteinuric kidney injury after bone marrow transplantation124. Membranous nephropathy, usually autoimmune in nature, can occur after allogeneic stem cell transplantation, and antibodies to protocadherin FAT1, as well as the FAT1 protein itself, have been detected in the kidney in this setting125.
Targets for immunotherapy
The management options for alloimmune GN focus on intensifying immunosuppression or adding intravenous immunoglobulin, with or without plasma exchange126. Therapies that induce immunological tolerance and those that more selectively target adaptive immunity, particularly humoral immunity, are likely to be useful (Table 4).
Autoinflammatory GN
Autoinflammatory GN disorders originate from inborn errors of innate immunity127. Only genetic testing can clarify the ultimate molecular diagnosis of an autoinflammatory GN (Table 2).
Immunopathology
Genetic abnormalities underlying overactivation of the alternative complement pathway are observed in 25% of patients with C3GN78,128 — that is, glomerular complement deposition without immunoglobulin deposits77. Such pathogenic variants in C3, CFB, CFH, CFI, DGKE and CFHR5 lower the threshold for spontaneous or induced C3 convertase activity129, possibly triggered by infections. Genomic rearrangements involving CFHR genes can also be identified130. Autoimmune GN and monoclonal gammopathy-related C3GN have a similar histological appearance but their respective immunopathogenesis warrants different treatments.
Genetic overactivation of Toll-like receptor 7 or direct overproduction of type I interferons induces a persistent antiviral immunity-like systemic inflammation, which may cause lupus-like immune complex GN17,131,132. Local effects of type I interferons promote podocytopathy-like lesions because type I interferons drive programmed cell death17,47. Accordingly, long-term exposure to type I interferons, such as during treatment for relapsing–remitting multiple sclerosis, can induce similar kidney lesions133.
Monogenic disorders of the NLRP3 inflammasome, pro-inflammatory cytokines or their receptors lead to spontaneous and persistent systemic and local tissue inflammation127 (Table 3). Usually, the kidney is only indirectly affected by AA amyloidosis, a glomerular deposition of β-fibrils of the acute phase protein serum amyloid A, which is constantly released by the liver in patients with hereditary fever syndromes134. The accumulating deposits interfere with the normal filtration process and trigger nonspecific local inflammation and glomerular cell injury, promoting an increasing degree of proteinuria and kidney failure over time.
Targets for immunotherapy
The selective overexpression of a single cytokine means that people with autoinflammatory GN are likely to respond to highly selective immunotherapies (Table 4), such as TNF blockers for TNF receptor-associated periodic syndrome, IL-1 blockers for cryopyrin-associated autoinflammatory syndromes130,135, interferon blockers for interferonopathies132 and complement inhibitors for hereditary C3GN77,136.
Monoclonal gammopathy-related GN
Risk factors and immunopathology
The production of nephrotoxic monoclonal immunoglobulins is the hallmark of monoclonal gammopathy-related GN137 (Table 3 and Fig. 3). Monoclonal immunoglobulins deposit as intact immunoglobulin or as fragments, for example in light chain deposition disease or immunoglobulin heavy chain amyloidosis. Such deposits can have various ultrastructural characteristics138. They can be amorphous (in monoclonal immunoglobulin deposition disease and proliferative GN with monoclonal immunoglobulin deposits) or organized, such as β-sheet amyloid fibrils (in amyloidosis) and microtubules (in immunotactoid glomerulopathy), or can form cryoglobulins (in cryoglobulin-associated GN) and microcrystals (in crystalloglobulin GN). Indirect modes of injury occur when monoclonal immunoglobulins activate complement, resulting in C3GN or thrombotic microangiopathy139,140.
Although a minority of individuals with monoclonal gammopathy-related GN also have overt multiple myeloma or high-grade lymphoma per se requiring immediate treatment, the source of the monoclonal immunoglobulin in most cases is small B cell clones or plasma cell clones137. These small clones are collectively designated as ‘monoclonal gammopathy of renal significance’141 and are the result of non-malignant somatic mutations in B cell or plasma cell precursors64. Nevertheless, the nephrotoxic effect of such monoclonal immunoglobulins is the result of the primary amino acid sequence142,143,144.
The monoclonal immunoglobulin deposits detected by immunostaining are the most common and most important pathological histological finding in monoclonal gammopathy-related GN138 (Fig. 2). Monotypic deposits are defined by κ or λ light chain restriction. In cases where the entire immunoglobulin is deposited, a heavy chain restriction (that is, IgG subclass) combined with light chain restriction strongly implies the deposits are monoclonal. Of note, heavy chain restriction by itself does not prove monoclonality unless it relates to a heavy chain disease such as AH amyloidosis or heavy chain deposition disease. Ultrastructural characteristics resolved by electron microscopy can help to distinguish one monoclonal gammopathy-related kidney disease from another (for example, fibrils in amyloidosis or microtubules in immunotactoid GN). Deposits of C3 with little immunoglobulin are seen in C3 glomerulopathy with monoclonal gammopathy. Finally, no immune deposits occur in monoclonal gammopathy-related thrombotic microangiopathy.
Targets for immunotherapy
Elimination of the B cell clones or plasma cell clones in the bone marrow and lymphoid organs is the main goal to deplete the monoclonal immunoglobulin to halt kidney injury and to preserve kidney function (Fig. 4). Standard immunosuppressive therapies are insufficient to induce a beneficial haematological response, which is usually a greater than 90% reduction in the level of monoclonal immunoglobulin145,146,147. To accomplish this, clone-directed therapy is required (Table 4). Chemotherapy is used often combined with immunotherapy137,148. For example, CD20-expressing B cell clones can be depleted by anti-CD20 monoclonal antibodies such as rituximab, obinutuzumab or ofatuzumab149. These can be used in combination with cyclophosphamide and corticosteroids. For plasma cell clones, anti-CD38 monoclonal antibodies such as daratumumab or isatuximab can be used alone or in combination with other targeted therapies (such as cyclophosphamide, proteasome inhibitors or immunomodulatory drugs148) to achieve the necessary response150,151,152,153. High-dose therapy followed by stem cell transplantation is sometimes necessary to achieve a sufficient reduction154.
Proteasome inhibitors are a successful class of drugs against myeloma cells155, with three currently approved (bortezomib, carfilzomib and ixazomib)156 (Fig. 4). They target the 20S unit of the proteasome, inhibiting the ubiquitin degradation pathway for proteins and activation of nuclear factor-κB157. However, as the 20S proteasome is ubiquitously expressed, proteasome inhibition has adverse effects. Immunoproteasomes, on the other hand, are predominantly expressed in lymphocytes and monocytes and are involved in cell-mediated immunity and production of MHC class I ligands during infections158. In addition, immunoproteasomes contribute to autoimmune diseases159. Theoretically, inhibition of immunoproteasomes should have fewer off-target side effects. Several immunoproteasome inhibitors have demonstrated promising in vitro results by targeting the large multifunctional peptidase 7 subunit160,161,162. Some immunoproteasome inhibitors may act synergistically with proteasome inhibitors156,157,163.
Concluding remarks
The growing awareness of the immunopathogenesis of GN has revealed the limitations of lesion-based classifications in identifying the ultimate cause to select appropriate treatments. The recent improvements in immune phenotyping, including the detection of autoantibodies to specific glomerular antigens, different complement subunits and immune complexes by immunofluorescence of kidney biopsies, have increased its value for diagnosis (Box 1). In addition, more immunotherapies are becoming available (Table 4). Extended genetic testing, with next-generation sequencing, is another evolving diagnostic tool to ultimately define a molecular diagnosis (Box 1). Indeed, mutations in complement genes128, in Toll-like receptors131 or in genes of the interferon pathways17 can directly trigger different types of immune-related GN or favour the production of autoantibodies17,128,129,131. Kidney biopsy can in many situations assist in diagnosis and remains the gold standard to determine disease activity and chronicity. Activity and chronicity are relevant parameters in determining treatment intensity, and these indices integrate with treatments targeting non-immune mechanisms of chronic kidney disease progression. Novel urinary biomarkers of immunological GN activity can be used to monitor treatment responses and be validated against repeated kidney biopsies.
An intuitive pathophysiology-based classification will facilitate the management of these conditions. We propose that individuals with GN be described under three headings: firstly, as having one of the five types of GN discussed in this Review (infection-related GN, autoimmune GN, alloimmune GN, autoinflammatory GN and monoclonal gammopathy-related GN); secondly, by detailing the exact disease type on the basis of the pathogenesis; and, thirdly, on the basis of the features of the histological lesion, if a kidney biopsy has been performed.
Despite the challenges in defining the pathogenesis of the large number of individual GN diseases, there has been significant progress in understanding the fundamental basis of immune glomerular disease. However, our understanding of GN is incomplete and requires greater depth if we are to develop more targeted immunotherapies. Among the infection-related GN disorders, the contribution of genetic susceptibilities in primary immunodeficiency and acquired complementopathies (genetic or acquired disorders of the complement system) driving clinically relevant GN deserves further study. The plethora of novel autoantigens in autoimmune GN mandates further investigation, not only to ensure accurate diagnostic tests are clinically available but also to determine the role of the related autoantibodies as biomarkers for diagnosis, disease activity and guidance of immunotherapy. In particular, the recently discovered anti-nephrin antibody-related podocytopathy is likely to have significant clinical implications89. Why alloimmune GN responds poorly to immunotherapy is not well understood. Prevention of priming of donor-specific antibodies early after transplantation is probably the key to preventing transplant glomerulopathy. Among autoinflammatory GN disorders, genotype–phenotype associations of the hereditary complementopathies encompass many unsolved questions. The wide spectrum of monoclonal gammopathy-related GN will benefit from plasma cell-targeted therapies under development.
GN requires an immunopathophysiology-based classification system to better connect with the growing number of available immunotherapy drugs. As GN comprises immune-mediated disorders, GN research needs more input from experts in immunology. Indeed, clinical immunologists may help to overcome traditional lesion-based concepts in this domain and improve the management of these important and at times difficult to treat diseases.
Change history
06 November 2023
In the version of the article initially published, affiliation 8 did not reflect a 2023 change in the institution’s name, which has now been updated.
References
Chadban, S. J. & Atkins, R. C. Glomerulonephritis. Lancet 365, 1797–1806 (2005).
Guo, Q., Wu, S., Xu, C., Wang, J. & Chen, J. Global disease burden from acute glomerulonephritis 1990–2019. Kidney Int. Rep. 6, 2212–2217 (2021).
GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 395, 709–733 (2020).
Wetmore, J. B., Guo, H., Liu, J., Collins, A. J. & Gilbertson, D. T. The incidence, prevalence, and outcomes of glomerulonephritis derived from a large retrospective analysis. Kidney Int. 90, 853–860 (2016).
United States Renal Data System. 2022 annual data report. https://usrds-adr.niddk.nih.gov/2022?dkrd=/about-niddk/strategic-plans-reports/usrds/annual-data-report (2022).
Titze, S. et al. Disease burden and risk profile in referred patients with moderate chronic kidney disease: composition of the German Chronic Kidney Disease (GCKD) cohort. Nephrol. Dial. Transplant. 30, 441–451 (2015).
Daehn, I. S. & Duffield, J. S. The glomerular filtration barrier: a structural target for novel kidney therapies. Nat. Rev. Drug Discov. 20, 770–788 (2021). This review explains the unique structure and function of the glomerular filtration barrier, and new ways by which its integrity might be preserved to treat glomerular disease.
Kopp, J. B. et al. Podocytopathies. Nat. Rev. Dis. Prim. 6, 68 (2020).
Grahammer, F. et al. A flexible, multilayered protein scaffold maintains the slit in between glomerular podocytes. JCI Insight 1, 86177 (2016).
Anguiano, L., Kain, R. & Anders, H. J. The glomerular crescent: triggers, evolution, resolution, and implications for therapy. Curr. Opin. Nephrol. Hypertens. 29, 302–309 (2020).
Kumar, S. V. et al. Neutrophil extracellular trap-related extracellular histones cause vascular necrosis in severe GN. J. Am. Soc. Nephrol. 26, 2399–2413 (2015). This study shows that severe immune glomerular injury is mediated by histones as products of NETs.
Kessenbrock, K. et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 15, 623–625 (2009).
Cortinovis, M., Perico, N., Ruggenenti, P., Remuzzi, A. & Remuzzi, G. Glomerular hyperfiltration. Nat. Rev. Nephrol. 18, 435–451 (2022).
Kitching, A. R. & Hickey, M. J. Immune cell behaviour and dynamics in the kidney-insights from in vivo imaging. Nat. Rev. Nephrol. 18, 22–37 (2022). This review details the unusual immune milieu within the glomerulus and how leukocytes behave within glomerular capillaries to mediate GN.
Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group. KDIGO 2021 clinical practice guideline for the management of glomerular diseases. Kidney Int. Suppl. 100, S1–S276 (2021).
Sethi, S. et al. Mayo Clinic/Renal Pathology Society consensus report on pathologic classification, diagnosis, and reporting of GN. J. Am. Soc. Nephrol. 27, 1278–1287 (2016).
Lodi, L. et al. Type I interferon-related kidney disorders. Kidney Int. 101, 1142–1159 (2022).
Ravaglia, F. et al. The pathology lesion patterns of podocytopathies: how and why? Front. Cell Dev. Biol. 10, 838272 (2022).
Lerner, G. B., Virmani, S., Henderson, J. M., Francis, J. M. & Beck, L. H. Jr. A conceptual framework linking immunology, pathology, and clinical features in primary membranous nephropathy. Kidney Int. 100, 289–300 (2021).
Kumar, R. et al. Case report: unusual aggregation of different glomerulopathies in a family resolved by genetic testing and reverse phenotyping. Front. Pediatrics 10, 826330 (2022).
Smith, K. D. et al. Digital spatial profiling of collapsing glomerulopathy. Kidney Int. 101, 1017–1026 (2022).
Anders, H. J., Jayne, D. R. & Rovin, B. H. Hurdles to the introduction of new therapies for immune-mediated kidney diseases. Nat. Rev. Nephrol. 12, 205–216 (2016).
Carapetis, J. R., Steer, A. C., Mulholland, E. K. & Weber, M. The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5, 685–694 (2005).
Rodriguez-Iturbe, B. & Musser, J. M. The current state of poststreptococcal glomerulonephritis. J. Am. Soc. Nephrol. 19, 1855–1864 (2008).
Boils, C. L., Nasr, S. H., Walker, P. D., Couser, W. G. & Larsen, C. P. Update on endocarditis-associated glomerulonephritis. Kidney Int. 87, 1241–1249 (2015).
Satoskar, A. A., Parikh, S. V. & Nadasdy, T. Epidemiology, pathogenesis, treatment and outcomes of infection-associated glomerulonephritis. Nat. Rev. Nephrol. 16, 32–50 (2020).
Genovese, G. et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329, 841–845 (2010).
Kopp, J. B. et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J. Am. Soc. Nephrol. 22, 2129–2137 (2011).
McGuire, B. M. et al. Brief communication: glomerulonephritis in patients with hepatitis C cirrhosis undergoing liver transplantation. Ann. Intern. Med. 144, 735–741 (2006).
Kupin, W. L. Viral-associated GN: hepatitis B and other viral infections. Clin. J. Am. Soc. Nephrol. 12, 1529–1533 (2017).
Barsoum, R. S. Tropical parasitic nephropathies. Nephrol. Dial. Transplant. 14, 79–91 (1999).
Neves, P. et al. Schistosomiasis-associated glomerulopathy: clinical aspects, pathological characteristics, and renal outcomes. Clin. Nephrol. 93, 251–261 (2020).
Walker, A. et al. Eosinophilic glomerulonephritis in children in southwestern Uganda. Kidney Int. 71, 569–573 (2007).
Sethi, S., D’Costa, M. R., Hermann, S. M., Nasr, S. H. & Fervenza, F. C. Immune-complex glomerulonephritis after COVID-19 infection. Kidney Int. Rep. 6, 1170–1173 (2021).
Rodriguez-Iturbe, B. Autoimmunity in acute poststreptococcal GN: a neglected aspect of the disease. J. Am. Soc. Nephrol. 32, 534–542 (2021).
Yoshizawa, N. et al. Nephritis-associated plasmin receptor and acute poststreptococcal glomerulonephritis: characterization of the antigen and associated immune response. J. Am. Soc. Nephrol. 15, 1785–1793 (2004).
Honda-Ogawa, M. et al. Cysteine proteinase from Streptococcus pyogenes enables evasion of innate immunity via degradation of complement factors. J. Biol. Chem. 288, 15854–15864 (2013).
Chauvet, S. et al. Anti-factor B antibodies and acute postinfectious GN in children. J. Am. Soc. Nephrol. 31, 829–840 (2020).
Nasr, S. H. et al. Postinfectious glomerulonephritis in the elderly. J. Am. Soc. Nephrol. 22, 187–195 (2011).
Couser, W. G. & Johnson, R. J. The etiology of glomerulonephritis: roles of infection and autoimmunity. Kidney Int. 86, 905–914 (2014).
Puelles, V. G. et al. Multiorgan and renal tropism of SARS-CoV-2. N. Engl. J. Med. 383, 590–592 (2020).
Li, J. et al. Transmembrane TNF-α facilitates HIV-1 infection of podocytes cultured from children with HIV-associated nephropathy. J. Am. Soc. Nephrol. 28, 862–875 (2017).
Araújo, S. A. et al. First report of collapsing variant of focal segmental glomerulosclerosis triggered by arbovirus: dengue and Zika virus infection. Clin. Kidney J. 12, 355–361 (2019).
Allam, R. et al. Viral RNA and DNA trigger common antiviral responses in mesangial cells. J. Am. Soc. Nephrol. 20, 1986–1996 (2009).
Gupta, S., Pepper, R. J., Ashman, N. & Walsh, S. B. Nephrotic syndrome: oedema formation and its treatment with diuretics. Front. Physiol. 9, 1868 (2018).
Fenaroli, P. et al. Collapsing glomerulopathy as a complication of type I interferon-mediated glomerulopathy in a patient with RNASEH2B-related Aicardi-Goutières syndrome. Am. J. Kidney Dis. 78, 750–754 (2021).
Migliorini, A. et al. The antiviral cytokines IFN-alpha and IFN-beta modulate parietal epithelial cells and promote podocyte loss: implications for IFN toxicity, viral glomerulonephritis, and glomerular regeneration. Am. J. Pathol. 183, 431–440 (2013).
Melica, M. E. et al. Differentiation of crescent-forming kidney progenitor cells into podocytes attenuates severe glomerulonephritis in mice. Sci. Transl. Med. 14, eabg3277 (2022). This study defines the participation of clonally expanded glomerular cells in severe glomerular injury and how tissue progenitors could assist in preserving glomerular structure and function in severe immune-mediated GN.
Nichols, B. et al. Innate immunity pathways regulate the nephropathy gene apolipoprotein L1. Kidney Int. 87, 332–342 (2015).
Wu, J. et al. The key role of NLRP3 and STING in APOL1-associated podocytopathy. J. Clin. Invest. 131, e136329 (2021).
Nystrom, S. E. et al. JAK inhibitor blocks COVID-19 cytokine-induced JAK/STAT/APOL1 signaling in glomerular cells and podocytopathy in human kidney organoids. JCI Insight 7, e157432 (2022).
Angeletti, A., Cantarelli, C. & Cravedi, P. HCV-associated nephropathies in the era of direct acting antiviral agents. Front. Med. 6, 20 (2019).
Razzak Chaudhary, S. et al. Trends in the outcomes of end-stage renal disease secondary to human immunodeficiency virus-associated nephropathy. Nephrol. Dial. Transplant. 30, 1734–1740 (2015).
Soriano, V. et al. Hepatitis C cure with antiviral therapy–benefits beyond the liver. Antivir. Ther. 21, 1–8 (2016).
Okumura, M. et al. Use of immunosuppressive therapy in the treatment of IgA-dominant infection-related glomerulonephritis. Intern. Med. 61, 697–701 (2022).
Arivazhagan, S. et al. Efficacy of corticosteroids in infection-related glomerulonephritis-a randomised controlled trial. Kidney Int. Rep. 7, 2160–2165 (2022).
Anders, H. J. et al. Lupus nephritis. Nat. Rev. Dis. Prim. 6, 7 (2020). This is a detailed review of the complexities and challenges in understanding and treating lupus nephritis.
Kitching, A. R. et al. ANCA-associated vasculitis. Nat. Rev. Dis. Prim. 6, 71 (2020). This article explains how in ANCA-associated vasculitis, activating autoantibodies and cellular immunity to neutrophil proteins affect severe microvascular injury.
Ronco, P. et al. Membranous nephropathy. Nat. Rev. Dis. Prim. 7, 69 (2021).
Schmidt, R. E., Grimbacher, B. & Witte, T. Autoimmunity and primary immunodeficiency: two sides of the same coin? Nat. Rev. Rheumatol. 14, 7–18 (2017).
Robson, K. J., Ooi, J. D., Holdsworth, S. R., Rossjohn, J. & Kitching, A. R. HLA and kidney disease: from associations to mechanisms. Nat. Rev. Nephrol. 14, 636–655 (2018). This article includes a comprehensive review of the associations of HLA with glomerular disease and the emerging HLA-functional links.
Stanescu, H. C. et al. Risk HLA-DQA1 and PLA2R1 alleles in idiopathic membranous nephropathy. N. Engl. J. Med. 364, 616–626 (2011).
Alriyami, M. & Polychronakos, C. Somatic mutations and autoimmunity. Cells 10, 2056 (2021).
Mustjoki, S. & Young, N. S. Somatic mutations in “benign” disease. N. Engl. J. Med. 384, 2039–2052 (2021).
Ooi, J. D. et al. Dominant protection from HLA-linked autoimmunity by antigen-specific regulatory T cells. Nature 545, 243–247 (2017). This study demonstrates the mechanism of HLA susceptibility and dominant protection and the key role of antigen-specific regulatory T cells in maintaining tolerance in autoimmune GN.
Lai, K. N. et al. IgA nephropathy. Nat. Rev. Dis. Prim. 2, 16001 (2016).
Muchtar, E., Magen, H. & Gertz, M. A. How I treat cryoglobulinemia. Blood 129, 289–298 (2017).
Audemard-Verger, A., Pillebout, E., Guillevin, L., Thervet, E. & Terrier, B. IgA vasculitis (Henoch-Shönlein purpura) in adults: diagnostic and therapeutic aspects. Autoimmun. Rev. 14, 579–585 (2015).
Kaul, A. et al. Systemic lupus erythematosus. Nat. Rev. Dis. Prim. 2, 16039 (2016).
Weening, J. J. et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. J. Am. Soc. Nephrol. 15, 241–250 (2004).
Kuligowski, M. P. et al. Antimyeloperoxidase antibodies rapidly induce alpha-4-integrin-dependent glomerular neutrophil adhesion. Blood 113, 6485–6494 (2009).
Dick, J. et al. C5a receptor 1 promotes autoimmunity, neutrophil dysfunction and injury in experimental anti-myeloperoxidase glomerulonephritis. Kidney Int. 93, 615–625 (2018).
Masuda, S. et al. Formation and disordered degradation of neutrophil extracellular traps in necrotizing lesions of anti-neutrophil cytoplasmic antibody-associated vasculitis. Am. J. Pathol. 189, 839–846 (2019).
Jayne, D. R. W., Merkel, P. A., Schall, T. J. & Bekker, P. Avacopan for the treatment of ANCA-associated vasculitis. N. Engl. J. Med. 384, 599–609 (2021).
Nakazawa, D., Masuda, S., Tomaru, U. & Ishizu, A. Pathogenesis and therapeutic interventions for ANCA-associated vasculitis. Nat. Rev. Rheumatol. 15, 91–101 (2019).
Iatropoulos, P. et al. Cluster analysis identifies distinct pathogenetic patterns in C3 glomerulopathies/immune complex-mediated membranoproliferative GN. J. Am. Soc. Nephrol. 29, 283–294 (2018).
Smith, R. J. H. et al. C3 glomerulopathy - understanding a rare complement-driven renal disease. Nat. Rev. Nephrol. 15, 129–143 (2019).
Heiderscheit, A. K., Hauer, J. J. & Smith, R. J. H. C3 glomerulopathy: understanding an ultra-rare complement-mediated renal disease. Am. J. Med. Genet. 190, 344–357 (2022).
Pedchenko, V., Kitching, A. R. & Hudson, B. G. Goodpasture’s autoimmune disease-a collagen IV disorder. Matrix Biol. 71-72, 240–249 (2018).
He, C. et al. Peroxidasin-mediated bromine enrichment of basement membranes. Proc. Natl Acad. Sci. USA 117, 15827–15836 (2020).
McCall, A. S. et al. Inhibitory anti-peroxidasin antibodies in pulmonary-renal syndromes. J. Am. Soc. Nephrol. 29, 2619–2625 (2018).
Beck, L. H. Jr. et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N. Engl. J. Med. 361, 11–21 (2009). This seminal study discovers the cause of most cases of membranous nephropathy, defining it as an autoimmune disease.
Tomas, N. M. et al. Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N. Engl. J. Med. 371, 2277–2287 (2014).
Sethi, S. et al. Protocadherin 7-associated membranous nephropathy. J. Am. Soc. Nephrol. 32, 1249–1261 (2021).
Al-Rabadi, L. F. et al. Serine protease HTRA1 as a novel target antigen in primary membranous nephropathy. J. Am. Soc. Nephrol. 32, 1666–1681 (2021).
Le Quintrec, M. et al. Contactin-1 is a novel target antigen in membranous nephropathy associated with chronic inflammatory demyelinating polyneuropathy. Kidney Int. 100, 1240–1249 (2021).
Reinhard, L. et al. Netrin G1 is a novel target antigen in primary membranous nephropathy. J. Am. Soc. Nephrol. 33, 1823–1831 (2022).
Sethi, S. et al. Neural epidermal growth factor-like 1 protein (NELL-1) associated membranous nephropathy. Kidney Int. 97, 163–174 (2020).
Watts, A. J. B. et al. Discovery of autoantibodies targeting nephrin in minimal change disease supports a novel autoimmune etiology. J. Am. Soc. Nephrol. 33, 238–252 (2022).
Mihai, S. & Nimmerjahn, F. The role of Fc receptors and complement in autoimmunity. Autoimmun. Rev. 12, 657–660 (2013).
Genest, D. S. et al. Comparison of complement pathway activation in autoimmune glomerulonephritis. Kidney Int. Rep. 7, 1027–1036 (2022).
Haddad, G. et al. Altered glycosylation of IgG4 promotes lectin complement pathway activation in anti-PLA2R1-associated membranous nephropathy. J. Clin. Invest. 131, e140453 (2021).
Bally, S. et al. Phospholipase A2 receptor-related membranous nephropathy and mannan-binding lectin deficiency. J. Am. Soc. Nephrol. 27, 3539–3544 (2016).
Kulkarni, O. et al. Anti-Ccl2 Spiegelmer permits 75% dose reduction of cyclophosphamide to control diffuse proliferative lupus nephritis and pneumonitis in MRL-Fas(lpr) mice. J. Pharmacol. Exp. Ther. 328, 371–377 (2009).
Westhorpe, C. L. V. et al. Effector CD4+ T cells recognize intravascular antigen presented by patrolling monocytes. Nat. Commun. 9, 747 (2018). This study discovers that antigens can be presented to effector CD4+ T cells within the microvasculature of the glomerulus by intravascular monocytes.
Summers, S. A. et al. Th1 and Th17 cells induce proliferative glomerulonephritis. J. Am. Soc. Nephrol. 20, 2518–2524 (2009).
Ooi, J. D. et al. The HLA-DRB1*15:01-restricted Goodpasture’s T cell epitope induces GN. J. Am. Soc. Nephrol. 24, 419–431 (2013).
Chen, A. et al. Bowman’s capsule provides a protective niche for podocytes from cytotoxic CD8+ T cells. J. Clin. Invest. 128, 3413–3424 (2018).
Krebs, C. F. et al. Pathogen-induced tissue-resident memory TH17 (TRM17) cells amplify autoimmune kidney disease. Sci. Immunol. 5, eaba4163 (2020). This study implicates CD4+ tissue-resident memory T cells within the kidney in immune glomerular disease.
Allam, R. et al. Viral 5’-triphosphate RNA and non-CpG DNA aggravate autoimmunity and lupus nephritis via distinct TLR-independent immune responses. Eur. J. Immunol. 38, 3487–3498 (2008).
Anders, H. J. et al. Bacterial CpG-DNA aggravates immune complex glomerulonephritis: role of TLR9-mediated expression of chemokines and chemokine receptors. J. Am. Soc. Nephrol. 14, 317–326 (2003).
Pawar, R. D. et al. Bacterial lipopeptide triggers massive albuminuria in murine lupus nephritis by activating Toll-like receptor 2 at the glomerular filtration barrier. Immunology 128, e206–e221 (2009).
Uhlin, F. et al. Endopeptidase cleavage of anti-glomerular basement membrane antibodies in vivo in severe kidney disease: an open-label phase 2a study. J. Am. Soc. Nephrol. 33, 829–838 (2022). This article reports a phase II trial demonstrating that an intravenous injection of endopeptidase to cleave nephritogenic autoantibodies may be as effective as or more effective than plasma exchange in treating acute GN.
Walsh, M. et al. The effects of plasma exchange in patients with ANCA-associated vasculitis: an updated systematic review and meta-analysis. BMJ 376, e064604 (2022).
Barratt, J. et al.Results from part A of the multi-center, double-blind, randomized, placebo-controlled NefIgArd trial, which evaluated targeted-release formulation of budesonide for the treatment of primary immunoglobulin A nephropathy. Kidney Int. https://doi.org/10.1016/j.kint.2022.09.017 (2022).
Rupanagudi, K. V. et al. Cathepsin S inhibition suppresses systemic lupus erythematosus and lupus nephritis because cathepsin S is essential for MHC class II-mediated CD4 T cell and B cell priming. Ann. Rheum. Dis. 74, 452–463 (2015).
Heo, Y. A. Voclosporin: first approval. Drugs 81, 605–610 (2021).
Rovin, B. H. et al. Efficacy and safety of voclosporin versus placebo for lupus nephritis (AURORA 1): a double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 397, 2070–2080 (2021).
Mougiakakos, D. et al. CD19-Targeted CAR T cells in refractory systemic lupus erythematosus. N. Engl. J. Med. 385, 567–569 (2021). This is a report of a single patient with refractory systemic lupus erythematosus, including nephritis, in whom conventional B cell-depleting therapies were ineffective, whose disease was treated with chimeric antigen receptor T cells specific for CD19.
Ostendorf, L. et al. Targeting CD38 with daratumumab in refractory systemic lupus erythematosus. N. Engl. J. Med. 383, 1149–1155 (2020).
Kurts, C., Panzer, U., Anders, H. J. & Rees, A. J. The immune system and kidney disease: basic concepts and clinical implications. Nat. Rev. Immunol. 13, 738–753 (2013).
Jayne, D. et al. Phase II randomised trial of type I interferon inhibitor anifrolumab in patients with active lupus nephritis. Ann. Rheum. Dis. 81, 496–506 (2022).
Hanf, W., Bonder, C. S. & Coates, P. T. Transplant glomerulopathy: the interaction of HLA antibodies and endothelium. J. Immunol. Res. 2014, 549315 (2014).
Lim, W. H., Shingde, M. & Wong, G. Recurrent and de novo glomerulonephritis after kidney transplantation. Front. Immunol. 10, 1944 (2019).
Hingorani, S. Renal complications of hematopoietic-cell transplantation. N. Engl. J. Med. 374, 2256–2267 (2016).
Roufosse, C. et al. A 2018 reference guide to the Banff classification of renal allograft pathology. Transplantation 102, 1795–1814 (2018).
Gloor, J. M. et al. Transplant glomerulopathy: subclinical incidence and association with alloantibody. Am. J. Transplant. 7, 2124–2132 (2007).
Cardinal, H., Dieudé, M. & Hébert, M. J. The emerging importance of non-HLA autoantibodies in kidney transplant complications. J. Am. Soc. Nephrol. 28, 400–406 (2017).
Koenig, A. et al. Missing self triggers NK cell-mediated chronic vascular rejection of solid organ transplants. Nat. Commun. 10, 5350 (2019).
Filippone, E. J. & Farber, J. L. Histologic antibody-mediated kidney allograft rejection in the absence of donor-specific HLA antibodies. Transplantation 105, e181–e190 (2021).
Van Buren, D. et al. De novo hemolytic uremic syndrome in renal transplant recipients immunosuppressed with cyclosporine. Surgery 98, 54–62 (1985).
Girsberger, M., Halter, J. P., Hopfer, H., Dickenmann, M. & Menter, T. Kidney pathology after hematologic cell transplantation-a single-center observation study of indication biopsies and autopsies. Biol. Blood Marrow Transpl. 24, 571–580 (2018).
Sadeghi, B. et al. Early-phase GVHD gene expression profile in target versus non-target tissues: kidney, a possible target? Bone Marrow Transpl. 48, 284–293 (2013).
Hingorani, S., Gooley, T., Pao, E., Sandmaier, B. & McDonald, G. Urinary cytokines after HCT: evidence for renal inflammation in the pathogenesis of proteinuria and kidney disease. Bone Marrow Transpl. 49, 403–409 (2014).
Sethi, S. et al. Hematopoietic stem cell transplant-membranous nephropathy is associated with protocadherin FAT1. J. Am. Soc. Nephrol. 33, 1033–1044 (2022).
Hart, A., Singh, D., Brown, S. J., Wang, J. H. & Kasiske, B. L. Incidence, risk factors, treatment, and consequences of antibody-mediated kidney transplant rejection: a systematic review. Clin. Transplant. 35, e14320 (2021).
Manthiram, K., Zhou, Q., Aksentijevich, I. & Kastner, D. L. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat. Immunol. 18, 832–842 (2017).
Lemaire, M., Noone, D., Lapeyraque, A. L., Licht, C. & Frémeaux-Bacchi, V. Inherited kidney complement diseases. Clin. J. Am. Soc. Nephrol. 16, 942–956 (2021). This study details the nature of genetic abnormalities of complement and its regulatory elements and how they can cause glomerular disease.
Moore, I. et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood 115, 379–387 (2010).
De Benedetti, F. et al. Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N. Engl. J. Med. 378, 1908–1919 (2018).
Brown, G. J. et al. TLR7 gain-of-function genetic variation causes human lupus. Nature 605, 349–356 (2022).
Crow, Y. J. & Stetson, D. B. The type I interferonopathies: 10 years on. Nat. Rev. Immunol. 22, 471–483 (2021).
Markowitz, G. S., Nasr, S. H., Stokes, M. B. & D’Agati, V. D. Treatment with IFN-α, -β, or -γ is associated with collapsing focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 5, 607–615 (2010).
Papa, R. & Lachmann, H. J. Secondary, AA, amyloidosis. Rheum. Dis. Clin. North Am. 44, 585–603 (2018).
Hansmann, S. et al. Consensus protocols for the diagnosis and management of the hereditary autoinflammatory syndromes CAPS, TRAPS and MKD/HIDS: a German PRO-KIND initiative. Pediatr. Rheumatol. Online J. 18, 17 (2020).
Gonzalez Suarez, M. L. et al. Treatment of C3 glomerulopathy in adult kidney transplant recipients: a systematic review. Med. Sci. 8, 44 (2020).
Leung, N., Bridoux, F. & Nasr, S. H. Monoclonal gammopathy of renal significance. N. Engl. J. Med. 384, 1931–1941 (2021). This study explains how a B cell or plasma cell secreting a single immunoglobulin clone can cause glomerular disease, even in the absence of formal diagnostic criteria for malignancy, and reviews its pathogenesis, features and management.
Bridoux, F. et al. Diagnosis of monoclonal gammopathy of renal significance. Kidney Int. 87, 698–711 (2015).
Chauvet, S. et al. Both monoclonal and polyclonal immunoglobulin contingents mediate complement activation in monoclonal gammopathy associated-C3 glomerulopathy. Front. Immunol. 9, 2260 (2018).
Yui, J. C. et al. Monoclonal gammopathy-associated thrombotic microangiopathy. Am. J. Hematol. 94, E250–E253 (2019).
Leung, N. et al. The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat. Rev. Nephrol. 15, 45–59 (2019).
Bridoux, F. et al. Unravelling the immunopathological mechanisms of heavy chain deposition disease with implications for clinical management. Kidney Int. 91, 423–434 (2017).
Sirac, C. et al. Role of the monoclonal kappa chain V domain and reversibility of renal damage in a transgenic model of acquired Fanconi syndrome. Blood 108, 536–543 (2006).
Sirac, C. et al. Animal models of monoclonal immunoglobulin-related renal diseases. Nat. Rev. Nephrol. 14, 246–264 (2018).
Chauvet, S. et al. Treatment of B-cell disorder improves renal outcome of patients with monoclonal gammopathy-associated C3 glomerulopathy. Blood 129, 1437–1447 (2017).
Kourelis, T. V. et al. Outcomes of patients with renal monoclonal immunoglobulin deposition disease. Am. J. Hematol. 91, 1123–1128 (2016).
Rezk, T. et al. Prolonged renal survival in light chain amyloidosis: speed and magnitude of light chain reduction is the crucial factor. Kidney Int. 92, 1476–1483 (2017).
Fermand, J. P. et al. How I treat monoclonal gammopathy of renal significance (MGRS). Blood 122, 3583–3590 (2013).
Fotiou, D., Theodorakakou, F. & Kastritis, E. Monoclonal antibody-based therapies for Waldenström’s macroglobulinemia. Leuk. Res. Rep. 17, 100324 (2022).
Milani, P. et al. Daratumumab in light chain deposition disease: rapid and profound hematologic response preserves kidney function. Blood Adv. 4, 1321–1324 (2020).
Milani, P. et al. High rate of profound clonal and renal responses with daratumumab treatment in heavily pre-treated patients with light chain (AL) amyloidosis and high bone marrow plasma cell infiltrate. Am. J. Hematol. 95, 900–905 (2020).
Palladini, G., Milani, P., Malavasi, F. & Merlini, G. Daratumumab in the treatment of light-chain (AL) amyloidosis. Cells 10, 545 (2021).
Zand, L. et al. Safety and efficacy of daratumumab in patients with proliferative GN with monoclonal immunoglobulin deposits. J. Am. Soc. Nephrol. 32, 1163–1173 (2021).
Gozzetti, A. et al. Monoclonal gammopathy of renal significance (MGRS): real-world data on outcomes and prognostic factors. Am. J. Hematol. 97, 877–884 (2022).
Engelhardt, M., Waldschmidt, J. M. & Wäsch, R. Proteasome inhibition: the dawn of novel therapies in multiple myeloma. Haematologica 107, 1018–1019 (2022).
Gavriatopoulou, M., Malandrakis, P., Ntanasis-Stathopoulos, I. & Dimopoulos, M. A. Nonselective proteasome inhibitors in multiple myeloma and future perspectives. Expert. Opin. Pharmacother. 23, 335–347 (2022).
Ettari, R. et al. Immunoproteasome-selective and non-selective inhibitors: a promising approach for the treatment of multiple myeloma. Pharmacol. Ther. 182, 176–192 (2018).
Basler, M., Kirk, C. J. & Groettrup, M. The immunoproteasome in antigen processing and other immunological functions. Curr. Opin. Immunol. 25, 74–80 (2013).
Basler, M., Mundt, S., Bitzer, A., Schmidt, C. & Groettrup, M. The immunoproteasome: a novel drug target for autoimmune diseases. Clin. Exp. Rheumatol. 33, S74–S79 (2015).
Klein, M. et al. Structure-based optimization and discovery of M3258, a specific inhibitor of the immunoproteasome subunit LMP7 (β5i). J. Med. Chem. 64, 10230–10245 (2021).
Sanderson, M. P. et al. M3258 is a selective inhibitor of the immunoproteasome subunit LMP7 (β5i) delivering efficacy in multiple myeloma models. Mol. Cancer Ther. 20, 1378–1387 (2021).
Singh, A. V. et al. PR-924, a selective inhibitor of the immunoproteasome subunit LMP-7, blocks multiple myeloma cell growth both in vitro and in vivo. Brit. J. Haematol. 152, 155–163 (2011).
Spencer, A. et al. A phase 1 clinical trial evaluating marizomib, pomalidomide and low-dose dexamethasone in relapsed and refractory multiple myeloma (NPI-0052-107): final study results. Brit. J. Haematol. 180, 41–51 (2018).
Acknowledgements
The authors thank F. Ravaglia for her help with the histopathology images.
Author information
Authors and Affiliations
Contributions
All the authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
H.-J.A. has received consultancy or lecture fees from Boehringer, Bayer, GlaxoSmithKline, AstraZeneca, Novartis, Otsuka, Janssen, Kezar, Lilly and PreviPharma. A.R.K. has received lecture fees from Vifor Pharma and research funding via consultancy and grants from Vifor, Visterra, Toleranzia, Variant Bio and CSL Limited. N.L. has received research funding from Omeros. P.R. has nothing to disclose.
Peer review
Peer review information
Nature Reviews Immunology thanks B. Seitz-Polski and the other, anonymous, reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Apolipoprotein L1 (APOL1) risk alleles
-
Genetic risk factors prevalent in people of West African ancestry that, in homozygosity or compound heterozygosity, lower the threshold for podocyte loss and faster progression towards end-stage kidney disease, especially in diseases involving type I interferon signalling.
- Bowman capsule
-
The outer capsule of the glomerular part of the nephron, which continues from the urinary pole to the tubular basement membrane. Fluids filtered from the blood in the glomerulus are drained into tubules along the Bowman capsule.
- Cryoglobulins
-
Autoantibodies (directed against IgG) that form immune complexes and cause small vessel vasculitis. They precipitate at low temperatures, which can be used as a diagnostic test.
- Glomerular filtration barrier
-
A semipermeable membrane in nephrons that is localized in a vascular network inside the glomerular tuft. The filtration barrier encompasses, from the inside to the outside, the endothelial glycocalyx, glomerular endothelial cells, glomerular basement membrane and visceral epithelial cells (podocytes).
- Glomerulus
-
The corpuscular part of the nephron encompassing the glomerular tuft, which is a capillary network formed by arterioles. Each glomerulus has a single afferent arteriole and a single efferent arteriole entering and leaving the glomerulus at the vascular pole. To direct the filtrate towards the draining tubule, the Bowman capsule surrounds the glomerular tuft and represents the outer border of the glomerulus.
- Glomerulosclerosis
-
Irreversible glomerular scarring leading to waning of filtration. Usually, podocyte loss is the starting point for glomerulosclerosis.
- Haematuria
-
The presence of blood in the urine, a consequence of vascular injury, for example rupture of the glomerular basement membrane in glomerulonephritis, due to complement activation or local release of proteolytic enzymes. Nephritic syndrome is manifested as haematuria and proteinuria, often with hypertension and impaired kidney function, and is a common clinical presentation of glomerulonephritis.
- Mesangium
-
A component of the glomerulus formed of smooth muscle cell-like or pericyte-like mesenchymal cells in between the glomerular capillary network. Glomerular endothelial cell dysfunction during local or systemic inflammation promotes the passage and entrapment of plasma proteins in the mesangium, which activates or injures mesangial cells.
- Nephrons
-
Independent functional units of the kidney. The sum of all nephrons defines kidney function. Nephron number is set at birth and declines with ageing. Injury-related nephron loss induces compensatory hypertrophy of the remaining nephrons to meet the unchanged haemodynamic and metabolic demands. This adaptive capacity differs across species and is much lower in humans than in rodents, which represents a hurdle in translational research.
- Nephrotic syndrome
-
Massive loss of plasma proteins into the urine due to diffuse podocyte injury. The clinical definition includes a massive proteinuria, hypoalbuminaemia (urinary losses exceed plasma protein production by the liver), hyperlipidaemia (compensatory increase in lipoprotein production, lipase dysfunction) and oedema (retention of sodium by the diseased kidney). Urinary losses of IgG and coagulation inhibitors can cause life-threatening secondary immunodeficiency and thromboembolism.
- Parietal epithelial cells
-
Epithelial cells lining the inner aspect of the Bowman capsule. Parietal cells are usually a relatively quiescent lining, but severe glomerulonephritis-related leakage of plasma proteins can drive local hyperplasia (as glomerular crescent formation). The parietal epithelial cell monolayer hosts immature progenitors with a dedicated capacity to differentiate and replace lost podocytes on the glomerular tuft.
- Plasma exchange
-
A therapeutic modality used to eliminate circulating pathogenic agents via an extracorporal circulation, separation of plasma proteins (for example, by ultrafiltration of serum proteins or by columns that bind and retain specific proteins) and reinfusion of the remainder.
- Podocytes
-
Cells comprising the epithelial layer on the outside of the glomerular capillaries and gatekeepers of protein selectivity of the glomerular filter. Selective podocyte injury is the central pathological mechanism of ‘podocytopathies’ presenting as nephrotic syndrome. Podocytopathies can be genetic, immunological, metabolic, monoclonal gammopathy related or shear stress related, presenting as nephrotic syndrome.
- Proteinuria
-
Excessive protein in the urine, a urinary biomarker of glomerular dysfunction or podocyte dysfunction when albumin is the predominating urinary protein. Other plasma proteins prevail in tubular or overflow types of proteinuria. Albuminuria is a general biomarker of systemic endothelial dysfunction, for example, in sepsis.
- Thrombotic microangiopathy
-
A lesion pattern characterized by immunothrombosis in arterioles, capillaries and venules and frequently caused by humoral or toxic triggers of endothelial injury, including antiphospholipid antibodies or inherited and acquired dysregulation of the complement cascade.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Anders, HJ., Kitching, A.R., Leung, N. et al. Glomerulonephritis: immunopathogenesis and immunotherapy. Nat Rev Immunol 23, 453–471 (2023). https://doi.org/10.1038/s41577-022-00816-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-022-00816-y
This article is cited by
-
RNAi-based drug design: considerations and future directions
Nature Reviews Drug Discovery (2024)
-
The current use of proteomics and metabolomics in glomerulonephritis: a systematic literature review
Journal of Nephrology (2024)
-
A guide to complement biology, pathology and therapeutic opportunity
Nature Reviews Immunology (2023)
-
Lupus Nephritis: New and Emerging Biologic and Targeted Therapies
BioDrugs (2023)
-
Infektassoziierte Glomerulonephritis (IRGN)
Die Nephrologie (2023)
