
Abstract
Cardiovascular disease is the leading cause of death globally. An advanced understanding of cardiovascular disease mechanisms is required to improve therapeutic strategies and patient risk stratification. State-of-the-art, large-scale, single-cell and single-nucleus transcriptomics facilitate the exploration of the cardiac cellular landscape at an unprecedented level, beyond its descriptive features, and can further our understanding of the mechanisms of disease and guide functional studies. In this Review, we provide an overview of the technical challenges in the experimental design of single-cell and single-nucleus transcriptomics studies, as well as a discussion of the type of inferences that can be made from the data derived from these studies. Furthermore, we describe novel findings derived from transcriptomics studies for each major cardiac cell type in both health and disease, and from development to adulthood. This Review also provides a guide to interpreting the exhaustive list of newly identified cardiac cell types and states, and highlights the consensus and discordances in annotation, indicating an urgent need for standardization. We describe advanced applications such as integration of single-cell data with spatial transcriptomics to map genes and cells on tissue and define cellular microenvironments that regulate homeostasis and disease progression. Finally, we discuss current and future translational and clinical implications of novel transcriptomics approaches, and provide an outlook of how these technologies will change the way we diagnose and treat heart disease.
Key points
-
A good experimental design requires a matching of the protocol workflow to the cells of interest and scientific goals.
-
The generation of reference heart cell atlases and standardized annotations is necessary for cross-study comparisons and accurate data interpretation.
-
Emerging disease gene signatures reveal cell-state-specific changes, which will facilitate the generation of novel putative biomarkers and therapeutic targets.
-
The definition of cellular microenvironments requires deconvolution with spatial and multi-omics approaches.
-
The density of information from these novel omics approaches will contribute to the design of computational models to predict disease, stratify patients and facilitate drug discovery.
Similar content being viewed by others

Introduction
The human heart is a dynamic organ composed of four morphologically and functionally distinct chambers, as well as highly specialized subdomains including the conduction system and valvular apparatus, all working in synchrony due to the perfectly coordinated actions of billions of cells. In each chamber and anatomical subdomain, diverse stimuli converge on the local cells, priming gene expression patterns that drive phenotypic and functional adaptations, cumulatively determining organ function. Indeed, this fine cellular orchestration facilitates not only continuous contraction and relaxation of cardiomyocytes, but also the effective responses to haemodynamic changes during prenatal and postnatal development, and adulthood.
During embryonic development, the cellular progenies of first and second heart fields are primed in utero, and exposure to haemodynamic changes contributes to their gene expression programmes and maturation after birth1,2. In disease states, perturbations to the normal cellular repertoire and microenvironments caused by noxious stimuli, such as mechanical, electrical, chemical or ischaemic damage, can disrupt the transcriptional landscape3,4,5,6. Gene expression is not merely an epiphenomenon, but rather an essential step in the implementation and amplification of pathogenetic circuits3,7. Therefore, studying cellular transcriptional signatures is essential for gaining a robust understanding of organ and tissue function.
Cardiovascular disease remains the leading cause of death globally8. Ischaemic heart disease, more specifically, is the foremost cause of death in both men and women, despite robust efforts in the field of preventative cardiology. Furthermore, heart failure (HF), a cardiac functional impairment secondary to many aetiologies, is a rising global epidemic and, despite a growing number of treatments9,10,11, transplantation remains the only definitive cure. Therefore, an urgent need exists for novel effective and targeted therapies with more precise risk stratification, which necessitates a deeper understanding of the underlying molecular mechanisms driving the progression of cardiac disease.
Single-cell omics technologies, and especially transcriptomics, have revolutionized the way we investigate organs and organisms, allowing an unprecedented level of resolution in the assessment of cell demographics during both health and disease12. Single-cell transcriptomics provides information on gene expression prevalence and heterogeneity as well as co-expression of genes at the individual cell level to facilitate a cell-centric outlook. This approach involves the definition of novel cell markers and transcriptional signatures to delineate cell types (well-established cellular lineages) and cell states (encompassing subtypes or transient, functional cellular transcriptomics signatures) with high accuracy, either manually or using automated computational pipelines13,14,15. Analysis of gene sets enriched in cell types or states allows functional inferences on intracellular regulatory networks and on intercellular pathways across specific cells by focusing on genes encoding ligand–receptor pairs16,17. Such analyses highlight the processes underlying coordinated cellular communication that are otherwise masked in bulk analysis.
In this Review, we discuss the latest findings obtained using single-cell transcriptomics that advance our understanding of cardiac biology, development and disease. We explain how we can maximize the implementation of these technologies to study cardiac disease and provide an outlook on multimodal integration with spatial transcriptomics and epigenetics.
Successes and challenges in experimental design
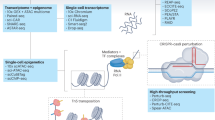
The implementation of single-cell transcriptomics consists of several steps, from tissue processing for cell isolation to single-cell capture, reverse transcription, complementary DNA amplification and library construction, which are followed by sequencing and computational analysis12,18,19. In this section, we describe the most successful experimental designs utilized to profile cardiac cells, discuss their advantages and disadvantages, and highlight the need for careful experimental planning and validation options.
Cell types of interest dictate protocol choice
Most single-cell capturing methods including flow cytometry, microfluidics and microdroplet-based systems have an upper size limit for cells of ~25–40 µm20,21. Therefore, if the study focuses on cardiac stromal, vascular and immune cell compartments with no major size limitations, these capturing techniques can be used to select single cells for RNA sequencing22,23,24,25,26 (Fig. 1). However, in the mammalian adult heart, cardiomyocytes pose a major challenge because they are large, rod-shaped cells of approximatively 20 µm in width and 100 µm in length27. Micromanipulation or laser capture of cardiomyocytes is possible, but these approaches are limited by low throughput and are operator-dependent7. Flow cytometry sorting of cardiomyocytes has been reported, although the length of sorted cardiomyocytes was ~50 µm, suggesting a bias for smaller cells or contracted cells during processing28,29. The sorting of cardiomyocytes with preserved RNA quality and function has been achieved using a specialized fluorescence-activated cell sorting (FACS) instrument with a large 500 µm nozzle. However, this type of instrument is not routinely available. Furthermore, the complexity and length of the cardiomyocyte isolation process can potentially affect the resulting transcriptional signature30. Single-cell RNA sequencing (scRNA-seq) technologies are limited not only by inefficiency in capturing adult cardiomyocytes, but also by the fact that adipocytes cannot be captured using single-cell isolation protocols. Moreover, an under-representation of cells such as pericytes and fibroblasts is also common in scRNA-seq27,31. Therefore, if the aim of the study is to assess and capture all cardiac cell types, profiling single nuclei by using single-nucleus RNA sequencing (snRNA-seq) is the most effective method27,32,33,34,35,36,37. Importantly, snRNA-seq generates largely overlapping molecular signatures with scRNA-seq in the adult human heart27 and during cardiomyocyte differentiation in vitro38. Furthermore, unlike snRNA-seq, single-cell isolation for scRNAseq requires fresh tissue, and enzymatic and mechanical methods used for single-cell dissociation can risk inducing stress-related transcriptional artefacts, especially when performed at 37 °C for >1 h39. The testing of RNA quality from a fresh or frozen tissue sample is recommended. Of note, there are also some caveats in the use of snRNA-seq. snRNA-seq is not ideal for cells such as neurons, in which crucial mRNA trafficking along the axon to the synapse would be missed40. Likewise, with snRNA-seq, data from binucleated or multinucleated cardiomyocyte nuclei and data from single-nucleated cardiomyocyte nuclei cannot be distinguished. Additionally, under-representation of endothelial cells in snRNA-seq studies has been reported, which varies according to the protocol used27,31. Taken together, snRNA-seq is an effective tool for obtaining comprehensive data from cardiac tissue, and its integration with scRNA-seq maximizes the coverage of cell types and states27.

The design and execution of single-cell experiments involves a multistep process that requires careful planning. The assessment of cardiomyocytes is challenging given that they are too large to be selected as single cells using fluorescence-activated cell sorting (FACS) or most microfluidics-based methods. If all cardiac cell types need to be analysed, the isolation of single nuclei from frozen samples is currently the most commonly used approach, given that this technique allows capture of all known cardiac cell types at the highest throughput. Nucleus isolation requires enzymatic and mechanical dissociation, often with the use of a Dounce homogenizer to help release the nuclei. Before proceeding to library preparation, nuclei can be purified by filtering and then by FACS, or less-stringent protocols include differential centrifugation before filtering and alternatively density gradient centrifugation. If only non-cardiomyocyte cells are needed, mechanical and enzymatic tissue dissociation of fresh samples allows the recovery of stromal and interstitial cells. For rare cell types of interest, the potential enrichment for specific populations can be achieved via antibody labelling of cell surface markers, followed by magnetic bead-based enrichment or flow cytometry sorting. These enriching methods can also be used to select cells expressing fluorescent proteins, such as in lineage-tracing experiments. After the isolation and purification steps, single cells and nuclei are captured, labelled (by barcoding) and incorporated into a library preparation using a variety of single-cell platforms: droplet-based approaches, such as the Chromium Controller from 10× Genomics, which have a high throughput; nanowell-based methods, such as: the ICELL8 instrument from Takara Bio, which allows the selection of a wide range of cell sizes with low-to-medium throughput; microfluidics approaches, such as the Fluidigm C1 platform; multiwell plate-based protocols, such as Smart-seq3xpress; and methods based on multiple rounds of splitting and pooling of cells, such as SPLiT-seq, which allow the barcoding of single cells without the need for physical separation. scRNA-seq, single-cell RNA sequencing; snRNA-seq, single-nucleus RNA sequencing.
Purification and enrichment
The assessment of large numbers of cells that are present at low frequency, such as immune cells, requires antibody-based approaches for enrichment by flow cytometry or immunomagnetic beads. In animal models, specific cell lineages or rare cells, including epicardial cells, can be enriched using methods involving cell tracers such as genetic fate mapping, which involves insertion of a heritable gene encoding a fluorescent protein41,42,43. After isolation, the nuclei are purified to remove the cytoplasmic debris resulting from deliberate disruption of the cytoplasm to release the nucleus. This filtering step can be performed by FACS27,37,44,45,46, differential centrifugation or straining33,47, or by density gradient separation48 (Fig. 1). The effectiveness of differential centrifugation depends on pipetting skills, and the use of filters with a pore size as small as 10 µm can introduce bias47. Indeed, the nuclei of mammalian cells have an estimated size of 8–12 µm37, or even larger under certain conditions such as in hypertrophic cardiomyocytes49. The use of a strainer alone can let through small tissue fragments and debris that can increase background noise33. Density gradient centrifugation is also highly operator-dependent and is unsuitable for small volumes with few nuclei50. Although FACS purification requires specialized instruments and expertise, this approach ensures good purification of nuclei given that the nozzles are typically ≥70 µm32,33,47. Visual inspection of nuclei to confirm nuclear membrane integrity and absence of blebbing and tissue debris is recommended after purification37. Of note, a FACS protocol based on immunolabelling of the cardiomyocyte-specific protein pericentriolar material 1 and Hoechst staining for DNA content allow the isolation and profiling of diploid versus tetraploid nuclei, a key step in defining the transcriptional changes occurring in neonatal proliferating cardiomyocytes48,51. However, certain dyes used for nuclei sorting can intercalate between DNA base pairs and disrupt chromatin structure52. Therefore, when downstream analysis includes single-nucleus assay for transposase-accessible chromatin sequencing (snATAC-seq), a recommended dye such as 7-aminoactinomycin D should be used. In addition, during FACS, nuclei shear stress, high hydrodynamic pressure and osmotic changes could induce chromatin rearrangement53. By contrast, these effects are unlikely to influence gene expression when the nuclei are isolated from frozen tissue and experiments are performed at 4 °C, which prevents the activation of transcription27,37,44,45,46.
Available single-cell or single-nucleus platforms
After choosing the best protocol for cell and nucleus isolation, the next crucial decision is the optimal method for single-cell and single-nucleus capture and library construction20,54,55 (Fig. 1). Microfluidic systems (such as the Fluidigm C1 platform) were among the first systems used to study heart cells; however, a low-to-medium throughput and high costs have limited their use20,23. In droplet-based platforms (such as 10× Genomics and Drop-seq technologies), thousands of isolated single cells or nuclei are moved through advanced microfluidic devices, where they are individually partitioned with uniquely barcoded beads into nanolitre-sized gel emulsions56,57. These platforms allow the capture of typically 5,000–10,000 cells or nuclei per sample with low costs, enabling a wide representation of cell populations, including rare cell types. Technologies based on full-length transcriptome sequencing, such as Smart-seq3, allow the characterization of transcript isoforms facilitating the detection of a larger number of transcripts per cell than droplet-based approaches, but at higher costs per cell and with lower throughput58. However, protocol updates in the past year (Smart-seq3xpress) have substantially increased the throughput while maintaining high sensitivity59. Nanowell-based technologies include ICELL8 (Takara), a mid-throughput platform that combines imaging and dispensing of single cells into nanowells to capture hundreds of cells with a wide range of sizes, including cardiomyocytes60,61,62. Finally, SPLiT-seq is an alternative affordable method based on combinatorial barcoding that does not require single-cell capture, is compatible with fixed cells and nuclei, and can be used for large cardiomyocytes63.
Capturing anatomical diversity
Designing cardiac single-cell or single-nucleus transcriptomics experiments requires careful consideration of the multiple (sub)anatomical regions of the heart, which consists of highly specialized structures with a diverse cellular composition. The most comprehensive human heart reference cell atlas involved the collection and analysis of six anatomical regions: the right ventricle (RV) and left ventricle (LV) free wall, apex, interventricular septum, and right and left atria27. According to snRNA-seq data, the atria and ventricles of donor hearts have different cellular compositions, with an inverse correlation between the proportion of fibroblasts and cardiomyocytes, in accordance with the role of the ventricles as the primary pumping chambers27. Analysis of the aortic valve and aorta was challenging, owing to the paucicellular nature of the tissue and its richness in extracellular matrix (ECM)64,65. Although some studies have explored anatomical differences during mouse development to define progenitor cells beyond the known first and second heart fields23,41,42,66, an unmet need exists for a systematic analysis of chamber differences in the adult mouse heart. Indeed, analyses with higher precision and broader spectrum of anatomical regions are needed for consistent comparisons across studies, species and disease phenotypes.
Power calculations
Two additional aspects are crucial in the design of scRNA-seq and snRNA-seq experiments: the number of single cells or nuclei to be analysed per sample and the depth of sequencing, which influences the number of genes obtained per cell. Most experiments are designed to obtain data from thousands of single cells with relatively shallow sequencing, or from hundreds of cells with deeper sequencing. The choice of approach can be qualitatively assessed in exploratory studies or on the basis of emerging tools for power calculations. The web-based single-cell one-sided probability interactive tool (SCOPIT) estimates the necessary number of cells to be sequenced to resolve cell types present at different frequencies67, whereas the statistical framework scPower models the relationship between the number of cells per individual, sequencing depth, sample size and power of differentially expressed genes within cell types to compare a multitude of experimental designs and to optimize the design within a limited budget68. In general, the sequencing of a large number of cells at a lower depth leads to higher power compared with sequencing fewer cells at a higher depth68. Nevertheless, deeper sequencing of hundreds of cells can be useful to characterize prospectively sorted rare populations. Regarding biological replicates, proof-of-feasibility experiments have largely been conducted with a low number of replicates22,26, but now the field has reached a phase in which variability can be predicted for both animal and human studies. In human cardiomyopathies involving pathogenic variants affecting the same gene, a cohort of five individuals has been shown to be sufficient to highlight statistically significant differences in gene expression and other related parameters, including cellular composition69.
The importance of metadata
For best practice, to reduce the risk of technical bias, studies should, if possible, incorporate cells or nuclei obtained with the same protocol, especially when performing comparisons across different treatment groups or diseases and controls. Indeed, the various protocols for the isolation and purification of cells and nuclei can have different effects on the proportions of the types and states of cells retrieved, as well as on their transcriptional and epigenetic signatures. Moreover, substantial structural changes in pathological tissue or analysis of different anatomical regions of the heart, such as highly fibrotic tissue, could affect the release of certain cell types, even when using the same protocol. Therefore, reporting technical metadata and histopathological evaluation of the tissue microstructure for each sample is important to facilitate accurate data interpretation and integration across studies. Consequently, cell annotation and compositional analysis need to be evaluated in the context of the experimental design, and require validation with multiple platforms, including high-resolution spatial transcriptomics.
Overview of the computational workflow
Computational analysis of scRNA-seq or snRNA-seq data is a complex multistep process that requires specialized expertise (Fig. 2). In this section, we provide an overview of the crucial steps70.

After the sequencing of libraries, the overall bioinformatics pipeline is largely the same, regardless of which sampling procedure was chosen. After aligning the reads to a reference genome, comprehensive single-cell packages (such as Scanpy and Seurat) are used for quality control, batch correction and data integration. A key step in this process is the clustering and annotation of cell types and states, which is performed on the basis of the expression of known marker genes and the interpretation of the transcriptional signatures of each cluster. Of note, deep learning-based approaches are emerging for automated annotation. Comprehensive annotation facilitates increased resolution in complex downstream analyses, such as: compositional analysis (the proportion of various cell types present in a tissue across various conditions); differential expression analysis between different diseases or treatments; and inferences on intercellular communication on the basis of the expression of genes encoding ligand receptors in different cell types and states. FC, fold change; SMCs, smooth muscle cells.
Quality control and data integration
Analysis pipelines for processing raw data, such as Cell Ranger57, SEQC71 and zUMIs72, perform initial quality checks73 on the sequencing reads, demultiplex data by assigning reads to their cellular barcodes and mRNA molecules of origin, and facilitate genome alignment and quantification. In droplet-based scRNA-seq and snRNA-seq analyses, the degree of contaminating ambient transcripts released into the cell or nucleus suspension during tissue dissociation can vary according to the dominant cell types and might lead to misinterpretation of the results. In the human heart, cardiomyocyte nuclei are the major contributors to ambient contamination. Software tools, such as CellBender, SoupX and DecontX, can minimize technical artefacts in data74,75,76. For example, CellBender can estimate ambient RNA from empty droplets and correct the expression metrics by removing counts related to ambient RNA molecules and even random barcode swapping74,75,76.
Standard quality control includes establishing minimum and maximum numbers of reads and genes per cell or nucleus, and determining a threshold for the highest percentage of genes encoding ribosomal proteins and mitochondria per cell or nucleus, above which a cell is defined as poor quality or unhealthy73. Single-cell analysis of cardiomyocytes needs to take into consideration the high proportion of mitochondria normally present in cardiomyocytes compared with other cells77. Two or more cells or nuclei that are attached, captured within the same droplet or microwell, and represented by the same barcode can generate a hybrid transcriptome, an artefact that violates the fundamental principle of single-cell technology and results in incorrect inferences. Doublet or multiplet detection is best performed using unbiased methods such as Scrublet and SOLO, which involve simulation of doublets or multiplets to create a training set for a machine learning classifier70,78,79. Correct and precise identification of doublets is necessary to avoid the risk of confusing artefactual chimeric hybrids derived from two (or more) cells adhered together with transitional cell states.
To overcome batch effects and unwanted technical variation while retaining biological differences, multiple integration methods can be applied80,81. The assembly of one of the largest human donor heart cell atlases involved the successful integration of data from single nuclei and cells from 14 donor hearts, in which differences between ventricles and atria were retained, as well as left and right specificities27. Successful data integration was also observed in two studies of human hearts from individuals with HF that included analyses of >800,000 nuclei47,69.
Defining cell types and states
The annotation of cell types and states is a complex task that is necessary for data interpretation. The major cardiovascular cell types include cardiomyocytes, fibroblasts, endothelial cells, smooth muscle cells (SMCs), pericytes, immune cells, neuronal and glial cells, and adipocytes. Most of these cell types have multiple identifiable cell states with anatomical specificities22,23,24,27,33,82. Cell annotation requires unbiased clustering and the analysis of the expression of known marker genes and novel gene signatures. Unbiased clustering is a key step, the results of which depend on algorithm resolution and the number of droplets analysed: a high resolution will determine a high number of cell states or types, whereas lower granularity will be obtained with lower resolution. Each study will apply a given resolution with a subjective final decision that influences the results, which together with the effectiveness of data integration, doublet exclusion and ambient contamination subtraction can affect the accuracy of comparisons across studies. Computational tools that can be used to estimate the best resolution include SCCAF83 and MultiK84. However, it is imperative to assess whether differentially expressed genes across the various clusters are clearly defined and are biologically meaningful. The annotation of the identified cell clusters (each of which represents a cell type or state) is mostly performed manually on the basis of transcriptional signatures. However, owing to the availability of open reference data, label transfer approaches are possible for some tissue systems such as immune cells. CellTypist and scNym can automatically annotate cell types or states in a query dataset by mapping them onto a reference13,85. The creation of databases and collaborative initiatives will be needed to form a consensus on the optimal approach to the annotation of cardiac cell types and states for accurate comparisons across studies. Furthermore, the use of novel deep learning strategies such as scArches, which is based on transfer learning, enables efficient building and sharing of reference atlasing data and to retain disease variation when mapping to a health reference, as shown for coronavirus disease 2019 (COVID-19) datasets86. Such approaches will be key to ensuring the efficient use of the human cardiac reference atlas to improve our understanding of disease and to create model organism atlases that will facilitate functional validations.
Integration of multimodal omics approaches
The integration of data across multiple modalities such as scRNA-seq and snRNA-seq, ATAC-seq or proteomics can generate standardized cell state labels87,88,89. Computational tools such as cell2location and Giotto allow mapping of newly defined cell types and states onto the physical 2D space by combined analysis of scRNA-seq or snRNA-seq data with spatial transcriptomics (analysis of gene expression on tissue sections)27,90,91,92. This integration is needed because of the intrinsic nature of current spatial transcriptomics technologies that are based on the analysis of microtiles of tissue typically encompassing 5–15 cells, although this number varies depending on the size of the tile and the size of the cells captured, resulting in microbulk gene expression data. Integration tools allow the deconvolution of this microbulk information and the mapping of specific cells in space, which guides the definition of cellular niches. One typical spatial transcriptomics method, such as the commercially available Visium by 10x Genomics, is based on positioning tissue samples on slides covered with unique barcoded mRNA-binding oligonucleotides, which facilitates the capture of RNA from the tissue with high spatial resolution (protocols are available for frozen sections and formalin-fixed paraffin-embedded (FFPE) sections)93,94. Alternatively, individual RNA molecules can be directly profiled using the Nanostring GeoMx Digital Spatial Profiler, which assigns fluorescently labelled barcoded probes to genes of interest that are hybridized on the tissue and subsequently counted by a computerized optical lens without the need for amplification (compatible with FFPE sections)95. Non-commercially available protocols such as Slide-seq have also been developed, which involve the use of barcodes to capture RNA with a resolution of 10 µm94.
Together, the assembly of large atlases from publicly available datasets representing tens to hundreds of individuals will contribute to the definition of consensus cardiac cellular maps by characterizing common signatures across multiple studies96. Larger multi-organ studies such as the Tabula Sapiens97 overcame difficulties of integrating data from diverse organs, including the heart, paving the way to a new series of studies focusing on the definition of shared and organ-specific molecular signatures of cells present across the whole body, such as fibroblasts, vascular cells and immune cells98. This step is crucial for the definition of putative organ-specific and tissue-specific cellular therapeutic targets. Likewise, the computational analysis of scRNA-seq and snRNA-seq data and its integration with other modalities is a fundamental and demanding step that requires interdisciplinary expertise for accurate hypothesis generation and functional inferences.
Cardiomyocyte profiling in health and disease
Isolating large numbers of rod-shaped cardiomyocytes from cardiac tissue is inherently difficult and requires manual micropipetting or dispenser approaches using a large nozzle size. Consequently, only a few studies have characterized gene expression by scRNA-seq in single, freshly isolated adult human cardiomyocytes7,61.
Cardiomyocytes in the healthy heart
snRNA-seq has emerged as a successful high-throughput transcriptomics approach for profiling adult human cardiomyocytes27,32,33 and has revealed previously unknown inter-compartmental and intra-compartmental cardiomyocyte heterogeneity between cardiac regions of donor hearts27,33 (Table 1). Although the transcriptional diversity between atrial and ventricular cardiomyocytes probably reflects different developmental origins and electromechanical stimulations, distinct genomic signatures of cardiomyocyte subpopulations within anatomical regions suggest additional functional diversity that might correspond to specific tissue microenvironments. Such subpopulations include cardiomyocytes enriched for retinoic acid-responsive genes and stress-response-related genes, as well as cardiomyocytes enriched for nuclear-encoded mitochondrial genes indicative of a high energetic state, which suggests that these cardiomyocytes are equipped for a higher workload27. Interestingly, these cardiomyocyte states have been found in both atrial and ventricular cardiomyocytes27. However, whether their localization is enriched in areas of increased wall stress within the ventricle or atrium and how they vary under different pathological conditions, such as mitral or tricuspid valve disease, remains unknown. Likewise, the differences between the transcriptional signatures specific to the cardiomyocytes of the spirally oriented ventricular muscle fibres that constrict the chamber and those of the trabecular cardiomyocytes remain unclear99. A novel cardiomyocyte state enriched for Myoz2, which encodes the calcineurin inhibitor myozenin 2, was localized just below the epicardial surface of the mouse heart28, supporting the hypothesis that specific cardiomyocyte states localize to defined microenvironments.
Prenatal cardiomyocyte development
Analysis of human fetal hearts at 5–25 weeks of gestation has led to the identification of cell clusters composed of trabecular, ventricular and atrial compact myocardium100. Key transcription factors, such as HAND2 and NR2F1 (encoding heart and neural crest derivative-expressed protein 2 and COUP transcription factor 1, respectively), were identified in atrial cardiomyocytes, whereas HAND1 and HEY2 (encoding hairy/enhancer-of-split related with YRPW motif protein 2) were found in ventricular muscle cells. At 5 weeks, proliferating cardiomyocytes were observed, as well as muscle cells with key left-side specification genes (IRX3, encoding iroquois-class homeodomain protein IRX 3 in ventricular compartments, and PITX2 encoding pituitary homeobox 2 in atrial compartments)100. At 19–22 weeks, a population of proliferating cardiomyocytes showed high expression of TOP2A (encoding DNA topoisomerase 2α) and MKI67 (encoding proliferation marker protein Ki-67), as well as a lack of expression of TCAP (encoding telethonin, a protein needed for the assembly of mature myofibrils)101. An understanding of the gene expression signatures present in proliferating cells is key to identifying cardiomyocytes with a predisposition to divide and to define potential therapeutic strategies for heart regeneration. In this regard, a comparison of gene expression signatures across different time points in prenatal and postnatal periods is needed.
Of note, a comparison of the cardiac cellular landscape in human versus that in mouse fetal hearts suggested differences in gene expression signatures between the two species23,100,102,103. Although cardiomyocytes were the most similar among all cell types, ventricular cardiomyocytes in humans expressed ECM-encoding genes earlier and more abundantly than those in mice. RELN (encoding reelin) expression was specific to human atrial trabecular cardiomyocytes and was absent in mice23,100,102,103. Furthermore, CITED2 (encoding Cbp/p300-interacting transactivator 2) and CITED4 were enriched in developing human cardiomyocytes, whereas Cited1 was enriched in the mouse counterpart23,100,102,103 (Fig. 3).

Fibroblasts are key in homeostasis as well as in disease progression, given that fibrosis is a common feature of the response to injury. Single-cell and single-nucleus RNA sequencing have provided insights into the role of fibroblasts in the ageing heart and in different disease settings, such as myocardial infarction (MI) and dilated cardiomyopathy (DCM). A list of fibroblast populations annotated using single-cell and single-nucleus studies is shown in Table 2. COVID-19, coronavirus disease 2019; TAC, transverse aortic constriction.
Cardiomyocytes in disease
Myocardial infarction
In a mouse model of myocardial infarction (MI), scRNA-seq identified a cardiomyocyte subset with upregulated expression of β2 microglobulin in response to ischaemic damage29. In vitro experiments suggested that this upregulation drives fibroblast activation. Furthermore, a multimodal approach combining snRNA-seq, snATAC-seq and spatial transcriptomics was used to study cardiac samples from hearts explanted 2–5 days after the onset of clinical symptoms of MI, before the patients received a total artificial heart104. This integrated method allowed the investigators to map putative enhancers controlling gene expression within distinct cardiomyocyte niches of injury, repair and remodelling, revealing a regional influx of immune cells in response to localized induction of cytokines, and defined RUNX1 (encoding runt-related transcription factor 1) as a potential driver of myofibroblast differentiation104. The inclusion of spatial transcriptomics helped to determine localized differences at the site of ischaemic injury. ANKRD1 (encoding the transcriptional repressor ankyrin repeat domain 1, the overexpression of which has been shown to impair cardiomyocyte function105) and NPPB (encoding natriuretic peptide B, which is widely used as a marker for cardiac disease and is upregulated in the border zone after MI in mice106,107) were defined as markers for two niches of stressed cardiomyocytes104. Importantly, integration with snATAC-seq facilitated the identification of T-box protein 3 (TBX3) and myocyte-specific enhancer factor 2D as regulators of an ANKRD1+NPPB− pre-stressed cell state, and cyclic AMP-dependent transcription factor ATF3 as a driver of the ANKRD1+NPPB+ state104.
Cardiac hypertrophy
A multimodal approach including epigenetic, morphological and functional assessment and co-expression network analysis of mouse and human single cardiomyocyte transcriptomes has facilitated the delineation of conserved mechanisms of pressure overload responses7. In early hypertrophy, cardiomyocytes were found to activate mitochondrial translation and oxidative phosphorylation, which correlated with morphological hypertrophy7. Subsequently, sustained overload induced p53 activation, leading to the disruption of adaptive hypertrophy transcriptional programmes and the promotion of HF7. This observation confirms that cardiomyocyte identity and morphological and functional phenotypes are encoded in transcriptional programmes.
A snRNA-seq study on pathological cardiac hypertrophy caused by aortic stenosis highlighted the downregulation of ephrins, the largest family of receptor tyrosine kinases, in cardiomyocyte hypertrophy46. In particular, a downregulation of EPHB1 (encoding ephrin type B receptor 1) observed in the human hypertrophic heart was confirmed in a transverse aortic constriction (TAC) mouse model of pressure overload46. In vitro treatment of cells with EFNB2, the ligand for EPHB1, rescued the hypertrophic phenotype46. A TAC mouse model was used to mimic the progression towards pathological hypertrophy and led to the identification of early metabolic cardiomyocyte adaptation, which was evidenced by an upregulation in the expression of glycolysis-related genes, a continuous increase in the expression of the hypertrophy-related genes Nppa and Nppb, and a decrease in calcium handling-related genes108. In addition, the number of cardiomyocytes enriched for ERBB4 (encoding receptor tyrosine protein kinase erbB4) and FGF12 (encoding fibroblast growth factor 12) was reduced in hypertrophied human hearts compared with healthy hearts46. To advance our understanding of pathological cardiac hypertrophy, the cardiomyocyte profile from patients with cardiac hypertrophy of different aetiologies, such as hypertension and hypertrophic cardiomyopathy, should be assessed. Similarly, the specific cardiomyocyte states present in these diseases and their distribution within the tissue remains largely unexplored.
Cardiomyopathy
A convergence of cardiomyocyte transcriptomic changes is thought to be present in the setting of HF associated with dilated cardiomyopathy (DCM)45. An analysis of 61 patients with DCM or arrhythmogenic cardiomyopathy (ACM) with defined genetic mutations showed a substantial proportion (20–40%) of differentially expressed genes, suggesting a degree of divergence even in late-stage failing hearts69. Specific expression profiles included a general downregulation of MYH6 (encoding myosin 6) and cell-state-specific upregulation of SH3RF2 (encoding the anti-apoptotic protein E3 ubiquitin protein ligase SH3RF2) in patients with no known pathological gene variant, and an upregulation of FNIP2 (encoding folliculin-interacting protein 2, which is involved in the inhibition of oxidative metabolism) in patients with variants in LMNA (encoding prelamin A/C) or PKP2 (encoding plakophilin 2), but not in patients with variants in TTN (encoding titin) or RBM20 (encoding RNA-binding protein 20)69. Different HF aetiologies also lead to different cardiomyocyte transcriptomic signatures, suggesting a divergence of pathogenic mechanisms. Cardiomyocytes from failing hearts due to ischaemic cardiomyopathy (ICM) show dysregulation of different gene ontologies from cardiomyocytes from hearts with DCM62. For example, ICM causes cardiomyocyte changes in energy metabolism and protein targeting to the endoplasmic reticulum, whereas DCM causes cardiomyocyte changes in muscle contraction62.
Congenital heart disease
Single-cell transcriptomics has also provided new insights into congenital heart disease. A study of hearts from nine paediatric patients with different aetiologies of congenital heart disease and four control individuals found that age had a minimal contribution to the cardiomyocyte transcriptome compared with disease status109. A disease-specific cell state was identified in patients with congenital heart disease, which was characterized by increased EGF receptor (EGFR) and forkhead box protein O signalling, as well as insulin resistance. These transcriptomic changes were validated by scATAC-seq data that showed an increase in chromatin accessibility in 84–90% of differentially expressed genes. The expression of CORIN (encoding atrial natriuretic peptide-converting enzyme) was also strongly associated with healthy cardiomyocytes, whereas CRIM1 (encoding cysteine-rich motor neuron 1 protein) was enriched in diseased cardiomyocytes. This observation was validated by RNA in situ hybridization, which showed that the CRIM1 to CORIN ratio was higher in cardiomyocytes from patients with congenital heart disease than in those from controls109.
Cardiac regeneration
The regeneration potential of adult mammalian hearts is very limited and the response after injury is inadequate to reconstitute ventricular cardiomyocyte numbers110. However, an analysis of mouse and human failing and non-failing adult hearts revealed subpopulations of cardiomyocytes with cardiac regeneration potential, underpinned by a capacity to dedifferentiate and upregulate cell cycle regulators after stress34. In a zebrafish model of heart regeneration after cryoinjury, scRNA-seq defined a subset of cardiomyocytes from the border zone that could partially dedifferentiate, shift their metabolism towards glycolysis and proliferate111. Neuregulin 1–ErbB2 signalling contributed to the switch to glycolysis and promotion of cell division in mice and zebrafish111. Subsequent snRNA-seq studies of mouse hearts during the early postnatal regenerative window identified immature cardiomyocytes that enter the cell cycle after injury but disappear as the heart loses its regenerative capacity48. According to findings from gain-of-function experiments, the unique transcriptional signature of these proliferative cardiomyocytes is related to the activity of nuclear transcription factor Y subunit α and serum response factor48. In addition, the endoplasmic reticulum membrane sensors NFE2L1 and NFEL2 drive cardiomyocyte protection48. The identification of proliferative cardiomyocytes provides novel inroads for future regenerative therapies.
Fibroblasts in health and disease
Cardiac fibroblasts are dynamic components of the heart’s cellular ecosystem that act as lineage progenitors, master conductors of ECM synthesis and remodelling, intercellular signalling hubs and electromechanical transducers112. Fibroblasts are also the central component in cardiac fibrosis, which is observed in most forms of cardiac pathology (Fig. 3a). Although initially protective, unresolved fibrosis leads to chamber stiffening and HF, as well as sudden death due to arrhythmias112. Many anti-fibrotic drugs have not been successful in improving end points in clinical trials, probably owing to factors such as biological differences between rodent and human hearts, and therapeutic target pleiotropy leading to adverse effects. Therefore, the identification of novel therapeutic targets in cardiac fibrosis is a priority113,114,115,116,117,118. Single-cell genomics have begun to unravel diverse quiescent and activated fibroblast populations in adult hearts22,24,27,28,119,120,121 (Table 2).
Human fibroblasts
In the human donor heart cell atlas, in a study with one of the highest number of cells and nuclei assembled to date, five fibroblast populations were identified, including chamber-specific ECM-producing fibroblasts and fibroblasts enriched for cytokine receptor genes such as OSMR (encoding oncostatin-M-specific receptor subunit β) and Il6ST (encoding IL-6 receptor subunit β), which might modulate immune responses27. This cell atlas identified activated fibroblasts that were shown to express pro-fibrotic genes27; however, other studies did not show activated fibroblasts in healthy hearts33,47. Shifts in the proportion of specific fibroblast populations and their gene signatures in disease states have been observed, including an increase in myofibroblasts enriched for ELN (encoding elastin) in patients with DCM45,122 or ICM122, as well as an increase in lipogenic fibroblasts enriched for DLK1 (encoding protein delta homologue 1) in the RV of patients with DCM and in non-ischaemic areas of the LV in patients with ICM122. In addition, a decrease in ‘resting’ fibroblasts (enriched for PLA2G2A, encoding membrane-associated phospholipase A2) was observed in patients with DCM45,122 or ICM122. Activated fibroblasts, which are characterized by the expression of POSTN (encoding periostin) and FAP (encoding fibroblast activation protein), are also consistently increased in HF45,47,69,122. However, the extent of the increase in activated fibroblasts might depend on patient genotype, given that the increase was less evident in patients with variants in LMNA and RBM20 compared with patients with TTN variants or no known pathogenic variants69. In a study of patients with DCM or HCM47, the best marker of activated fibroblasts was expression of COL22A1 (encoding collagen α1 (XXII) chain), which was variably detected in patients with DCM or HCM47. This finding should be interpreted cautiously, considering that a separate study of patients with DCM or ACM showed genotype-specific upregulation of different collagens, such as COL4A1 and COL4A2, in patients with pathogenic variants in LMNA, TTN or PKP2, and COL4A5 and COL24A1 in patients with no known pathogenic variants69. Overall, these findings highlight unexpected nuances in the gene signatures of activated fibroblasts, and a need to further understand the relationship between different fibroblast states and their distribution in cardiovascular diseases. A CRISPR-based knockout screen of several genes expressed in cardiomyopathy-associated fibroblast populations uncovered several new regulators of fibrosis, such as PRELP and JAZF1 (encoding prolargin and juxtaposed with another zinc finger protein 1, respectively)47.
Single-cell and single-nucleus transcriptomics studies of developing, healthy, diseased and partially recovered (unloaded) adult human hearts have confirmed the presence of fibroblast cell state heterogeneity and have highlighted additional regulators of fibroblast differentiation27,61,100,104,112,123. Co-localization of myofibroblasts with phagocytic macrophages after MI has also been demonstrated using spatial transcriptomics104. Finally, increased expression of ACE2 (encoding angiotensin-converting enzyme 2) was observed in the fibroblasts of hearts from patients with COVID-19 compared with healthy controls124, in addition to an enrichment in fibroblasts with pro-thrombotic, ECM-producing, ECM-organizing and immune cell-related signatures27,124,125,126,127,128,129.
Mouse fibroblasts
In mouse ventricles, a predominant quiescent fibroblast subtype (PDGFRA+SCA1high fibroblasts, termed F-SH24; also described as progenitor-like state fibroblasts25) exists across all organs130,131,132,133,134, and is enriched in cells with progenitor characteristics such as hypoxic niche and potential for multilineage differentiation, suggesting that these fibroblasts might be a lineage reserve for mobilization after injury131,132,133,135,136. F-Act fibroblasts24,132, an activated fibroblast population expressing Meox1 (encoding homeobox protein MOX1 (MEOX1), a transcriptional regulator involved in fibroblast activation during cardiac dysfunction137) and Cilp (encoding cartilage intermediate layer protein 1), are present at low levels in uninjured hearts, but expand to account for 20–50% of all cardiac fibroblasts by day 3 after MI24,132, seemingly deriving from more quiescent fibroblasts, or their pre-proliferative and proliferating counterparts24. F-Act fibroblasts lack expression of the α-smooth muscle actin gene Acta2, and thus might correspond more closely to previously characterized ‘proto-myofibroblasts’112. Interestingly, Cilp1-knockout mice have better cardiac function after MI and a reduced number of myofibroblasts than control mice138, supporting a key role of Cilp1 in fibroblast activation and differentiation. F-Act fibroblasts resemble the human POSTN+FAP+ fibroblasts mentioned above, but their functional similarity needs validation112. Another minor activated fibroblast population in mice, F-WntX24, expresses genes encoding secreted antagonists of WNT signalling and other pro-fibrotic regulators, one of which, WIF1, is essential for restricting the response of inflammatory monocytes after MI24,139.
scRNA-seq has enabled investigators to define fibroblast-dependent cytokine pathways that contribute to the inflammatory phase of MI repair140. The levels of a fibroblast subpopulation known as injury response cells (IR) peak on day 1 after MI, before decreasing rapidly, and this cell subtype is likely to be involved in the early inflammatory response25. IR cells might be similar to pro-inflammatory fibroblasts, previously described in a cardiac pressure overload model, which recruit Ly6Chigh monocytes via NF-κB signalling and expression of genes encoding monocyte chemoattractants such as CC-motif chemokine 2 (CCL2) and CCL5 (ref.141).
The number of myofibroblasts, the most distinctive injury-related fibroblast subtype, peaks at about day 7 after MI in mice25. Myofibroblasts can derive from the F-Act pool and/or other activated populations and as they differentiate show downregulation of stem cell markers and massive upregulation of ECM-related and contraction-related genes25,142. However, myofibroblast subgroups have different pro-fibrotic or anti-fibrotic regulatory gene signatures, suggesting that fibrosis is self-limiting24. Accordingly, myofibroblasts transition to a deactivated, post-proliferative cell type known as matrifibrocytes143, which are associated with osteogenic and chondrogenic ECM signatures and persist within the scar, probably to direct its maintenance and remodelling25,143. Activated fibroblasts that accumulate late in angiotensin II-induced cardiac hypertrophy also resemble matrifibrocytes144, suggesting a role for matrifibrocytes in non-ischaemic heart disease, in which perivascular and interstitial fibrosis predominates.
scRNA-seq analyses of other mouse models of heart disease have highlighted the presence of inflammatory, angiogenic and osteogenic fibroblast signatures82,121,134,137,145,146, which are more abundant with advanced age and might compromise fibroblast–endothelial intercellular signalling82. In mice, deletion of Hif1a (encoding hypoxia-inducible factor 1α) in fibroblasts leads to excessive fibrosis after MI132, whereas conditional deletion of Lats1 and Lats2 (encoding mechanosensitive Hippo pathway-related serine/threonine protein kinases) in fibroblasts leads to their spontaneous transition to myofibroblasts in uninjured hearts, and formation of a pervasive, non-compacted scar after MI via a mechanism involving the transcription factors YAP and TEAD121. Underlining the context-specific effect of the Hippo pathway, epicardial-specific deletion of Lats1 and Lats2 in mice was embryonically lethal, leading to defective coronary vasculature remodelling and an impairment in the differentiation of epicardial progenitors into cardiac fibroblasts121. scRNA-seq and scATAC-seq analyses in a mouse model of pressure overload, with and without pharmacological inhibition of epigenetic factors, have demonstrated that activated fibroblasts are capable of reverting to quiescent fibroblasts, and MEOX1 was revealed as a key regulator of fibroblast activation137. Taken together, these findings show that the annotation of cardiac fibroblast states and inferences of their function is at present more granular in mouse hearts than in human hearts, and that for several mouse cell states, a human equivalent has not yet been identified.
Vascular cell signatures in health and disease
Cardiac vascular cell compartments include those within coronary arteries, capillaries and the endocardium, and have different developmental origins, structures and functions. Importantly, the myocardial microvasculature is the most abundant and capillary endothelial cells are in close contact with cardiomyocytes147. Although this microarchitecture infers a close regulatory relationship between endothelial cells and cardiomyocytes, much remains to be understood about the drivers of vascular remodelling in cardiovascular disease.
Endothelial cells
In adult organ donor hearts, the transcriptional signatures of all major cardiac endothelial cell populations have been reported, including capillary, arterial, venous, endocardial and lymphatic endothelial cells27. The highest heterogeneity was observed in capillary endothelial cells, which express RGCC (encoding a regulator of the cell cycle) and AQP1 (encoding aquaporin 1) (Fig. 4a), with several new cell states identified. One of these cell states was characterized by high expression of genes encoding transcription factors (ATF3, FOS and JUN)148, which could be induced by endoplasmic reticulum stress149 (as seen in atherosclerosis150) and other stimuli such as DNA damage151 and oxidative stress152. A second cell state showed enrichment for cytokine-related genes, such as CX3CL1, CCL2 and Il16, and interferon-related genes, suggesting that these cells regulate the local immune response27. This analysis remains the most in-depth study of endothelial cell populations, particularly for capillary endothelial cells, with other studies being limited by the numbers of cells and nuclei available, and conservative choices of clustering resolution33,47. Indeed, although lymphatic and endocardial endothelial cells have been identified33,47, the whole range of vascular endothelial cells have not yet been recapitulated47. Some of the inconsistencies in endothelial cell states between studies are related to differences in annotations or markers used. For example, the annotation of a DKK2+ population47 probably corresponds to arterial endothelial cells45. This example again highlights the importance of standardizing annotations to facilitate more accurate comparison across studies.

a, Illustration of the major vascular structures in the human heart, including the endocardium, and summary of key endothelial cell subsets and their gene markers described in the healthy human heart. b, Genes with different expression patterns in endothelial cells of healthy human and mouse hearts. ‘No cell state specificity’ indicates that the gene is enriched in several cell states without any clear specificity. c, Genes with differential expression in endothelial cells in diseased and healthy hearts, including human hearts with dilated cardiomyopathy (DCM) and ischaemic cardiomyopathy (ICM), and mouse hearts after myocardial infarction (MI). TGFβ, transforming growth factor-β.
SnRNA-seq analysis of patients with DCM have highlighted differences in the transcriptional signatures of venous, capillary and especially endocardial endothelial cells compared with healthy controls45. The transforming growth factor-β (TGFβ) signalling pathway is enriched in both venous and capillary endothelial cells45. Differential gene expression analysis identified major changes in the endocardium with upregulation of BMP4 and BMP6 (encoding bone morphogenetic protein 4 (BMP4) and BMP6, respectively) between healthy hearts and DCM hearts and a shift in expression from NRG3 (encoding membrane-bound pro-neuregulin 3 (NRG3)) in healthy hearts to NRG1 in DCM hearts45,69, which mediates the compensatory response to stress153,154. In addition, a genotype stratification study of samples of hearts with DCM and ACM demonstrated that BMP and NRG signalling does not change uniformly in all cardiomyopathies, but that genotype-specific and chamber-specific disruptions of these intercellular signalling pathways are driven by the endocardium69. Indeed, in the LV, NRG1 and BMP6 are upregulated in the genotypes associated with DCM (variants in LMNA, TTN or RBM20, or no pathogenic variants), but not in ACM (variants in PKP2)69. However, patients with variants in PKP2 showed upregulation of NRG1 and BMP6 in the RV, whereas patients with no known pathogenic variants did not69. Of note, Nrg1 was described as an endocardial marker in healthy mouse hearts155, suggesting potential species-specific differences in the role and transcriptional profile of the endocardium (Fig. 4b). By contrast, NRP3 expression is conserved in both the human and mouse endocardium28,118,122. Finally, a potentially angiogenic capillary endothelial cell population that expressed TMEM163 (encoding transmembrane protein 163) and KIT (encoding mast/stem cell growth factor receptor KIT) was shown to increase in both DCM and HCM, suggesting an increase in the formation of small vessels to compensate for impaired cardiac function47.
In mice, endothelial cells showed increased expression of genes encoding cytokines such as CXCL2 and CCL9 on day 3 after MI, and a proliferative signature on day 7 after MI156 (Fig. 4c), which was accompanied by an increase in cells enriched for interferon signalling157. Upregulation of Plvap (encoding plasmalemma vesicle-associated protein (PLVAP), a membrane protein that contributes to permeability) and an increase in endothelial fenestration, stomata of caveolae and transendothelial channel formation was also detected157. In human hearts, increased PLVAP expression was found in venous endothelial cells and the endocardium, and widespread localization of PLVAP was observed in ischaemic hearts, especially in fibrotic areas27,157. Together, these data suggest a potential phenotypic shift in endothelial cells after injury away from the previously described continuous capillary cardiac endothelium147. In addition, in a TAC mouse model, scRNA-seq of cadherin 5 lineage-traced cells showed an increase in VEGF, WNT, EGFR and MAPK pathways in a cell state that was enriched for genes related to angiogenesis158. Of note, in the adult human heart, expression of VWF (encoding von Willebrand factor (vWF)) is detected in most endothelial cell states except lymphatic endothelial cells27. By contrast, in the mouse heart, Vwf is enriched only in endocardial and venous endothelial cells155,159 (Fig. 4b). Given the role of vWF in haemostasis, inflammation, vascular permeability and angiogenesis, this finding could have important implications in the interpretation of mouse models of disease for clinical translational purposes160,161. Likewise, single-cell and single-nucleus data identified ACKR1 (encoding a chemokine-scavenging receptor) as a venous endothelial cell marker in the human heart. However, this gene has not been reported in the mouse heart, suggesting a different modulation of chemokine bioavailability and, consequently, leukocyte recruitment across species.
Finally, a single-cell analysis of 4,000 cardiac cells from human fetuses (aged 5–25 weeks) identified four main states of endothelial cells: endocardial, valvular, coronary and vascular100. Of note, CDH11 (encoding cadherin 11) is broadly expressed in human endocardial cells, whereas in equivalent stages of mouse development, its expression is restricted to the endocardium of the valves64,100. Expression of RNASE1 (encoding ribonuclease pancreatic, shown to protect endothelial cells during inflammation162) was found in human cardiac fetal endothelial cells only, whereas expression of Icam2 (encoding intercellular adhesion molecule 2, which is involved in regulating vascular permeability163) was specific to the mouse embryo100 (Fig. 4b). A separate study that analysed 17,000 cardiac from cells human fetuses (aged 19–22 weeks) defined six different endothelial states, including two endocardial populations enriched in NPR3 (encoding natriuretic peptide receptor 3)101. One of these populations co-expressed INHBA (encoding inhibin-βa chain, which is selectively expressed in adult endocardium27,64,100) and MEIS2 (encoding meis homeobox 2, a transcriptional regulator essential for cardiac neural crest development164). The expression of MEIS2 might reflect a more immature subset of endocardial cells or a different subset of cells that disappears in adulthood.
Smooth muscle cells and pericytes
SMCs from human adult donor hearts include cells expressing classic gene markers such as MYH11 and ACTA2 and a second population with high expression of CNN1 (encoding calponin 1), which probably represents arterial SMCs27,165. Pericyte subtypes include typical ABCC9+ and KCNJ8+ cells, and a population enriched for AGT (encoding angiotensinogen), the expression of which is downregulated in DCM and indicative of dysregulated vasoconstriction27,45. scRNA-seq analysis of vascular SMCs (VSMCs) defined a synthetic state enriched in coronary artery SMCs compared with aorta and pulmonary artery SMCs166, which might indicate a specific adaptation of coronary arteries that are constantly exposed to local pressure changes due to cardiac contraction and relaxation. A proliferative VSMC state expressing FABP4 (encoding a fatty acid-binding protein) was identified and the numbers were found to be expanded in atherosclerosis166. scRNA-seq of atherosclerotic lesions showed that VSMCs undergo phenotypic modulation to transform into unique fibroblast-like cells (fibromyocytes) that contribute to the lesion and fibrous cap in both humans and mice167. Loss-of-function and gain-of-function validation studies in mouse models showed that TCF21 (encoding transcription factor 21) is essential for this phenotypic modulation and that higher levels of TCF21 are associated with decreased risk of coronary artery disease in humans167. Interestingly, in a study of human hearts with DCM or ACM, SMCs and pericytes showed an upregulation in the long non-coding RNA ADAMTS9-AS2 and simultaneous downregulation in ADAMTS9 (encoding disintegrin and metalloproteinase with thrombospondin motifs 9, which is involved in ECM remodelling)69. In addition, genotype-specific changes were also observed. For example, NOTCH3 (encoding neurogenic locus notch homologue protein 3) was downregulated in the pericytes of patients with pathogenic variants in TTN or PKP2, whereas SLIT3 (encoding slit homologue 3 protein, a ligand that regulates fibroblast activity) was upregulated in ELN+ SMCs from patients with variants in PKP2 or LMNA69.
The integration of fate mapping with Wnt1–Cre and scRNA-seq helped establish the heterogeneity of cardiac neural crest cell (CNCC)-derived cardiac cell populations from embryonic day (E) 10.5 to postnatal day 7 in mice168. As expected, on postnatal day 7, most of the CNCCs localized to the aorta, pulmonary arteries and the coronary vasculature. Nine VSMC populations were identified, in addition to microvascular SMC (mVSMC) and pericyte states. An analysis of lineage trajectories (computational inference of differentiation paths) using RNA velocity revealed a transition from pericytes to mVSMCs with Notch3, Tbx2, Fosb (encoding protein FoxB) and Klf2 (encoding Krueppel-like factor 2 (KLF2)) as potential key regulators168,169,170. Importantly, when analysing potential differentiation paths, the inferred latent time based on RNA velocity was similar to the developmental time. The identification of several differentiation paths included one rooted on a Crabp1+ (encoding cellular retinoic acid-binding protein 1) cell population, branching into both mesenchymal cells and VSMCs168, and highlighting previously unknown lineage relationships.
Dynamic adaptations of immune cells
Previous studies have shown that healthy adult human and mouse hearts contain major immune cell populations of both lymphocytes and myeloid cells27,171. Resident immune cells, such as cardiac macrophages, are interspersed across the heart between cardiomyocytes, the epicardium, the endocardium, valves and the nodes, where they contribute to organ homeostasis.
Myeloid cells
Myeloid cells in the healthy adult human heart comprise monocytes, dendritic cells and several subtypes of macrophages, such as LYVE1+ macrophages (which include MHC-IIhigh and MHC-IIlow populations), MHC-II+TREM2+ macrophages, fibroblast-interacting macrophages and monocyte-derived macrophages27. In addition, comparisons with skeletal muscle and kidneys revealed that cardiac myeloid cells, including LYVE1+ macrophages, have cardiac-specific features, which might be attributable to the tissue-specific adaptability of myeloid cells27,31,172. scRNA-seq findings in mice showed that cardiac macrophages are enriched for ion channels and facilitate electrical conduction through the distal atrioventricular node173.
Several cardiac monocyte or macrophage populations have been described, with different origins, mechanisms of self-replenishment and distinct functions in the context of inflammation, fibrosis and tissue repair174,175,176. scRNA-seq studies based on mouse models of MI defined a gene expression signature to discriminate a self-renewing macrophage population that originated partly from monocytes (Ccr2−MHCIIhigh), in addition to the previously described monocyte-derived macrophages (Ccr2+MHC-IIhigh) and self-renewing tissue-resident macrophages (Ccr2−MHC-IIlowLyve1+Timd4+)177,178. Further analysis revealed a homeostatic and reparative function of tissue-resident macrophages, whereas Ccr2+ macrophages are enriched for classic inflammatory pathways178. In mouse models, monocytes and monocyte-derived macrophages present at the early stages after MI were found to be recruited via pro-inflammatory circuits24,177. Loss-of-function and scRNA-seq studies demonstrated a crucial homeostatic and anti-inflammatory role of tissue-resident macrophages, whereby depletion of these macrophages increased monocyte recruitment and worsened ventricular dysfunction after cardiac injury and hypertension177,178,179. In addition, a putative self-renewing population of tissue-resident macrophages (MHC-IIlowLyve1+) was shown to contribute to the formation of the lymphatic network in a pressure-overload HF model and during heart development in mice180,181.
scRNA-seq also revealed immune biphasic cell recruitment in mice after MI24. Diffusion map analysis showed a trajectory from early infiltrating monocyte-derived macrophages to inflammatory macrophages, which led to a later peak of tissue-resident reparative macrophages with a pro-regenerative upregulation of Igf1 (encoding insulin-like growth factor 1)24. Combining snRNA-seq and spatial transcriptomics to study samples from patients with MI-induced cardiogenic shock helped to uncover an accumulation of CCR2+ macrophages in the injury zone, which predicted the localization of specific fibroblasts and TGFβ2 pathway activity104. Specifically, SPP1+ (encoding osteopontin) macrophages were enriched in ischaemic tissue samples and colocalized with myofibroblasts, whereas CCL18+ macrophages were enriched in fibrotic tissue samples. Finally, in a TAC mouse model, activation of pro-inflammatory macrophages was identified as a key event during the transition from normal to reduced cardiac function108. At 5 weeks, macrophages showed upregulation of several chemokines of the CCL subclass, and treating cardiomyocytes in vitro with the conditioned medium of macrophages from the 5-week post-MI samples led to an increase in Nppa and Nppb expression, typical stress-related genes that are upregulated in HF.
Lymphoid cells
Lymphocytes comprise various subtypes in the healthy adult human heart, including T cell subsets, natural killer (NK) cells and B cells27. CD4+ T cells can be classified into naive, effector and regulatory CD4+ T cells, whereas CD8+ T cells include a population of cytotoxic cells characterized by high expression of granzymes and perforin. Cell-mediated cytotoxicity and CD4+ effector T cells have been implicated in the pathogenesis of cardiomyopathy and myocarditis182,183,184. In a snRNA-seq study of HCM and DCM hearts, an increase in lymphocytes expressing LINGO2 (encoding leucine-rich repeat and immunoglobulin-like domain-containing nogo receptor-interacting protein 2) and several known NK cell markers was identified, but their function remains unclear47. NK cells have been shown to have a protective effect against cardiac fibrosis185, suggesting a potential immune-mediated protective mechanisms during HF. To summarize, enrichment of immune cells before scRNA-seq analysis will help to capture these highly diverse and dynamic cells and improve our understanding of their role in homeostasis and disease.
Functionally important rare cell types
Conduction system and neuronal cells
Neuronal cells represent approximately 1% of human cardiac nuclei and are defined by expression of NRXN1 (encoding neurexin 1) which is involved in the formation of synaptic contacts27. Given that these cells also express PLP1 (encoding myelin proteolipid protein, a well-known Schwann cell marker), they might plausibly include glial cells or represent doublets of neurons and glial cells27,186. In mice, scRNA-seq identified neuronal-like cells based on the expression of Kcna1 (encoding potassium voltage-gated channel subfamily A member 1) which is involved in regulating nerve signalling25. An analysis of 3,961 human cardiac neuronal cells revealed that 80% of these cells are prototypic neurons27. Furthermore, a cell state enriched for the WNT signalling receptor gene LGR5 (encoding leucine-rich repeat-containing G protein coupled receptor 5) and genes encoding myelin constituents was identified as potential Schwann cells27.
To define the transcriptional signature of the conduction system, specific anatomical regions should be profiled. A multi-species study involving mice, rabbits and cynomolgus monkeys characterized the cells of the mammalian sinoatrial node187. Vsnl1 (encoding the calcium-sensing protein visinin-like protein 1) was defined as a core marker of pacemaker cells in the three species studied. Indeed, disruption of this gene led to reduced beating rates in human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) and decreased heart rate in mice187. Of note, Kcnj8 was highly expressed in pacemaker cells of cynomolgus monkeys, but not in mice or rabbits. Moreover, analysis of the sinoatrial node from mouse hearts at E16.5 showed that 25% of genes previously reported to be sinoatrial node-specific by bulk RNA sequencing were in fact expressed in cells other than pacemaker cells188, confirming the value of single-cell approaches188. Studying the human conduction system at the single-cell level is a crucial next step that is currently hampered by limited availability of tissue.
Adipose cells
Adipocytes identified by snRNA-seq analysis using classic markers such as ADIPOQ (encoding an adipokine involved in the control of fat metabolism and insulin sensitivity) and GPAM (encoding mitochondrial glycerol-3-phosphate acyltransferase 1) represent 0.2–0.5% of the droplets obtained from human hearts27. A potential fibrogenic adipocyte precursor expressing ECM-related genes, as well as a cytokine-enriched population were reported; however, the distinction between white and brown cardiac adipocytes cannot yet be made27. A disease-specific population of adipocytes identified in DCM and ACM samples is characterized by changes in the expression of genes related to fatty acid metabolism and is enriched in patients with pathogenic variants in PKP2, LMNA or RBM20 (ref.69). Given the potential role of adipocytes in diseases such as metabolic syndrome and arrhythmia, efforts should be intensified to study this cell population.
Epicardial cells
Epicardial cells are identifiable by their expression of known markers such as MSLN and WT1 (encoding mesothelin and Wilms tumour protein, respectively27), and are one of the rarest cell populations in the human heart, thus necessitating enrichment for a full exploration of its complexity. Using a Wt1 lineage tracing system in a mouse model of MI, two Wt1+ epicardial cell populations were identified, one of which was enriched for Msln and keratin-encoding genes, whereas the other was characterized by a proliferation-associated gene signature43. Interestingly a Cd44+Wt1− epicardial population was also described, suggesting a heterogeneity that goes beyond Wt1+ cells43. Furthermore, in a mouse model of neonatal heart regeneration, a combination of scRNA-seq and scATAC-seq analyses revealed an increase in epicardial cells after MI that was accompanied by increased transcriptional activation155. Transcription factor motifs were preferentially enriched in cis-regulatory elements of epicardial MI-induced genes, including the KLF14 motif specific to the regenerative phase and FOS-related or JUN-related motifs enriched in the non-regenerative phase155.
Using scRNA-seq, it was possible to assess the cellular heterogeneity of the cardiac crescent in E7.5–E8.0 mouse embryos66. Findings from this study led to the identification of a new cardiac field, denoted as the juxtacardiac field, which is characterized by the expression of Mab21l2. Mab21l2 fate mapping showed that the juxtacardiac field contains a pool of progenitor cells for cardiomyocytes and epicardial cells66. Although enrichment of rare cell types can be easily achieved in mouse models using lineage tracing tools, in human hearts, it is necessary to select specific anatomical microdomains or identify novel surface markers for prospective sorting.
Cellular networks
Intercellular communication in the heart contributes to development, maintenance of homeostasis and disease progression. Novel computational tools facilitate the mapping of cell states identified using single-cell and single-nucleus transcriptomics integrated with spatial transcriptomics, allowing the unbiased discovery of cellular microenvironments and the prioritization of cell–cell interactions based on niches containing specific cell types or states90,189 (Fig. 5).

Single-cell RNA sequencing (scRNA-seq) and single-nucleus RNA sequencing (snRNA-seq) allow the definition of cell states and the prediction of cellular interactions based on the expression of genes encoding ligands and receptors. However, these data do not provide information about cellular proximity. Spatial transcriptomics allows the mapping of gene expression within tissue sections across microtiles that represent microbulk transcriptomics data. By mapping scRNA-seq and snRNA-seq data onto spatial data using computational tools such as cell2location and Giotto, it is possible to predict the localization of cell types and states within microtiles and microanatomical niches. The cellular proximity within the defined cellular niches facilitates inferences on the most probable cellular interactions and the identification of putative functional microenvironments.
In the adult human heart, CellPhoneDB, a public repository of ligand receptors and their interactions, was used to predict intercellular communications within NOTCH pathways between vascular endothelial cells and VSMCs, uncovering venous-specific and arterial-specific interactions27. Furthermore, interactions between fibroblasts and macrophages were predicted, including one that depended on the macrophage migration inhibitory factor (MIF)–CD74 receptor–ligand interaction, which was confirmed by co-expression in spatial transcriptomics27. A 2022 study of pressure overload-induced hypertrophy used CellPhoneDB and CellChat to investigate changes in signalling between cardiomyocytes and other cell types46. A downregulation in the expression of EPHB1 and EFNB2 was observed in cardiomyocytes and endothelial cells, respectively. A validation study using rat cardiomyocytes in 2D cultures and hiPSC-CM organoids with gain-of-function or loss-of-function approaches confirmed that EFNB2–EPHB1 signalling in cardiomyocytes regulates hypertrophy, contraction and stress responses, implicating EPHB1 as a putative therapeutic target in HCM46. In a large study of patients with DCM and ACM with different pathogenic variants, an analysis of cell–cell interactions was crucial in identifying genotype and chamber-dependent signalling pathways involved in disease pathogenesis69. For example, endothelin signalling originating from the endocardium was increased in the LV of patients with pathogenic variants in LMNA and in the RV of patients with pathogenic variants in PKP2, but not in patients with other genotypes. Of note, the dynamics of some pathways are cell type-dependent. For example, in the LV of patients with pathogenic variants in LMNA, the predicted BMP signalling from SMCs and pericytes to cardiomyocytes is decreased, whereas signalling from endothelial cells to cardiomyocytes is increased69.
In the adult mouse heart, analysis of non-cardiomyocyte cell populations identified fibroblasts as a crucial intercellular communication hub22. After MI, among the non-cardiomyocyte populations, myofibroblasts have the highest number of differentially expressed ligands (consisting mostly of ECM-related genes24,156), followed by macrophages24. Unexpectedly, endothelial cells express the highest number of differentially expressed receptors, suggesting a role as downstream effectors of paracrine and juxtacrine mechanisms after injury24,156. In neonatal mouse hearts, the epicardium was defined as a source of paracrine signalling during both regenerating and non-regenerating phases155. As confirmed by in vitro experiments, epicardial induction of Rspo1 (encoding R-spondin 1, a potent activator of the WNT–β-catenin signalling pathway) is a likely driver of pro-angiogenic signalling. Moreover, given the epicardial upregulation of Ltbp3 (encoding latent-transforming growth factor-β-binding protein 3 (LTBP3)) and the increased proportion of fibroblasts in the S-phase after treatment with recombinant LTBP3 in vitro, epicardium-derived signalling was suggested to contribute to fibroblast proliferation155. Finally, in a TAC mouse model, loss of protective signalling from fibroblasts to cardiomyocytes and increased macrophage–cardiomyocyte interactions were suggested to contribute to the initiation and progression of hypertrophy24,108,156.
During cardiac development in the human fetus (age 5–25 weeks), a cardiomyocyte-to-endothelial cell BMP paracrine signalling pattern was predicted on the basis of increased cardiomyocyte expression of BMP5 and BMP7 and enrichment for BMP receptor genes in endothelial cells, suggesting a potential role for BMP in driving endothelial-to-mesenchymal transition during endocardial cushion formation100. In mouse embryonic development, a MIF–CXCR2 interaction was predicted in the second heart field of embryos at E8.5 (ref.190). Inhibition of MIF and CXCR2 using small-molecule compounds impaired the elongation of the outflow tract and RV, probably due to defective cell migration. Taken together, to prioritize validation among hundreds of predicted receptor–ligand pairs, filtering criteria require computational tools, which take into consideration the activation of downstream cascades in the cells receiving the signalling cues90,189.
Future directions for omics approaches
Transforming diagnosis and therapy
Single-cell, single-nucleus and spatial transcriptomics accompanied by machine learning-based analysis is likely to contribute to the identification of markers for diagnostic and prognostic evaluation of cardiovascular diseases. In particular, these technologies can provide new insights into cell states (including those of rare cells) and cell-type composition of diseased hearts, and define cell-specific theraputic targets45,46,191. An early scRNA-seq study identified the source of IL-11, a novel anti-fibrotic target for the cardiovascular system145, demonstrating the potential of single-cell omics approaches in developing innovative therapies.
Capturing the earliest and most specific therapeutic cellular targets among the numerous maladaptive cardiomyocyte responses driving HF is crucial in hereditary DCM compared with HCM, in which variants in the same culprit gene can lead to opposite phenotypes192. Although a study of patients with end-stage HCM or DCM showed changes in cellular composition across the two groups, only minimal differences were detected at the transcriptional level47, highlighting a need to stratify patients on the basis of aetiology and to study earlier stages of disease progression. Indeed, analysis of genotype-stratified DCM samples identified specific transcriptional changes in different pathogenic variants groups, in addition to shared gene signatures. The number of differentially expressed genes was sufficient to develop a deep learning method that could predict the genotype on the basis of gene expression signatures of cardiomyocytes, endothelial cells, fibroblasts and myeloid cells69. This finding indicates that even at later stages of HF, the underlying cellular landscape and implicitly maladaptive mechanisms differ depending on the underlying cause. To understand the mechanisms involved in progression of DCM to HF, multimodal omics analysis should be performed in both early and late phases of disease, and patients should be stratified according to disease aetiology. Likewise, in ICM, understanding the cellular changes occurring before the onset of HF will be crucial to the stratification of patients in minimizing the current heterogeneity in response to therapy and towards personalized medicine approaches. Taken together, such information can improve our understanding of the pathobiological mechanisms underlying HF and guide the identification of prognostic biomarkers and novel druggable targets for cell-targeted interceptive medicine193.
hiPSC-derived cardiovascular cells
HiPSC-derived cardiovascular cells and cardiovascular cells derived from direct reprogramming hold great promise, not only for cardiovascular regeneration but also for drug screening and disease modelling, which is especially relevant for cardiomyocytes, given the lack of reliable cell lines and the challenges in working with large numbers of primary cells for an extended amount of time. To ensure the replicability of hiPSC-CMs as in vitro models, the maturation process needs to be improved and the phenotypic heterogeneity of the cells needs to be functionally evaluated. This latter step is necessary to establish precise models of the cellular subtypes identified in vivo by single-cell and single-nucleus approches. Indeed some of these cell states might be better therapeutic targets than others or even therapeutic products for cell therapy. The integration of single-cell and single-nucleus transcriptomics data from human hearts from all stages of development with those from hiPSC-derived cardiovascular cells contributes to our understanding of the variability in differentiation and maturity occurring in vitro compared with the processes occurring within the cardiac cellular landscape in vivo124,194,195,196,197,198. One such study described the role of the progesterone receptor in driving sex-specific metabolic programmes and maturation of cardiomyocytes199. Importantly, transcriptomics and epigenomic single-cell analyses have guided improvements in protocols for differentiation and maturation of cardiovascular lineages such as cardiomyocytes and endothelial cells in hiPSC-based 2D and 3D platforms, as well as in direct reprogramming approaches38,200,201,202,203. Furthermore, single-cell studies of hiPSC-derived cardiovascular cells were not only highly informative in reproducing the differentiation steps occurring in cardiac development, but also contributed to the modelling of congenital and other genetic disorders194,204,205,206. Such studies provide a comprehensive overview of the transcriptional changes that occur in specific cells at various stages of differentiation to facilitate precise mechanistic insights, as seen in studies showing a dosage-dependent effect of TBX5 in different cell states in the setting of congenital heart disease, as well as the convergence of cell cycle dysregualtion and autophagy as pathogenic mechanisms of hypoplastic heart disease204,207.
Age, sex and comorbidities
As presented throughout this Review, great effort has already been made to understand the complexity of the cardiac cellular landscape. However, some knowledge gaps remain. An urgent need exists for the development of a longitudinal heart atlas from the tissues of infants, children, adolescents and young adults (aged 20–40 years) that recapitulates the substantial morphological and haemodynamic changes occurring from birth until adulthood. Understanding sex-related differences in health and disease is a developing area in cardiology. A single-cell study showed differences in the prevalence of fibroblast subtypes and gene expression patterns between male and female mice22,144 and between inbred strains25. Furthermore, a study based on limited numbers of donor hearts identified the progesterone receptor as a key mediator of sex-dependent transcriptional programmes during cardiomyocyte maturation199. Therefore, given the effect of biological variables such as sex and ancestry on cardiovascular risk, it is imperative to integrate datasets from increasing numbers of donors to evaluate the influence of these variables on the human cardiac cellular landscape. Moreover, patients with cardiovascular disease, especially those of advanced age, often have comorbidities such diabetes mellitus, obesity and high blood pressure, which will need to be considered in the analysis and interpretation. The stratification of patients and organ donors will be possible through the integration of data from hundreds of individuals and the collection of appropriate clinical metadata.
Conclusions
Single-cell and single-nucleus omics technologies have provided invaluable novel insights into the cardiac cellular landscape to facilitate a better understanding of disease mechanisms and more tools for accurate risk stratification and precision medicine. However, newly defined cell states not only require thorough validation, but also cooperative and interdisciplinary efforts to standardize definitions and nomenclature within and across species, which will facilitate more accurate comparison between studies and the optimization of experimental designs for disease models. Finally, an urgent need exists to ensure the availability of computational resources and training for the new generation of scientists and clinicians, to allow improved interrogation of the biology and to accelerate the application of these transformative technologies to the study and treatment of cardiac disease.
References
Morton, S. U., Quiat, D., Seidman, J. G. & Seidman, C. E. Genomic frontiers in congenital heart disease. Nat. Rev. Cardiol. 19, 26–42 (2022).
Banjo, T. et al. Haemodynamically dependent valvulogenesis of zebrafish heart is mediated by flow-dependent expression of miR-21. Nat. Commun. 4, 1978 (2013).
van Heesch, S. et al. The translational landscape of the human heart. Cell https://doi.org/10.1016/j.cell.2019.05.010 (2019).
Saucerman, J. J., Tan, P. M., Buchholz, K. S., McCulloch, A. D. & Omens, J. H. Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat. Rev. Cardiol. 16, 361–378 (2019).
Liu, Y. et al. RNA-Seq identifies novel myocardial gene expression signatures of heart failure. Genomics 105, 83–89 (2015).
Burke, M. A. et al. Molecular profiling of dilated cardiomyopathy that progresses to heart failure. JCI Insight https://doi.org/10.1172/jci.insight.86898 (2016).
Nomura, S. et al. Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat. Commun. 9, 4435 (2018).
Roth, G. A. et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the GBD 2019 study. J. Am. Coll. Cardiol. 76, 2982–3021 (2020).
Groenewegen, A., Rutten, F. H., Mosterd, A. & Hoes, A. W. Epidemiology of heart failure. Eur. J. Heart Fail. 22, 1342–1356 (2020).
Murphy, S. P., Ibrahim, N. E. & Januzzi, J. L. Jr. Heart failure with reduced ejection fraction: a review. JAMA 324, 488–504 (2020).
Maddox, T. M. et al. 2021 Update to the 2017 ACC expert consensus decision pathway for optimization of heart failure treatment: answers to 10 pivotal issues about heart failure with reduced ejection fraction a report of the American College of Cardiology Solution Set Oversight Committee. J. Am. Coll. Cardiol. 77, 772–810 (2021).
Aldridge, S. & Teichmann, S. A. Single cell transcriptomics comes of age. Nat. Commun. 11, 4307 (2020).
Domínguez Conde, C. et al. Cross-tissue immune cell analysis reveals tissue-specific adaptations and clonal architecture in humans. Science 376, eabl5197 (2022).
Pasquini, G., Rojo Arias, J. E., Schafer, P. & Busskamp, V. Automated methods for cell type annotation on scRNA-seq data. Comput. Struct. Biotechnol. J. 19, 961–969 (2021).
Clarke, Z. A. et al. Tutorial: guidelines for annotating single-cell transcriptomic maps using automated and manual methods. Nat. Protoc. 16, 2749 (2021).
Efremova, M., Vento-Tormo, M., Teichmann, S. A. & Vento-Tormo, R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 15, 1484–1506 (2020).
Jin, S. Q. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. https://doi.org/10.1038/s41467-021-21246-9 (2021).
Svensson, V., Vento-Tormo, R. & Teichmann, S. A. Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protoc. 13, 599–604 (2018).
Efremova, M. & Teichmann, S. A. Computational methods for single-cell omics across modalities. Nat. Methods 17, 14–17 (2020).
Paik, D. T., Cho, S., Tian, L., Chang, H. Y. & Wu, J. C. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat. Rev. Cardiol. 17, 457–473 (2020).
Cossarizza, A. et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur. J. Immunol. 49, 1457–1973 (2019).
Skelly, D. A. et al. Single-cell transcriptional profiling reveals cellular diversity and intercommunication in the mouse heart. Cell Rep. 22, 600–610 (2018).
DeLaughter, D. M. et al. Single-cell resolution of temporal gene expression during heart development. Dev. Cell 39, 480–490 (2016).
Farbehi, N. et al. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife https://doi.org/10.7554/eLife.43882 (2019).
Forte, E. et al. Dynamic interstitial cell response during myocardial infarction predicts resilience to rupture in genetically diverse mice. Cell Rep. 30, 3149–3163.e6 (2020).
Tombor, L. S. et al. Single cell sequencing reveals endothelial plasticity with transient mesenchymal activation after myocardial infarction. Nat. Commun. 12, 681 (2021).
Litvinukova, M. et al. Cells of the adult human heart. Nature 588, 466–472 (2020).
Gladka, M. M. et al. Single-cell sequencing of the healthy and diseased heart reveals cytoskeleton-associated protein 4 as a new modulator of fibroblasts activation. Circulation 138, 166–180 (2018).
Molenaar, B. et al. Single-cell transcriptomics following ischemic injury identifies a role for B2M in cardiac repair. Commun. Biol. 4, 146 (2021).
Kannan, S. et al. Large particle fluorescence-activated cell sorting enables high-quality single-cell RNA sequencing and functional analysis of adult cardiomyocytes. Circ. Res. 125, 567–569 (2019).
Eraslan, G. et al. Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function. Science 376, eabl4290 (2022).
Tucker, N. R. et al. Myocyte-specific upregulation of ACE2 in cardiovascular disease: implications for SARS-CoV-2-mediated myocarditis. Circulation 142, 708–710 (2020).
Tucker, N. R. et al. Transcriptional and cellular diversity of the human heart. Circulation 142, 466–482 (2020).
See, K. et al. Single cardiomyocyte nuclear transcriptomes reveal a lincRNA-regulated de-differentiation and cell cycle stress-response in vivo. Nat. Commun. 8, 225 (2017).
Nicin, L. et al. Single nuclei sequencing reveals novel insights into the regulation of cellular signatures in children with dilated cardiomyopathy. Circulation 143, 1704–1719 (2021).
Hu, P. et al. Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev. 32, 1344–1357 (2018).
Nadelmann, E. R. et al. Isolation of nuclei from mammalian cells and tissues for single-nucleus molecular profiling. Curr. Protoc. 1, e132 (2021).
Selewa, A. et al. Systematic comparison of high-throughput single-cell and single-nucleus transcriptomes during cardiomyocyte differentiation. Sci. Rep. 10, 1535 (2020).
Denisenko, E. et al. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 21, 130 (2020).
Rodrigues, E. C., Grawenhoff, J., Baumann, S. J., Lorenzon, N. & Maurer, S. P. Mammalian neuronal mRNA transport complexes: the few knowns and the many unknowns. Front. Integr. Neurosci. 15, 692948 (2021).
Ivanovitch, K. et al. Ventricular, atrial, and outflow tract heart progenitors arise from spatially and molecularly distinct regions of the primitive streak. PLoS Biol. 19, e3001200 (2021).
Lescroart, F. et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 359, 1177–1181 (2018).
Hesse, J. et al. Single-cell transcriptomics defines heterogeneity of epicardial cells and fibroblasts within the infarcted murine heart. Elife https://doi.org/10.7554/eLife.65921 (2021).
Axelsson Raja, A. et al. Ablation of lysophosphatidic acid receptor 1 attenuates hypertrophic cardiomyopathy in a mouse model. Proc. Natl Acad. Sci. USA 119, e2204174119 (2022).
Koenig, A. L. et al. Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure. Nat. Cardiovasc. Res. 1, 263–280 (2022).
Nicin, L. et al. A human cell atlas of the pressure-induced hypertrophic heart. Nat. Cardiovasc. Res. 1, 174–185 (2022).
Chaffin, M. et al. Single-nucleus profiling of human dilated and hypertrophic cardiomyopathy. Nature https://doi.org/10.1038/s41586-022-04817-8 (2022).
Cui, M. et al. Dynamic transcriptional responses to injury of regenerative and non-regenerative cardiomyocytes revealed by single-nucleus RNA sequencing. Dev. Cell 53, 102–116.e8 (2020).
Koda, M. et al. Nuclear hypertrophy reflects increased biosynthetic activities in myocytes of human hypertrophic hearts. Circ. J. 70, 710–718 (2006).
Chongtham, M. C., Todorov, H., Wettschereck, J. E., Gerber, S. & Winter, J. Isolation of nuclei and downstream processing of cell-type-specific nuclei from micro-dissected mouse brain regions – techniques and caveats. bioRxiv https://doi.org/10.1101/2020.11.18.374223 (2020).
Cui, M. & Olson, E. N. Protocol for single-nucleus transcriptomics of diploid and tetraploid cardiomyocytes in murine hearts. STAR Protoc. 1, 100049 (2020).
Wojcik, K. & Dobrucki, J. W. Interaction of a DNA intercalator DRAQ5, and a minor groove binder SYTO17, with chromatin in live cells – influence on chromatin organization and histone–DNA interactions. Cytom. A 73, 555–562 (2008).
Chongtham, M. C., Butto, T., Mungikar, K., Gerber, S. & Winter, J. INTACT vs. FANS for cell-type-specific nuclei sorting: a comprehensive qualitative and quantitative comparison. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22105335 (2021).
Zhang, X. et al. Comparative analysis of droplet-based ultra-high-throughput single-cell RNA-seq systems. Mol. Cell 73, 130–142.e5 (2019).
Kolodziejczyk, A. A., Kim, J. K., Svensson, V., Marioni, J. C. & Teichmann, S. A. The technology and biology of single-cell RNA sequencing. Mol. Cell 58, 610–620 (2015).
Macosko, E. Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Zheng, G. X. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Hagemann-Jensen, M. et al. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 38, 708–714 (2020).
Hagemann-Jensen, M., Ziegenhain, C. & Sandberg, R. Scalable single-cell RNA sequencing from full transcripts with Smart-seq3xpress. Nat. Biotechnol. https://doi.org/10.1038/s41587-022-01311-4 (2022).
Yekelchyk, M., Guenther, S., Preussner, J. & Braun, T. Mono- and multi-nucleated ventricular cardiomyocytes constitute a transcriptionally homogenous cell population. Basic. Res. Cardiol. 114, 36 (2019).
Wang, Y. et al. Single-cell analysis of murine fibroblasts identifies neonatal to adult switching that regulates cardiomyocyte maturation. Nat. Commun. 11, 2585 (2020).
Wang, L. et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat. Cell Biol. 22, 108–119 (2020).
Rosenberg, A. B. et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182 (2018).
Xu, K. et al. Cell-type transcriptome atlas of human aortic valves reveal cell heterogeneity and endothelial to mesenchymal transition involved in calcific aortic valve disease. Arterioscler. Thromb. Vasc. Biol. 40, 2910–2921 (2020).
Pirruccello, J. P. et al. Deep learning enables genetic analysis of the human thoracic aorta. Nat. Genet. 54, 40–51 (2022).
Tyser, R. C. V. et al. Characterization of a common progenitor pool of the epicardium and myocardium. Science https://doi.org/10.1126/science.abb2986 (2021).
Davis, A., Gao, R. & Navin, N. E. SCOPIT: sample size calculations for single-cell sequencing experiments. BMC Bioinformatics 20, 566 (2019).
Schmid, K. T. et al. scPower accelerates and optimizes the design of multi-sample single cell transcriptomic studies. Nat. Commun. 12, 6625 (2021).
Reichart, D. et al. Pathogenic variants damage cell composition and single cell transcription in cardiomyopathies. Science 377, eabo1984 (2022).
Luecken, M. D. & Theis, F. J. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 15, e8746 (2019).
Azizi, E. et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 174, 1293–1308.e36 (2018).
Parekh, S., Ziegenhain, C., Vieth, B., Enard, W. & Hellmann, I. zUMIs – a fast and flexible pipeline to process RNA sequencing data with UMIs. Gigascience https://doi.org/10.1093/gigascience/giy059 (2018).
Ilicic, T. et al. Classification of low quality cells from single-cell RNA-seq data. Genome Biol. 17, 29 (2016).
Fleming, S. J. et al. Unsupervised removal of systematic background noise from droplet-based single-cell experiments using CellBender. bioRxiv https://doi.org/10.1101/791699 (2022).
Young, M. D. & Behjati, S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience https://doi.org/10.1093/gigascience/giaa151 (2020).
Yang, S. et al. Decontamination of ambient RNA in single-cell RNA-seq with DecontX. Genome Biol. 21, 57 (2020).
Osorio, D. & Cai, J. J. Systematic determination of the mitochondrial proportion in human and mice tissues for single-cell RNA-sequencing data quality control. Bioinformatics 37, 963–967 (2021).
Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291.e9 (2019).
Bernstein, N. J. et al. Solo: doublet identification in single-cell RNA-seq via semi-supervised deep learning. Cell Syst. 11, 95–101.e5 (2020).
Luecken, M. D. et al. Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods 19, 41–50 (2022).
Tran, H. T. N. et al. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 21, 12 (2020).
Vidal, R. et al. Transcriptional heterogeneity of fibroblasts is a hallmark of the aging heart. JCI Insight https://doi.org/10.1172/jci.insight.131092 (2019).
Miao, Z. et al. Putative cell type discovery from single-cell gene expression data. Nat. Methods 17, 621–628 (2020).
Liu, S., Thennavan, A., Garay, J. P., Marron, J. S. & Perou, C. M. MultiK: an automated tool to determine optimal cluster numbers in single-cell RNA sequencing data. Genome Biol. 22, 232 (2021).
Kimmel, J. C. & Kelley, D. R. Semisupervised adversarial neural networks for single-cell classification. Genome Res. 31, 1781–1793 (2021).
Lotfollahi, M. et al. Mapping single-cell data to reference atlases by transfer learning. Nat. Biotechnol. 40, 121–130 (2022).
Jain, M. S. et al. MultiMAP: dimensionality reduction and integration of multimodal data. Genome Biol. 22, 346 (2021).
Gayoso, A. et al. Joint probabilistic modeling of single-cell multi-omic data with totalVI. Nat. Methods 18, 272–282 (2021).
Ashuach, T., Gabitto, M. I., Jordan, M. I. & Yosef, N. MultiVI: deep generative model for the integration of multi-modal data. bioRxiv https://doi.org/10.1101/2021.08.20.457057 (2021).
Kleshchevnikov, V. et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat. Biotechnol. https://doi.org/10.1038/s41587-021-01139-4 (2022).
Andersson, A. et al. Single-cell and spatial transcriptomics enables probabilistic inference of cell type topography. Commun. Biol. 3, 565 (2020).
Dries, R. et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 22, 78 (2021).
Stahl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016).
Rodriques, S. G. et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019).
Merritt, C. R. et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 38, 586–599 (2020).
Sikkema, L. et al. An integrated cell atlas of the human lung in health and disease. bioRxiv https://doi.org/10.1101/2022.03.10.483747 (2022).
Tabula Sapiens, C. et al. The Tabula Sapiens: a multiple-organ, single-cell transcriptomic atlas of humans. Science 376, eabl4896 (2022).
Elmentaite, R., Dominguez Conde, C., Yang, L. & Teichmann, S. A. Single-cell atlases: shared and tissue-specific cell types across human organs. Nat. Rev. Genet. 23, 395–410 (2022).
Nielles-Vallespin, S. et al. Cardiac diffusion: technique and practical applications. J. Magn. Reson. Imaging 52, 348–368 (2020).
Cui, Y. et al. Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 26, 1934–1950.e5 (2019).
Suryawanshi, H. et al. Cell atlas of the foetal human heart and implications for autoimmune-mediated congenital heart block. Cardiovasc. Res. 116, 1446–1457 (2020).
Li, G. et al. Transcriptomic profiling maps anatomically patterned subpopulations among single embryonic cardiac cells. Dev. Cell 39, 491–507 (2016).
Xiao, Y. et al. Hippo signaling plays an essential role in cell state transitions during cardiac fibroblast development. Dev. Cell 45, 153–169.e6 (2018).
Kuppe, C. et al. Spatial multi-omic map of human myocardial infarction. Nature 608, 766–777 (2022).
Piroddi, N. et al. Myocardial overexpression of ANKRD1 causes sinus venosus defects and progressive diastolic dysfunction. Cardiovasc. Res. 116, 1458–1472 (2020).
Sergeeva, I. A. & Christoffels, V. M. Regulation of expression of atrial and brain natriuretic peptide, biomarkers for heart development and disease. Biochim. Biophys. Acta 1832, 2403–2413 (2013).
Sergeeva, I. A. et al. A transgenic mouse model for the simultaneous monitoring of ANF and BNP gene activity during heart development and disease. Cardiovasc. Res. 101, 78–86 (2014).
Ren, Z. et al. Single-cell reconstruction of progression trajectory reveals intervention principles in pathological cardiac hypertrophy. Circulation 141, 1704–1719 (2020).
Hill, M. C. et al. Integrated multi-omic characterization of congenital heart disease. Nature 608, 181–191 (2022).
Riching, A. S. & Song, K. Cardiac regeneration: new insights into the frontier of ischemic heart failure therapy. Front. Bioeng. Biotechnol. 8, 637538 (2020).
Honkoop, H. et al. Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. Elife https://doi.org/10.7554/eLife.50163 (2019).
Frangogiannis, N. G. Cardiac fibrosis. Cardiovasc. Res. 117, 1450–1488 (2021).
Aghajanian, H. et al. Targeting cardiac fibrosis with engineered T cells. Nature 573, 430–433 (2019).
Duan, Q. et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aah5084 (2017).
Fang, L., Murphy, A. J. & Dart, A. M. A clinical perspective of anti-fibrotic therapies for cardiovascular disease. Front. Pharmacol. 8, 186 (2017).
Gourdie, R. G., Dimmeler, S. & Kohl, P. Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat. Rev. Drug Discov. 15, 620–638 (2016).
Rog-Zielinska, E. A., Norris, R. A., Kohl, P. & Markwald, R. The living scar – cardiac fibroblasts and the injured heart. Trends Mol. Med. 22, 99–114 (2016).
Luo, S. et al. SAIL: a new conserved anti-fibrotic lncRNA in the heart. Basic Res. Cardiol. 116, 15 (2021).
Pinto, A. R. et al. Revisiting cardiac cellular composition. Circ. Res. 118, 400–409 (2016).
Soliman, H. et al. Multipotent stromal cells: one name, multiple identities. Cell Stem Cell 28, 1690–1707 (2021).
Xiao, Y. et al. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev. 33, 1491–1505 (2019).
Rao, M. et al. Resolving the intertwining of inflammation and fibrosis in human heart failure at single-cell level. Basic Res. Cardiol. 116, 55 (2021).
Sim, W. S., Park, B. W., Ban, K. & Park, H. J. In situ preconditioning of human mesenchymal stem cells elicits comprehensive cardiac repair following myocardial infarction. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22031449 (2021).
McCracken, I. R. et al. Lack of evidence of angiotensin-converting enzyme 2 expression and replicative infection by SARS-CoV-2 in human endothelial cells. Circulation 143, 865–868 (2021).
Brener, M. I. et al. Clinico-histopathologic and single-nuclei RNA-sequencing insights into cardiac injury and microthrombi in critical COVID-19. JCI Insight https://doi.org/10.1172/jci.insight.154633 (2022).
Zou, X. et al. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 14, 185–192 (2020).
Muus, C. et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat. Med. 27, 546–559 (2021).
Sungnak, W. et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 26, 681–687 (2020).
Delorey, T. M. et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 595, 107–113 (2021).
Buechler, M. B. et al. Cross-tissue organization of the fibroblast lineage. Nature 593, 575–579 (2021).
Chong, J. J. et al. Adult cardiac-resident MSC-like stem cells with a proepicardial origin. Cell Stem Cell 9, 527–540 (2011).
Janbandhu, V. et al. Hif-1a suppresses ROS-induced proliferation of cardiac fibroblasts following myocardial infarction. Cell Stem Cell 29, 281–297.e12 (2022).
Noseda, M. et al. PDGFRα demarcates the cardiogenic clonogenic Sca1+ stem/progenitor cell in adult murine myocardium. Nat. Commun. 6, 6930 (2015).
Soliman, H. et al. Pathogenic potential of hic1-expressing cardiac stromal progenitors. Cell Stem Cell 26, 205–220.e8 (2020).
Soliman, H. & Rossi, F. M. V. Cardiac fibroblast diversity in health and disease. Matrix Biol. 91-92, 75–91 (2020).
Pillai, I. C. L. et al. Cardiac fibroblasts adopt osteogenic fates and can be targeted to attenuate pathological heart calcification. Cell Stem Cell 20, 218–232.e5 (2017).
Alexanian, M. et al. A transcriptional switch governs fibroblast activation in heart disease. Nature 595, 438–443 (2021).
Zhang, Q. J. et al. Matricellular protein cilp1 promotes myocardial fibrosis in response to myocardial infarction. Circ. Res. 129, 1021–1035 (2021).
Meyer, I. S. et al. The cardiac microenvironment uses non-canonical WNT signaling to activate monocytes after myocardial infarction. EMBO Mol. Med. 9, 1279–1293 (2017).
Saxena, A. et al. IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J. Immunol. 191, 4838–4848 (2013).
Abe, H. et al. NF-κB activation in cardiac fibroblasts results in the recruitment of inflammatory Ly6C(hi) monocytes in pressure-overloaded hearts. Sci. Signal. 14, eabe4932 (2021).
Tallquist, M. D. & Molkentin, J. D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 14, 484–491 (2017).
Fu, X. et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Invest. 128, 2127–2143 (2018).
McLellan, M. A. et al. High-resolution transcriptomic profiling of the heart during chronic stress reveals cellular drivers of cardiac fibrosis and hypertrophy. Circulation 142, 1448–1463 (2020).
Schafer, S. et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 552, 110–115 (2017).
Ruiz-Villalba, A. et al. Single-cell RNA sequencing analysis reveals a crucial role for CTHRC1 (collagen triple helix repeat containing 1) cardiac fibroblasts after myocardial infarction. Circulation 142, 1831–1847 (2020).
Aird, W. C. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ. Res. 100, 174–190 (2007).
Hai, T. & Curran, T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc. Natl Acad. Sci. USA 88, 3720–3724 (1991).
Jiang, H. Y. et al. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol. Cell Biol. 24, 1365–1377 (2004).
Nawa, T. et al. Expression of transcriptional repressor ATF3/LRF1 in human atherosclerosis: colocalization and possible involvement in cell death of vascular endothelial cells. Atherosclerosis 161, 281–291 (2002).
Fan, F. et al. ATF3 induction following DNA damage is regulated by distinct signaling pathways and over-expression of ATF3 protein suppresses cells growth. Oncogene 21, 7488–7496 (2002).
Hoetzenecker, W. et al. ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat. Med. 18, 128–134 (2011).
Odiete, O., Hill, M. F. & Sawyer, D. B. Neuregulin in cardiovascular development and disease. Circ. Res. 111, 1376–1385 (2012).
De Keulenaer, G. W. et al. Mechanisms of the multitasking endothelial protein NRG-1 as a compensatory factor during chronic heart failure. Circ. Heart Fail. 12, e006288 (2019).
Wang, Z. N. et al. Cell-type-specific gene regulatory networks underlying murine neonatal heart regeneration at single-cell resolution (vol 33, 108472-1.e1, 2020). Cell Rep. 35, 109211 (2021).
Zhuang, L., Lu, L., Zhang, R., Chen, K. & Yan, X. Comprehensive integration of single-cell transcriptional profiling reveals the heterogeneities of non-cardiomyocytes in healthy and ischemic hearts. Front. Cardiovasc. Med. 7, 615161 (2020).
Li, Z. et al. Single-cell transcriptome analyses reveal novel targets modulating cardiac neovascularization by resident endothelial cells following myocardial infarction. Eur. Heart J. 40, 2507–2520 (2019).
Peisker, F. et al. Mapping the cardiac vascular niche in heart failure. Nat. Commun. 13, 3027 (2022).
Kalucka, J. et al. Single-cell transcriptome atlas of murine endothelial cells. Cell 180, 764–779.e20 (2020).
Rauch, A. et al. On the versatility of von Willebrand factor. Mediterr. J. Hematol. Infect. Dis. 5, e2013046 (2013).
Randi, A. M., Smith, K. E. & Castaman, G. von Willebrand factor regulation of blood vessel formation. Blood 132, 132–140 (2018).
Bedenbender, K. & Schmeck, B. T. Endothelial ribonuclease 1 in cardiovascular and systemic inflammation. Front. Cell Dev. Biol. 8, 576491 (2020).
Amsellem, V. et al. ICAM-2 regulates vascular permeability and N-cadherin localization through ezrin-radixin-moesin (ERM) proteins and Rac-1 signalling. Cell Commun. Signal. 12, 12 (2014).
Machon, O., Masek, J., Machonova, O., Krauss, S. & Kozmik, Z. Meis2 is essential for cranial and cardiac neural crest development. BMC Dev. Biol. 15, 40 (2015).
Vanlandewijck, M. et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480 (2018).
Hu, Z. et al. Single-cell transcriptomic atlas of different human cardiac arteries identifies cell types associated with vascular physiology. Arterioscler. Thromb. Vasc. Biol. 41, 1408–1427 (2021).
Wirka, R. C. et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 25, 1280–1289 (2019).
Chen, W. et al. Single-cell transcriptomic landscape of cardiac neural crest cell derivatives during development. EMBO Rep. 22, e52389 (2021).
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020).
Swirski, F. K. & Nahrendorf, M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 18, 733–744 (2018).
Chakarov, S. et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science https://doi.org/10.1126/science.aau0964 (2019).
Hulsmans, M. et al. Macrophages facilitate electrical conduction in the heart. Cell 169, 510–522.e20 (2017).
Lavine, K. J. et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl Acad. Sci. USA 111, 16029–16034 (2014).
Lavine, K. J. et al. The macrophage in cardiac homeostasis and disease: JACC macrophage in CVD series (Part 4). J. Am. Coll. Cardiol. 72, 2213–2230 (2018).
Bajpai, G. et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 24, 1234–1245 (2018).
Bajpai, G. et al. Tissue resident CCR2− and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ. Res. 124, 263–278 (2019).
Dick, S. A. et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 20, 29–39 (2019).
Zaman, R. et al. Selective loss of resident macrophage-derived insulin-like growth factor-1 abolishes adaptive cardiac growth to stress. Immunity 54, 2057–2071.e6 (2021).
Bizou, M. et al. Cardiac macrophage subsets differentially regulate lymphatic network remodeling during pressure overload. Sci. Rep. 11, 16801 (2021).
Cahill, T. J. et al. Tissue-resident macrophages regulate lymphatic vessel growth and patterning in the developing heart. Development https://doi.org/10.1242/dev.194563 (2021).
Badorff, C., Noutsias, M., Kuhl, U. & Schultheiss, H. P. Cell-mediated cytotoxicity in hearts with dilated cardiomyopathy: correlation with interstitial fibrosis and foci of activated T lymphocytes. J. Am. Coll. Cardiol. 29, 429–434 (1997).
Barin, J. G. & Cihakova, D. Control of inflammatory heart disease by CD4+ T cells. Ann. N. Y. Acad. Sci. 1285, 80–96 (2013).
Vdovenko, D. & Eriksson, U. Regulatory role of CD4(+) T cells in myocarditis. J. Immunol. Res. 2018, 4396351 (2018).
Ong, S. et al. Natural killer cells limit cardiac inflammation and fibrosis by halting eosinophil infiltration. Am. J. Pathol. 185, 847–861 (2015).
Nagy, C. et al. Single-nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat. Neurosci. 23, 771–781 (2020).
Liang, D. et al. Cellular and molecular landscape of mammalian sinoatrial node revealed by single-cell RNA sequencing. Nat. Commun. 12, 287 (2021).
Goodyer, W. R. et al. Transcriptomic profiling of the developing cardiac conduction system at single-cell resolution. Circ. Res. 125, 379–397 (2019).
Raredon, M. S. B. et al. Comprehensive visualization of cell-cell interactions in single-cell and spatial transcriptomics with NICHES. bioRxiv https://doi.org/10.1101/2022.01.23.477401 (2022).
Xiong, H. et al. Single-cell transcriptomics reveals chemotaxis-mediated intraorgan crosstalk during cardiogenesis. Circ. Res. 125, 398–410 (2019).
Dann, E., Henderson, N. C., Teichmann, S. A., Morgan, M. D. & Marioni, J. C. Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat. Biotechnol. 40, 245–253 (2022).
Dellefave, L. & McNally, E. M. The genetics of dilated cardiomyopathy. Curr. Opin. Cardiol. 25, 198–204 (2010).
Rajewsky, N. et al. Publisher correction: LifeTime and improving European healthcare through cell-based interceptive medicine. Nature 592, E8 (2021).
Churko, J. M. et al. Defining human cardiac transcription factor hierarchies using integrated single-cell heterogeneity analysis. Nat. Commun. 9, 4906 (2018).
Ruan, H. et al. Single-cell reconstruction of differentiation trajectory reveals a critical role of ETS1 in human cardiac lineage commitment. BMC Biol. 17, 89 (2019).
Paik, D. T. et al. Large-scale single-cell RNA-seq reveals molecular signatures of heterogeneous populations of human induced pluripotent stem cell-derived endothelial cells. Circ. Res. 123, 443–450 (2018).
Friedman, C. E. et al. Single-cell transcriptomic analysis of cardiac differentiation from human PSCs reveals HOPX-dependent cardiomyocyte maturation. Cell Stem Cell 23, 586–598.e8 (2018).
Wang, H., Yang, Y., Qian, Y., Liu, J. & Qian, L. Delineating chromatin accessibility re-patterning at single cell level during early stage of direct cardiac reprogramming. J. Mol. Cell Cardiol. 162, 62–71 (2022).
Sim, C. B. et al. Sex-specific control of human heart maturation by the progesterone receptor. Circulation 143, 1614–1628 (2021).
Ni, X. et al. Single-cell analysis reveals the purification and maturation effects of glucose starvation in hiPSC-CMs. Biochem. Biophys. Res. Commun. 534, 367–373 (2021).
Giacomelli, E. et al. Human-iPSC-derived cardiac stromal cells enhance maturation in 3D cardiac microtissues and reveal non-cardiomyocyte contributions to heart disease. Cell Stem Cell 26, 862–879.e11 (2020).
Zhou, Y. et al. Single-cell transcriptomic analyses of cell fate transitions during human cardiac reprogramming. Cell Stem Cell 25, 149–164.e9 (2019).
Liu, Z. et al. Single-cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte. Nature 551, 100–104 (2017).
Krane, M. et al. Sequential defects in cardiac lineage commitment and maturation cause hypoplastic left heart syndrome. Circulation 144, 1409–1428 (2021).
Lam, Y. Y. et al. Single-cell transcriptomics of engineered cardiac tissues from patient-specific induced pluripotent stem cell-derived cardiomyocytes reveals abnormal developmental trajectory and intrinsic contractile defects in hypoplastic right heart syndrome. J. Am. Heart Assoc. 9, e016528 (2020).
Mehrabi, M. et al. A study of gene expression, structure, and contractility of iPSC-derived cardiac myocytes from a family with heart disease due to LMNA mutation. Ann. Biomed. Eng. 49, 3524–3539 (2021).
Kathiriya, I. S. et al. Modeling human TBX5 haploinsufficiency predicts regulatory networks for congenital heart disease. Dev. Cell 56, 292–309.e9 (2021).
Acknowledgements
S.A.T. is supported by a Wellcome Sanger Institute grant (WT206194) and the Wellcome Science Strategic Support for a Pilot for the Human Cell Atlas (WT211276/Z/18/Z). N.H. is the recipient of an ERC Advanced Grant under the European Union Horizon 2020 Research and Innovation Program (AdG788970) and the Federal Ministry of Education and Research of Germany in the framework of CaRNAtion (031L0075A). M.D.S. received funding from the British Heart Foundation (CH/08/002/292257, RE/13/4/30184, RG/15/1/31165) and the European Research Council (233158). R.P.H. is supported by a National Health and Medical Research Council of Australia Investigator Grant (2021/GNT20087443) and Ideas Grant (2020/GNT2000615). M.N. received funding from the British Heart Foundation (PG/16/47/32156). S.A.T., N.H. and M.N. have received funding from a BHF/DZHK grant (SP/19/1/34461) and from the Chan Zuckerberg Initiative (2021-237882 and 2019-202666). The authors thank E. Adami (Max-Delbrück-Center for Molecular Medicine in the Helmholtz Association, Berlin, Germany) for her help with Table 1.
Author information
Authors and Affiliations
Contributions
A.M.A.M., V.J., H.M., K.K., J.C., M.D.S. R.P.H. and M.N. researched data for the article. and wrote the manuscript. A.M.A.M., V.J., H.M., K.K., S.A.T., N.H., M.D.S., R.P.H. and M.N. contributed to the discussion of content. All authors reviewed and edited the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
S.A.T. has consulted for or has been a member of scientific advisory boards at Biogen, ForeSite Labs, Genentech, GlaxoSmithKline, Qiagen and Roche, and is an equity holder of Transition Bio. All other authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Cardiology thanks David Paik and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Miranda, A.M.A., Janbandhu, V., Maatz, H. et al. Single-cell transcriptomics for the assessment of cardiac disease. Nat Rev Cardiol 20, 289–308 (2023). https://doi.org/10.1038/s41569-022-00805-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41569-022-00805-7
This article is cited by
-
Plasma fibroblast activation protein is decreased in acute heart failure despite cardiac tissue upregulation
Journal of Translational Medicine (2024)
-
Macrophages in cardiovascular diseases: molecular mechanisms and therapeutic targets
Signal Transduction and Targeted Therapy (2024)
-
The Genetic Factors Influencing Cardiomyopathies and Heart Failure across the Allele Frequency Spectrum
Journal of Cardiovascular Translational Research (2024)
-
Immunology of human fibrosis
Nature Immunology (2023)
-
The technological landscape and applications of single-cell multi-omics
Nature Reviews Molecular Cell Biology (2023)
