Abstract
Current electrolyte design for Li metal anodes emphasizes fluorination as the pre-eminent guiding principle for high Coulombic efficiency (CE) based largely on perceived benefits of LiF in the solid–electrolyte interphase (SEI). However, the lack of experimental techniques that accurately quantify SEI compositional breakdown impedes rigorous scrutiny of other potentially key phases. Here we demonstrate a quantitative titration approach to reveal Li2O content in cycled Li anodes, enabling this previously titration-silent phase to be compared statistically with a wide range of other leading SEI constituents including LiF. Across diverse electrolytes, CE correlates most strongly with Li2O above other phases, reaching its highest values when Li2O particles order along the SEI, demonstrating integrated chemical–structural function. The beneficial role of Li2O was exploited to create entirely fluorine-free electrolytes that breach >99% CE, highlighting electrolyte/SEI oxygenation as an underexplored design strategy.
Similar content being viewed by others
Main
Replacing the graphite anode in Li-ion batteries with Li metal is one of the most compelling strategies to meet targets of >750 Wh l−1 for electric vehicles1,2, but Li cannot yet achieve Coulombic efficiencies (CEs) > 99.95% required for >1,000 cycle life3,4,5. During cycling, active Li inventory is lost through several pathways: first, as solubilized material at ultralow CE (< ~10%); second, via the evolution of electronically isolated Li0 at intermediate CE (~ 10–95%); and third, through the irreversible formation of the solid–electrolyte interphase (SEI), which dominates losses as CE reaches 99% (refs. 5,6,7). To support design of very high-CE electrolytes, it is becoming crucial to understand the ideal target SEI composition and gain precise control over SEI formation so that only the most beneficial phases are formed.
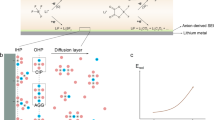
Even after extensive research, SEI chemical and functional understanding is remarkably incomplete and reliant upon qualitative models8,9. Broadly, it is accepted that the SEI comprises inorganic and organic phases, with the former including lithium fluoride (LiF), lithium oxide (Li2O), lithium carbonate (Li2CO3) and lithium nitride (Li3N) depending on salt; and the latter including semi-carbonates, alkoxides (ROLi) and poly/oligomers depending on solvent10. More accurately determining SEI composition remains a major challenge due to its exceedingly low amounts (sub-μmolLi cm−2 per cycle at >99% CE), instability and susceptibility to contamination during analysis, and limitations of spectroscopy techniques. Much understanding derives from X-ray photoelectron spectroscopy9, which has low depth sensitivity (~10 nm), resulting in widespread use of sputter depth profiling that can create compositional artefacts such as fictitious LiF enrichment11,12,13 from beam-induced decomposition of electrolyte salts and fragments.
Despite this, LiF has been proposed as a leading SEI descriptor, resulting in fluorination becoming the principal electrolyte design strategy (Supplementary Fig. 1)14,15,16,17. On the basis of bulk properties, LiF has been rationalized to be a desirable SEI phase given its chemical inertness, mechanical strength, low electronic conductivity and high interfacial energy16. Yet it is unclear whether bulk properties are relevant in the nanoscopic, complex SEI, and LiF is among the more resistive phases for Li+ transport18,19,20,21. Additionally, mounting evidence questions whether LiF has a clear chemical benefit in the SEI16,21,22 or instead plays the role of an inert building block, motivating a deeper look into which phases provide the major SEI functionality.
Lithium oxide (Li2O) is the second major ionic phase present in all model descriptions of the SEI9,10. In Li-ion batteries, Li2O is the fully reduced form of typical carbonate solvents and is thus presumed to play a role in the passivation of graphite23,24. On Li, however, Li2O has received less focus, especially compared with LiF. We previously observed that nanostructured all-Li2O SEI grown on Li foil possesses an approximately two times higher Li+ conductivity than all-LiF SEI, thus supporting comparatively more homogeneous Li+ flux, and rendering Li2O more beneficial to SEI transport18. This hypothesis is consistent with observed O enrichment of many high-CE SEI25,26 and in some advanced electrolytes27,28. Weighing the relative significance of Li2O, LiF and other phases is however challenging without an ability to accurately determine their proportions throughout the SEI. Titration-based analysis of cycled electrode materials has emerged in recent years as a powerful technique to quantify inactive Li0 and a growing number of SEI phases7,29,30,31,32, but the lack of a selective technique appropriate for cycled Li anodes has left Li2O as a titration-silent phase. Salt-derived phases such as Li3N, S- and B-containing phases have also not yet been studied in titration frameworks.
To fill this gap, this work reports an alcohol-based titration followed by Karl Fischer analysis that selectively quantifies total Li2O in cycled Li anodes. The method is integrated into a broader workflow designed to quantify other key SEI phases in parallel. These include LiF; Li3N; S-, P-, B-containing phases; Li2CO3/semi-carbonates (collectively, ROCO2Li); Li2C2; RLi and inactive Li0, providing a methodology to probe the rich SEI composition and resolve key building blocks in the limit of high CE. Across ten diverse electrolytes spanning broad composition and CE range, a major fraction of capacity loss is allocated, substantially exceeding prior quantification benchmarks6. Contrary to conventional understanding, Li2O is the most consistently abundant phase at high CE and the strongest CE descriptor, surpassing LiF, even in highly fluorinated electrolytes. Cryogenic high resolution transmission electron microscopy (Cryo-HRTEM) characterization further reveals that the distribution of Li2O within the SEI affects CE, providing both chemical and morphological function. Leveraging these findings, we demonstrate the possibility to achieve highly competitive >99% Li CE using oxygenated, rather than fluorinated, solvents and salts. The results indicate that LiF enrichment is not strictly requisite for high-Li CE and highlight SEI oxygenation as a compelling but underexplored pathway to expand versatility of electrolyte design.
Quantitative titration of Li2O in cycled anodes
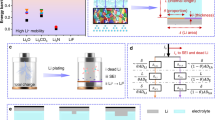
Samples for titration analysis were generated over several plating/stripping cycles of Li on Cu to accumulate irreversible materials (herein termed SEI/Li0 residuals), ending with a full stripping step. The Li2O titration (Fig. 1a) is a two-step reaction in which residuals on Cu are reacted with 2-butoxyethanol to form LiOH from any present Li2O (Li2O + BuOC2H4OH → LiOH + BuOC2H4OLi)33,34. The LiOH solution is injected into a Karl Fischer (KF) titrator and reacted to completion to produce H2O (Supplementary Fig. 2), which is related back to the original amount of SEI Li2O by charge balance (2 e– detected at the KF electrode per unit LiOH = unit Li2O; Fig. 1b and Methods). The 2-butoxyethanol/KF titration series is selective to Li2O and LiOH over other SEI phases such as Li2CO3, Li3N, LiH, LiF and metallic Li0 (Fig. 1c). The latter reacts with 2-butoxyethanol to form an alkoxide rather than LiOH (Li0 + BuOEtOH → BuOEtOLi + 0.5 H2, as verified in Fig. 1c and Supplementary Fig. 3), thus is invisible to KF titration34. In this regard, using alcohol as the titrant provides an important advantage over acid-base titration of Li2O35, because aqueous titrations yield LiOH from both Li2O and Li0, which cannot be differentiated (Supplementary Note 1). LiOH was confirmed to be negligible in the initial SEI using infrared spectroscopy and low-water-content electrolytes (Supplementary Figs. 4 and 5 and Supplementary Note 2), and thus was produced exclusively upon butoxyethanol reaction with SEI Li2O.

a, Illustration of the experimental workflow used for Li2O quantification based on conversion of Li2O into LiOH by reaction with 2-butoxyethanol, followed by titration of the resulting hydroxide using a coulometric KF method. b, Charge measured by the coulometric KF method after titration of standard solutions of LiOH and Li2O in 2-butoxyethanol. The dashed line confirms the typical expected titrated 2 e− charge for each unit Li2O/LiOH. Slight deviation from the expected value at higher μmol values is due to residual water in the powders. c, Sensitivity of the KF method to other SEI phases and Li0 in which 10 mg of each material was dissolved/reacted with 1 ml of 2-butoxyethanol and subsequently titrated by the KF method after 24 h. Error bars in b and c correspond to the error of the balance used to weigh Li2O and LiOH samples (Supplementary Fig. 2) and not from statistics over multiple samples. d, Capacity loss in the 1 M LiPF6 EC/DEC electrolyte attributed to individual phases, measured after 5–10 full cycles (0.5 mA cm−2, 1.5 mAh cm−2), including Li2O in addition to Li0, ROCO2Li, Li2C2, RLi, LiF and P-containing (P-cont.) phases. e, Average partitioning of total capacity loss for each phase in which the absolute measured quantities of each phase in d are normalized by each cell’s measured capacity loss. Data presented as mean ± standard deviation, and open markers denote individual data points (n = 3 for Li0, ROCO2Li, Li2C2, RLi; n = 4 for P, LiF; n = 5 for Li2O). The inset indicates a cumulative representation of the average of each SEI phase and Li0 from d.
The Li2O titration was integrated into a parallelized workflow to quantify capacity loss partitioning in a first exemplar electrolyte, 1 M LiPF6 in ethylene carbonate (EC)/diethyl carbonate (DEC) (50/50 vol%), examined previously by titration but with no information on Li2O6. A three-pronged titration scheme was adopted wherein batches of Cu/Li cells were cycled galvanostatically to a targeted capacity loss and diverted to either HCl, water or 2-butoxyethanol titration (Supplementary Figs. 6–8). Following prior work6, the first two are used to quantify inactive Li0, ROCO2Li, Li2C2, RLi (HCl) and LiF, P-containing phases (H2O). Samples used for those titrations were rinsed with anhydrous dimethyl carbonate (DMC) to remove electrolyte, which was confirmed to not alter the detected SEI composition or amount of Li0 (Supplementary Fig. 9). The targeted capacity loss was ~1 mAh for acid/water and ~2 mAh for butoxyethanol, determined by minimum sample requirements for limiting phases in each method (Supplementary Notes 3 and 4 and Supplementary Fig. 10).
Titration results (Fig. 1d) were normalized to each cell’s capacity loss and averaged across replicates, yielding the capacity loss partitioning (%) of the electrolyte shown in Fig. 1e (Supplementary Note 5). As observed previously6, a major capacity loss mechanism in 1 M LiPF6 EC/DEC forms inactive Li0 (46.1% of total loss). However, among the remaining 53.9% comprising SEI phases, strong Li2O prevalence could now be confirmed (yellow bars). Moreover, Li2O was the most abundant detectable SEI phase (14.6% of total loss or 27.1% of SEI Li+), followed more remotely by ROCO2Li (5.4% or 10.0%, respectively) and yet more minor phases: RLi, Li2C2, P-containing phases and LiF, each comprising ≤1% of total capacity loss. The remaining unidentified SEI losses (~32%) are shown in grey and correspond to solvent-derived phases (for example, poly/oligomeric species, polycarbonates or alkoxides), salt fragments besides LiF, or soluble phases; the former two currently elude analysis because they do not produce unambiguously measurable phases upon titration. To better understand the relationship between SEI composition and performance, the workflow is next applied to a broader series of electrolytes.
Li2O and SEI quantification across diverse electrolytes
A range of electrolyte compositions, spanning conventional carbonate/ether classes with diverse salts and localized high-concentration electrolytes (LHCE), were selected to bridge low to high CE (Fig. 2a and Table 1). To account for different salt products in the SEI/Li0 residuals (Fig. 2b), S- and B-containing phases were measured by inductively coupled plasma-atomic emission spectroscopy (ICP-AES), and Li3N was analysed by a salicylate assay method36 (Supplementary Fig. 11) in addition to the above-noted workflow after cells were cycled to their target capacity loss over 1–10 cycles (Supplementary Figs. 12–14). Fig. 2c shows the compositional breakdowns by rank in select electrolytes, with the complete data for all electrolytes in Fig. 2d (titration data: see Source Data, Supplementary Figs. 15–24, Supplementary Tables 1–9; SEI breakdown: Supplementary Fig. 25). Key findings in each electrolyte are first highlighted before a cross comparison by phase.

a, Coulombic efficiency across diverse electrolytes, determined by the same cycling protocol used to generate the titration samples (Supplementary Note 4). Data presented as mean ± standard deviation, and open markers denote individual cells (n = 10, 13, 11, 11, 12, 12, 12, 12, 15, 13 for electrolytes in order of increasing CE). Lower-CE electrolytes consistently displayed larger scatter than high-CE electrolytes. b, Examples of solvent species and salts present in the electrolytes considered herein, annotated with the instrument/titration techniques used to measure decomposition products from the indicated chemical moieties. Techniques include gas chromatography with thermal conductivity (GC-TCD) and flame ionization (GC-FID) detectors, 19F nuclear magnetic resonance spectroscopy (NMR), ultraviolet-visible spectroscopy (UV-Vis), ICP-AES, and KF titration. c, Measured SEI/Li0 residuals normalized by capacity loss for select electrolytes in the series. Data are presented as mean ± standard deviation. Open markers denote individual data points (top frame: n = 6 for Li0, S, ROCO2Li, Li2C2, RLi; n = 4 for Li3N, LiF; n = 3 for Li2O; middle frame: n = 4 for all phases; bottom frame: n = 3 for all phases). d, Cumulative representation of the average of each SEI/Li0 residual normalized by capacity loss for all electrolytes in the series, organized by CE. Colours as in c. The error bars denote the estimated cumulative standard deviation of all phases, calculated from the root square sum of the error bars in c and centred around the sum of the mean of all phases. Sample number ‘n’ for each electrolyte equal to a. Raw data for each electrolyte can be found in Source Data and Supplementary Figs. 15–24. In electrolytes for which there is extensive anion decomposition, >100% cumulative SEI/Li0 residuals can occur due to contributions from charged salt ions to the Li inventory on the cycled electrode (Supplementary Note 6 for LiNO3 as an example). Supplementary Fig. 25 shows the data for the SEI phases in d but normalized by SEI, rather than total, capacity loss (that is, total capacity loss minus inactive Li0).
A first electrolyte examined was 1.37 M lithium bis(trifluoromethane)sulfonimide (LiTFSI) in 1,1,2,2-tetrafluoroethyl 2,2,3,3-tetrafluoropropyl ether (TTE)/DMC (70/30 vol%, CE = 40.4%), the composition of which was based on leading LHCEs37,38, but replaces the usual lithium bis(fluorosulfonyl)imide (LiFSI) salt with LiTFSI. This electrolyte exhibited low CE consistent with inferiority of non-additive containing LiTFSI electrolytes compared with those based on LiFSI (Supplementary Fig. 1). Expectedly for very low CE, capacity loss was dominated by Li0 (81.9% of total loss)7. The SEI comprised S-containing (S-cont.) phases from LiTFSI followed by minor amounts of other SEI phases, such as Li2O and LiF. Organofluorine (R-CFx) species, consistent with TFSI−/TTE decomposition, were also observed by 19F-NMR (Supplementary Fig. 26; Supplementary Figs. 27–35 for other electrolytes), though could not be quantified due to unknown fluorine stoichiometry x. Upon changing the solvent (1 M LiTFSI in 1,3-dioxolane (DOL)/1,2-dimethoxyethane (DME)), CE increased (59.8%), Li0 decreased and Li2O increased, though was still relatively minor. Whereas S- and F-containing phases in this electrolyte arise unambiguously from salt decomposition, Li2O could also form from solvent reduction. Notably, this electrolyte yields the largest proportion of unresolved phases (grey region, 44.8% of total capacity loss) consistent with a poly/oligomeric-rich SEI39. A third electrolyte (1.37 M LiFSI 5TTE/5DMC) replaces LiTFSI with LiFSI but uses a blend of 5TTE/5DMC with suboptimal contact-ion pairing due to excess DMC compared with a high-CE LHCE analogue38. Still, it achieves higher CE (76.8%) and a higher SEI proportion of S-containing phases (35.8% of capacity loss) from participation of FSI− in SEI formation. Li2O and LiF are present in lower amounts than S-containing phases, indicating that FSI− fragments in the SEI do not undergo complete reduction at these cycle conditions.
The next electrolytes comprise a series of conventional carbonates: 1 M LiPF6 EC/DEC + 10 vol% vinylene carbonate (VC) (CE = 90.1%); and the same without additive (CE = 92.5%) or with 10 vol% fluoroethylene carbonate (FEC) (CE = 95.1%). All have marked presence of ROCO2Li, as expected40. However, increasing CE corresponds to increasing preponderance of Li2O, from being nearly comparable to ROCO2Li for the VC-based electrolyte (7.0% of total loss) to well-exceeding ROCO2Li with FEC (26.4%). With fluorinated LiPF6, Li2O can only originate from solvent-derived phases such as ROCO2Li41 and oligo/polycarbonates from VC or FEC42. Reasonably, LiF content was highest with FEC.
Electrolytes with higher CE (generally >90%) exhibit SEI increasingly dominated by Li2O. In 1.2 M LiBF4 and 0.6 M LiBF4/0.6 M lithium difluoro(oxalato)borate (LiDFOB) in 1FEC/2DEC (CE = 96.0% and 97.3%, respectively), Li2O is followed by notable contributions from B-containing (B-cont.) phases (Supplementary Figs. 32 and 33) and LiF resulting from salt and FEC breakdown. Meanwhile, the LHCE 1.37 M LiFSI 7TTE/3DMC (CE = 98.8%) has a more chemically diverse SEI38 with major contributions from S-containing phases (for example, -SO2F; Supplementary Fig. 34), followed by Li2O/LiF and more minor contributions of ROCO2Li and Li3N.
Finally, an electrolyte containing LiNO3, an additive known to promote Li2O formation28,43, was examined (1 M LiTFSI DOL/DME + 3 wt% LiNO3, CE = 99.0%). This electrolyte yielded SEI capacity losses overwhelmingly dominated by Li2O (80.8%), indicating that the relationship between Li2O and CE, examined more rigorously below, can be nonlinear. LiF, which in this electrolyte can only originate from C–F in LiTFSI, was proportionally much lower (4.3%). The same electrolyte was also tested with only 1% LiNO3 and yielded similar CE and Li2O (Supplementary Fig. 36). Notably, this electrolyte’s cumulative quantification exceeded 100% of capacity loss when normalized to measured charge. This scenario indicates an SEI-forming contribution from the salt, here Li+ NO3−, that can form Li2O without electrons being counted by the potentiostat (Supplementary Note 6). Across all electrolytes with N-containing salts (LiTFSI, LiFSI, LiNO3), this electrolyte exhibited the highest amount of fully reduced Li3N (4.8%). Together, the above results provide substantially improved resolution to SEI composition, enabling further scrutiny of compositional correlations with CE.
Statistical correlations among SEI phases and CE
The relationship of a given phase with CE is generally not monotonic (Supplementary Fig. 37). To rigorously identify statistical correlations within the dataset, the Spearman rank correlation coefficient (ρ) was utilized (Supplementary Note 7)44. Electrolytes were ranked in order of increasing CE and in proportion of each phase (%), and the linear coefficient of correlation between ranks, ρ, was calculated (Fig. 3a and Supplementary Fig. 38), with ρ = 1/−1/0 for strictly positive/negative/neutral correlations. Associated with each ρ, a degree of statistical significance σ quantifies the null hypothesis probability that an observed correlation could have originated by chance from uncorrelated variables (σ > 2.5 corresponds to <1%; Fig. 3b). Confidence intervals around each ρ were also calculated to account for the effect of potential outliers in the dataset (Fig. 3c).

a, Rank CE and rank phase (%) for inactive Li0, LiF and Li2O. The coefficient of correlation ρ measures whether the relationship between CE and a given phase is monotonic, with ρ = 1 perfectly monotonic and positively correlated and ρ = −1 perfectly monotonic and negatively correlated. Each data point indicates a different electrolyte. b, Statistical significance σ associated with each phase-CE correlation. c, Frequency histograms of ρ, calculated from all possible combinations of (ten choose seven) data points (that is, excluding up to three data points from the dataset). The solid vertical line indicates ρ calculated from all ten data points, and the shaded region indicates the 95% confidence interval calculated from the frequency histogram. d, CE vs Li2O relationship, now including eight additional electrolytes (18 total; additional electrolytes are 1.37 M LiClO4 7TTE/3DMC, 0.5 M and 0.75 M LiFSI 7TTE/3DMC, 1 M LiClO4 propylene carbonate (PC), 1.5 M LiFSI dimethoxydimethylsilane (DMMS), 1 M LiTFSI 2,2,2-trifluoro-N,N-dimethylacetamide (FDMA)/FEC, 0.8 M LiPF6 1FEC/3DMC + 5 wt% 4 M LiNO3/dimethyl sulfoxide (DMSO) and 2 M LiFSI/1 M LiTFSI DOL/DME + 3 wt% LiNO3), for a total of n = 81 samples. CE and Li2O were measured using cycling conditions that meet the capacity loss requirements for 2-butoxyethanol titration. Error bars indicate the standard deviation of each electrolyte, over at least three cells. e, Rank correlation from d.
Expectedly7, a clear inverse relationship between CE and inactive Li0 was confirmed (Fig. 3a,b, \(\rho\) = –0.818, σ = 2.77), and LiF exhibited a positive albeit weaker correlation with CE (\(\rho\) = 0.758, σ = 2.42). However, Li2O showed the strongest and most statistically significant correlation across all capacity loss contributions (\(\rho\) = 0.903, σ = 3.29), notably stronger than the well-established negative relationship with Li0 (ref. 7). Furthermore, when phases are normalized to SEI loss only, rather than total capacity loss (Supplementary Fig. 25), Li2O remains the most correlated with CE, whereas the positive correlation of LiF became less significant (Supplementary Figs. 39 and 40). All other SEI phases showed weaker statistical significance for CE (σ < 1; Fig. 3b). However, it is emphasized that trends in Li3N, P-, S- and B-content could not be as rigorously examined because the electrolytes considered do not uniformly span all elemental diversity equally, leading to wide confidence intervals in ρ for these phases (Fig. 3c). Regardless, excepting S-containing phases, these were not typically seen to be major SEI components, and none (including S) was required to achieve high CE, unlike Li2O. Similarly, the remaining solvent-derived phases RLi, ROCO2Li and Li2C2 showed weak correlation with CE and wide confidence intervals.
To further test the relationship between Li2O and CE, Li2O was measured for four additional lower-CE and four other >99% CE electrolytes selected from Supplementary Fig. 145,46,47,48. Fig. 3d shows the CE vs Li2O relationship for each cycled cell (in grey) and, in yellow, the Li2O and CE per electrolyte, averaged over at least three cells per electrolyte. The rank correlation over all cells (Fig. 3e) confirms the high coefficient of correlation between CE and Li2O (ρ = 0.88) but now with even stronger statistical significance (σ = 3.7).
Li2O microstructure within the SEI
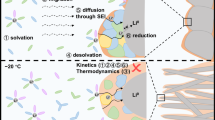
Titration-based correlations capture the ensemble composition of the SEI, but deciphering the role of Li2O in improving CE requires closer scrutiny of its distribution and function. Cryo-HRTEM was leveraged to localize crystalline phases in low- (1.37 M LiTFSI 7TTE/3DMC) and high-CE (1.37 M LiFSI 7TTE/3DMC, 1 M LiTFSI DOL/DME + 3 wt% LiNO3) SEIs from the prior dataset (Fig. 4). These experiments were first conducted at low capacity (0.1 mAh cm−2) to ensure samples remained electron transparent. Varied Li morphology was observed, from needle-like to hexagonally faceted to oblate, respectively, in order of increasing CE (Fig. 4a). The faceted deposition morphology is evidence of unimpeded Li+ diffusion through a thin and/or permissive SEI49 and is consistent with the lowest SEI thickness (7 nm) in the LiFSI 7TTE/3DMC electrolyte (Fig. 4b). On the other hand, the highest-CE electrolyte exhibited the thickest SEI (27 nm), which is consistent with the high degree of LiNO3 reactivity observed in Fig. 2d and indicates that the relationship between CE and SEI thickness need not be strictly inverse. Further characterization was performed by selective area electron diffraction (SAED) and energy dispersive spectroscopy (EDS). SAED revealed diffraction rings attributed to Li2O exclusively, and notably no LiF reflections were found in any electrolyte at this plated capacity (Fig. 4c). However, F-, N- and S-containing phases were detected on all samples by EDS (Supplementary Fig. 41), consistent with titration (Fig. 2), thus indicating that the chemically diverse phases seen by these techniques are not crystalline at this plated capacity. HRTEM revealed Li2O particles in all samples but with notably distinct spatial distribution between the low- and high-CE SEIs (Fig. 4d). In the two high-CE electrolytes, a uniform ~5–10 nm Li2O layer was found oriented parallel to the SEI surface. Between the outer Li2O layer and the metallic Li core, larger, randomly oriented crystalline lattices were also found within an amorphous SEI matrix for the DOL/DME electrolyte, which agrees with the very high Li2O content (Fig. 2d). In the low-CE electrolyte, on the other hand, a primarily mosaic-like SEI nanostructure50 was observed, with few Li2O particles dispersed discontinuously at the outer SEI region. Thus, the benefits imparted to the SEI by Li2O content are most strongly associated with a continuous localization facing the electrolyte51, with the additional mosaic-like inclusions in the DOL/DME SEI boosting Li2O content but not substantially improving CE.

a, Low magnification images of the plated Li deposit morphology (0.1 mAh cm−2, no stripping) in low- and high-CE electrolytes, chosen from the quantification analysis in Fig. 2. b, Magnification of the interface between the Li deposit and vacuum, highlighting the thickness of the SEI. c, SAED measurements near the interface. Enlarged and non-annotated versions are shown in Supplementary Fig. 50. d, HRTEM images of the crystalline lattices within the SEI. e, Low magnification image of a Li deposit in the 1.37 M LiFSI 7TTE/3DMC LHCE electrolyte, now at 0.2 mAh cm−2 plated capacity. f, SAED patterns of the SEI. g, HRTEM images of the crystalline phases in the higher-capacity SEI. Inner frame indicates the Fourier transform of the outer SEI region with only Li2O reflections. h, Fourier transform of the white region indicated in g, with both Li2O and LiF reflections. i, 19F-NMR measurements of -SO2F and LiF in plated Li electrodes.
To better understand the apparent absence of crystallized LiF, further imaging was conducted on the 1.37 M LiFSI 7TTE/3DMC LHCE electrolyte. At a higher capacity (0.2 mAh cm−2), high aspect ratio deposits were now observed (Fig. 4e), and the SEI showed evidence of emerging crystalline LiF superposed with Li2O (Fig. 4f–h). This delayed onset of LiF formation during Li deposition indicates that phases containing F, S and N from the FSI− anion remain amorphous in the SEI matrix and are only later reduced and crystallized, possibly due to contact with subsequently plated Li0. These findings were further verified by 19F-NMR, which showed a greater abundance of -SO2F at low capacity relative to LiF, the amounts of which inverted once more Li was plated (Fig. 4i and Supplementary Fig. 42–44). Thus, whereas LiF is a product of SEI evolution during Li plating, the initial SEI and morphology of Li are governed primarily by organofluorine and readily formed Li2O.
Li2O formation in localized high-concentration electrolytes
To further interrogate the role of Li2O, an electrolyte series based on LiFSI 7TTE/3DMC was examined as a function of salt concentration, spanning below (0.25–1.25 M) and up to (1.37 M) the LHCE threshold. LiFSI is the most widely used salt used in high-CE electrolytes (Supplementary Fig. 1), where the higher salt concentrations enhance Li+-FSI− pairing, confirmed here via diffusion-ordered spectroscopy (DOSY)-NMR (Fig. 5a), that promote anion-derived SEI important for high CE38,52,53. The first-cycle CEs (4 mAh cm−2, 0.5 mA cm−2) are shown in Fig. 5b.

a, Diffusivity of all electrolyte components in 0.5 M and 1.37 M LiFSI TTE/DMC measured by DOSY-NMR, revealing the formation of DMC-Li+-FSI− coordination shells. b, Capacity loss and CE after the first cycle (4 mAh cm−2, 0.5 mA cm−2) in the LHCE-like electrolytes LiFSI 7TTE/3DMC, with salt concentrations increasing from 0.5 M to 1.37 M. Data presented as mean ± standard deviation (n = 3 per concentration). c, Salt-derived SEI phases (Li3N, S-containing, LiF and Li2O) and solvent-derived SEI phases (ROCO2Li, Li2C2, RLi) normalized to capacity loss, measured after the first cycle. d, 19F-NMR of electrode residuals dissolved in H2O after the first cycle in the 0.5 M and 1.37 M electrolytes.
Capacity loss breakdowns found Li0 to be the major contributor at lower CE (16.5–91.4%, 0.25–0.75 M; Supplementary Fig. 45). From 0.75 M to 1.00 M, CE increases to >97%, coinciding with a notable increase in Li2O and, more modestly, LiF (Fig. 5c). At 1.25–1.37 M, a substantial increase in -SO2F is seen by 19F-NMR and S in ICP, along with a suppression in CFx fragments from TTE decomposition (Fig. 5d and Supplementary Fig. 46), thus providing conclusive evidence that LiF derives primarily from anions in this regime. Li2O can, in principle, form from DMC, though a more plausible pathway involves fragmentation of LiFSI54, which is in accordance with increased contact-ion pairing and appearance of other salt byproducts (S, LiF, -SO2F). However, relatively little Li3N was observed, indicating that complete anion fragmentation and incorporation of N as a fully reduced phase occurs less readily than LiF/Li2O formation. These results reveal that LiFSI−-based LHCEs benefit from their ability to promote formation of Li2O as a key ionic phase resulting from elevated anion reactivity, especially at FSI− concentrations >0.75 M (Supplementary Fig. 45b,c) with correspondingly high CE. If true, other salts forming Li2O could in principle perform an equivalent function without the coupled formation of LiF, relaxing sole reliance on LiFSI for high Li CE.
To test this hypothesis, we designed an additional series of electrolytes to modulate Li2O and LiF proportions beyond those achievable with LiFSI (Fig. 6). The recipe for LHCE preparation described by Ren et al.38 was adapted, starting with a fixed 3:1 molar ratio of diluent:salt and adding DME until full salt dissolution, to produce LHCEs with selective F or O enrichment. TTE/DME electrolytes37,38,52 with fluorinated, O-free salts (1.52 M LiBF4 or 1.57 M LiPF6) yielded poor CE (~ 65% and ~91%, respectively; Fig. 6a,b), low Li2O (Fig. 6c) and greater amounts of LiF. On the other hand, an analogous LHCE based on oxygenated LiClO4, that is, an F-free salt, achieved higher CE (98.7%) and formed a larger amount of Li2O (Fig. 6c). To completely exclude any source of fluorine from the electrolyte, TTE was replaced with anisole55, forming an entirely F-free electrolyte, which led to a slightly higher CE (98.9%). This ‘defluorination’ strategy was then applied to the 1 M LiTFSI DOL/DME + 3 wt% LiNO3 tested previously in Figs. 2–4 but now replacing the LiTFSI with LiClO4, yielding yet another fluorine-free electrolyte with high CE (99.1%). Accordingly, addition of the Li2O-forming additive to the LiClO4 TTE/DME LHCE electrolyte also further increased its CE to 99.1% CE. These results indicate that electrolytes yielding Li2O-rich, LiF-free interphases represent a promising alternative to LiFSI-based LHCEs for high-performance Li anodes; such LiFSI-based electrolytes were measured here to have CE = ~99.3% (Supplementary Fig. 47) but are often corrosive and toxic due to their high fluorine content. Whereas the particular blends reported herein are not optimal for full cells due the instability of non-fluorinated ethers at cathode potentials (Supplementary Fig. 48), safety concerns associated with strongly oxidizing salts such as LiClO424, and limited high rate performance due to solvent viscosity (Supplementary Fig. 49), these challenges motivate further research on non-fluorinated but cathodically stable salts and solvents such as sulfates and carbonates24.

a, Cycling data used to measure CE in the LHCEs tested herein (0.4 mA cm−2, 4 mAh cm−2 formation cycle, 4 mAh cm−2 reservoir, 0.5 mAh cm−2 cycles), following the Pacific Northwestern National Laboratory (PNNL) protocol61. b, Summary of CEs measured for LHCEs containing LiBF4, LiPF6, LiClO4 and LiNO3 as salts. c, Total amounts of Li2O and LiF normalized by capacity loss in select electrolytes. In b–c, full electrolyte formulations correspond to, in order of CE: 1.52 M LiBF4 0.69-TTE/0.31-DME; 1.57 M LiPF6 0.71-TTE/0.29-DME; 1.7 M LiClO4 0.77-TTE/0.23-DME; 2.27 M LiClO4 0.74-anisole/0.26-DME; 1 M LiClO4 DOL/DME + 3 wt% LiNO3; 1.7 M LiClO4 0.77-TTE/0.23-DME + ~0.1 M LiNO3; solvent fractions given in vol/vol. Data presented as mean ± standard deviation, and open markers denote individual cells (n = 3 for all phases, except n = 5 for LiF in LiClO4 TTE/DME).
Thus, despite LiF being widely regarded as the major desirable SEI phase, LiF is not required to achieve >99% CE with Li anodes, as demonstrated by the designed oxygen-rich, fluorine-free electrolytes (Fig. 6). Such observation is not true for Li2O: not only did Li2O show a stronger correlation with CE than LiF (Fig. 3), but no examined electrolyte in this study could achieve such CEs without having Li2O as a major SEI phase. These findings highlight Li2O content as a powerful SEI-focused descriptor to guide broad electrolyte discovery, contrasting with existing electrolyte-focused descriptors15,25,26,52,56,57,58,59 that are effective when utilizing LiFSI salts, but which have not yet effectively described performance across broader electrolyte classes. Nonetheless, continued efforts are needed to understand intriguing questions raised by these results. These include whether and which electrolyte species, beyond anions, may source Li2O and the conditions that promote or hinder such Li2O-forming pathways to enable finer control over Li2O structure in the SEI. It is also critical to examine the role of phases beyond Li2O and LiF as noted herein; and insofar invisible SEI phases (for example, alkoxides, oligo/polymeric phases), as their possible contribution to high CE cannot yet be commented upon. Similarly, the results cannot exclude the significance of non-LiF fluorinated phases, which were detected by 19F-NMR. For example, when LiFSI is used as a salt, both Li2O- and SO2F-containing phases were seen to increase with CE and salt concentration, which indicates that there may be a relationship between these phases. Quantification of their effect on CE, along with needed advances in the chemical resolution and quantification of anion fragments more broadly, may be possible in the future through multidimensional NMR experiments and represents a frontier in improving SEI chemical resolution. Furthermore, the titration scheme presented herein is not limited to Li and can be extended to anode chemistries such as graphite and Na for which titration methods have started to be developed30,60 but which currently lack quantitative methods for oxide and fluoride characterization. Finally, we emphasize that electrolyte fluorination is still currently necessary for full Li-ion cells (Supplementary Fig. 48)14, as it aides the cathode by preventing Al corrosion and promoting oxidative stability. Future efforts should focus on integrating and balancing beneficial fluorination and oxygenation features to achieve both anodic and cathodic improvements while balancing parallel demands regarding corrosion, viscosity/wettability, cost and sustainability.
Conclusions
A titration methodology was developed to precisely measure total Li2O content in cycled Li anodes, adding to a growing suite of chemical titrations available for probing Li SEI composition encompassing LiF, Li3N, ROCO2Li, Li2C2, RLi, P-, S- and B-containing phases in addition to inactive Li0 formed during cycling. Among these, Li2O was found to be the most prevalent phase at high CE across a chemically diverse set of electrolytes. Consistently, statistical analyses on the titration data revealed that Li2O has the strongest positive correlation with CE with a correlation coefficient of ρ > 0.9, even exceeding that of the broadly regarded LiF (ρ = 0.758), countering the prevailing wisdom that LiF is a uniquely important SEI building block. Cryo-TEM analyses showed the importance of the morphology of Li2O nanostructures, where in low-CE electrolytes Li2O forms with seemingly arbitrary crystal planes, contrasting with the highly organized particles found at high CE that are oriented along the SEI interface. LiFSI, the current leading salt in modern electrolyte design, was shown to be a major driver of Li2O formation, especially in systems where contact-ion pairing is promoted. Finally, the critical role of Li2O vs LiF was leveraged to demonstrate that completely fluorine-free, oxygen-rich electrolytes can breach 99% Li CE. This insofar-neglected strategy of SEI oxygenation opens an unexplored design space for high-CE electrolytes, enabling an alternative route complementary to, and potentially competitive with, fluorination. Such a route is becoming increasingly important as industry seeks to move beyond costly, toxic fluorination schemes that governed the past decades of electrolyte development towards more sustainable paradigms for future battery designs.
Methods
Cell assembly and cycling
Coin cell components (CR2032, MTI) were cleaned with ethanol and deionized (DI) water, then dried in the vacuum oven at 70 °C for at least 12 hours and taken inside an Ar glovebox (MBraun, <0.1 ppm H2O, <0.1 ppm O2) without ambient exposure. Cu foil current collectors (15 mm diameter) were soaked in 1 M HCl for 1 hour, rinsed with DI water and similarly dried in a vacuum glass oven at 70 °C. Polymeric separators (typically Celgard 2325; 3501 used for 1 M LiClO4 PC due to electrolyte wetting) were punched to 20 mm and were similarly dried and transferred. Lithium foil discs (Alfa Aesar) were punched to 15 mm inside of the glovebox. Each cell was prepared with a total of 50 μl of electrolyte (25 μl added before each separator). After assembly, cells were sealed using an automatic crimper (MTI, 0.82 T load setting). The assembled cells were then taken outside of the glovebox, rested for 5 hours at open circuit voltage (OCV) and cycled galvanostatically at 0.5 mA cm−2 at room temperature in a battery cycler (BCS, MTI) for a number of cycles determined by the target capacity loss amounts (Supplementary Note 4), with a stripping cut-off voltage of 1 V. Between each plating or stripping half cycle, the cells rested for 5 minutes at OCV. Coulombic efficiency was determined as the average cycling efficiency over all galvanostatic cycles, unless explicitly noted otherwise (Fig. 6).
Sample preparation and titration
After cycling, cells were taken inside the glovebox and unsealed using an automated decrimper (MTI). The cycled Cu current collector containing SEI/Li0 residuals was carefully extracted from the cell and prepared according to the type of reactant used for titration. For acid/water titration, samples were soaked in anhydrous DME or DMC for 3 min before the reaction to remove excess electrolyte (Supplementary Fig. 9), dried under antechamber vacuum and taken outside of the glovebox in gas-tight vials. For Li2O quantification, 2-butoxyethanol was used for titration, and no rinsing was required. For LiF, P-containing phases and Li3N quantification, H2O was used for titration. For Li0, ROCO2Li, Li2C2, RLi, S-containing and B-containing phases, 3.5 M HCl was used for titration. Supplementary Methods provide detailed information on each analytical technique, their appropriate calibrations and sensitivities.
For HCl titration, 500 μl of 3.5 M HCl was injected into the vial using a gas-tight syringe. After >8 hours, 2.5 ml of gas was extracted from the headspace of the vial and injected into a GC (Agilent 7890 A) to quantify the amounts of H2, CO2, C2H2, CH4, C2H6, C2H4, which determine the total amount of Li0, ROCO2Li, Li2C2 and RLi. The vial was opened and another 500 μl of DI water was added, further diluting the liquid solution in a total volume of 1 ml. Using the resulting solution, samples were then prepared for ICP analysis to determine the concentration of S- and B-containing phases in solution. The acid titration scheme is depicted in Supplementary Fig. 6.
For H2O titration, 500 μl of DI H2O was injected into the vial using a gas-tight syringe. After >8 hours, 2.5 ml of gas was extracted from the headspace of the vial using a gas-tight syringe and injected into the GC for analysis. Then, the vial was opened and another 1 ml of DI water was added, further diluting the liquid solution in a total volume of 1.5 ml. Using the resulting solution, samples were then prepared for 19F-NMR to determine the concentration of LiF; for ICP-AES to determine the concentration of P-containing phases; and for UV-Vis analysis to determine the concentration of Li3N following a salicylate assay36. The water titration scheme is depicted in Supplementary Fig. 7.
For 2-butoxyethanol titration, special care was taken to further suppress contamination of the materials used in the analysis by water. As such, after regular vacuum drying at 70 °C, all materials and utensils, including vials, tweezers, syringes and needles, were dried in a vacuum glass oven at 70 °C for at least 12 hours and taken inside of the glovebox without any atmospheric exposure. Inside of the glovebox, the cycled Cu current collector and the separator closest to it were carefully extracted from the cell and closed inside a dry gas-tight vial without rinsing. Immediately after, 500 μl of dry butoxyethanol (typically ~3 ppm H2O) was injected into the vial through a septum using a dry gas-tight syringe. After ~24 hours, when the sample had fully reacted, another 1.5 ml of butoxyethanol was added to the vial, resulting in a 2 ml solution per sample. The solution was then analysed by KF titration to determine the concentration of Li2O in the solution. The 2-butoxyethanol titration scheme is depicted in Supplementary Fig. 8.
Coulometric KF titration of Li2O
Upon reaction of the Li residuals with 2-butoxyethanol, Li2O was converted into LiOH following the reaction ROH + Li2O → LiOH + ROLi (R = CH3(CH2)3O(CH2)2 for 2-butoxyethanol). The KF electrolyte (CombiCoulomat fritless) is methanol-based and contains a small amount of hydroiodic acid (HI) due to the presence of iodine (I2) in the solution, which is needed for KF titration. When added to the electrolyte, the hydroxide in the sample solution is consumed by HI, forming water stoichiometrically (LiOH + HI → H2O + LiI), which then follows the typical Karl Fischer reaction by consuming iodine (H2O + I2 + SO2 + 3 RbN + CH3OH → 2 RbN HI + RN HSO4CH3, where RbN is a buffering agent, typically pyridine). Iodine is then rebalanced coulometrically by the generator electrode in the KF titrator, and the charge needed for rebalancing is recorded (2 I+ + 2 e− → I2). Hence, there is a direct and stoichiometric relationship between the original amount of Li2O in the sample and the charge measured for rebalancing iodine (2 e−KF = 1 Li2O), as shown in Fig. 1b. More details on KF titration are in the Supplementary Methods.
Cryogenic transmission electron microscopy
Cu/Li cells used for Cryo-TEM were assembled as usual, with the only difference being the addition of three TEM Cu grids (Ted Pella, 1GC300) directly on top of the Cu current collector. Li was directly plated on the TEM grids at 0.5 mA cm−2 to a capacity of either 0.1 or 0.2 mAh cm−2 and immediately taken inside of the glovebox and disassembled for analysis. After disassembly, the grids were carefully rinsed by droplets of DMC or DME based on the solvent of the electrolyte, and subsequently grids were placed on a piece of Kimwipe in the glovebox for ten minutes to dry out the remaining rinsing solvents, then grids were sealed in Eppendorf tubes separately with Parafilm to ensure air-tight transfer. Afterwards, the Eppendorf tubes with grids inside were transferred outside of the glovebox, immediately immersed in a Styrofoam container filled with liquid nitrogen, and the Eppendorf tubes were crushed by a pair of pliers to immerse the grids in the liquid nitrogen (LN2). Grids were transferred into a grid box within LN2 and then stored in a Dewar for following cryo analysis.
Cryo analysis was conducted by an instrument in the Electron Imaging Center for Nanomachines at California NanoSystems Institute, using a FEI Titan 80–300 scanning transmission electron microscope operated at an accelerating voltage of 300 kV, which is equipped with a field emission gun, an Oxford X-MaxTEM 100 N TLE windowless SDD 100 mm2 for EDS and Ultrascan US1000 1 K digital camera. The grids were transferred into the TEM column with Gatan 626 cryo-transfer holder and the grids were immersed in LN2 during the whole transfer process to avoid potential air contamination. After insertion of the cryo holder, the temperature was maintained at −178 °C. The electron flux under low magnification was around 100 e Å−2 s−1 and 1,000 e Å−2 s−1 under high magnification. The acquisition time for SAED images was 0.05 s and for TEM images was 0.4 to 2 s.
Data availability
The data supporting the findings of this study are included within the article and its Supplementary Information files. Source data are provided with this paper.
References
Electrochemical Energy Storage Technical Team Roadmap (US DOE Vehicle Technologies Office, 2017).
Schmuch, R., Wagner, R., Hörpel, G., Placke, T. & Winter, M. Performance and cost of materials for lithium-based rechargeable automotive batteries. Nat. Energy 3, 267–278 (2018).
Batteries: 2021 Annual Progress Report (US DOE Vehicle Technologies Office, 2022).
Xiao, J. et al. Understanding and applying coulombic efficiency in lithium metal batteries. Nat. Energy 5, 561–568 (2020).
Hobold, G. M. et al. Moving beyond 99.9% Coulombic efficiency for lithium anodes in liquid electrolytes. Nat. Energy 6, 951–960 (2021).
Hobold, G. M. & Gallant, B. M. Quantifying capacity loss mechanisms of Li metal anodes beyond inactive Li0. ACS Energy Lett. 7, 3458–3466 (2022).
Fang, C. et al. Quantifying inactive lithium in lithium metal batteries. Nature 572, 511–515 (2019).
Meng, Y. S., Srinivasan, V. & Xu, K. Designing better electrolytes. Science 378, eabq3750 (2022).
Wu, H., Jia, H., Wang, C., Zhang, J. G. & Xu, W. Recent progress in understanding solid electrolyte interphase on lithium metal anodes. Adv. Energy Mater. 11, 2003092 (2020).
Peled, E. & Menkin, S. Review—SEI: past, present and future. J. Electrochem. Soc. 164, A1703–A1719 (2017).
Yu, W., Yu, Z., Cui, Y. & Bao, Z. Degradation and speciation of Li salts during XPS analysis for battery research. ACS Energy Lett. 7, 3270–3275 (2022).
Dedryvère, R. et al. XPS identification of the organic and inorganic components of the electrode/electrolyte interface formed on a metallic cathode. J. Electrochem. Soc. 152, A689 (2005).
Schechter, A., Aurbach, D. & Cohen, H. X-ray photoelectron spectroscopy study of surface films formed on Li electrodes freshly prepared in alkyl carbonate solutions. Langmuir 15, 3334–3342 (1999).
Wang, C., Meng, Y. S. & Xu, K. Perspective—fluorinating interphases. J. Electrochem. Soc. 166, A5184–A5186 (2018).
Suo, L. et al. Fluorine-donating electrolytes enable highly reversible 5-V-class Li metal batteries. Proc. Natl Acad. Sci. USA 115, 1156–1161 (2018).
Tan, J., Matz, J., Dong, P., Shen, J. & Ye, M. A growing appreciation for the role of LiF in the solid electrolyte interphase. Adv. Energy Mater. 11, 2100046 (2021).
Li, T., Zhang, X.-Q., Shi, P. & Zhang, Q. Fluorinated solid–electrolyte interphase in high-voltage lithium metal batteries. Joule 3, 2647–2661 (2019).
Guo, R. & Gallant, B. M. Li2O solid electrolyte interphase: probing transport properties at the chemical potential of lithium. Chem. Mater. 32, 5525–5533 (2020).
Smeu, M. & Leung, K. Electron leakage through heterogeneous LiF on lithium–metal battery anodes. Phys. Chem. Chem. Phys. 23, 3214–3218 (2021).
Chen, Y. C., Ouyang, C. Y., Song, L. J. & Sun, Z. L. Electrical and lithium ion dynamics in three main components of solid electrolyte interphase from density functional theory study. J. Phys. Chem. C. 115, 7044–7049 (2011).
He, M., Guo, R., Hobold, G. M., Gao, H. & Gallant, B. M. The intrinsic behavior of lithium fluoride in solid electrolyte interphases on lithium. Proc. Natl Acad. Sci. USA 117, 73–79 (2020).
Huang, W., Wang, H., Boyle, D. T., Li, Y. & Cui, Y. Resolving nanoscopic and mesoscopic heterogeneity of fluorinated species in battery solid-electrolyte interphases by cryogenic electron microscopy. ACS Energy Lett. 5, 1128–1135 (2020).
Heiskanen, S. K., Kim, J. & Lucht, B. L. Generation and evolution of the solid electrolyte interphase of lithium-ion batteries. Joule 3, 2322–2333 (2019).
Xu, K. Nonaqueous liquid electrolytes for lithium-based rechargeable batteries. Chem. Rev. 104, 4303–4418 (2004).
Zhao, Y. et al. Electrolyte engineering for highly inorganic solid electrolyte interphase in high-performance lithium metal batteries. Chem 9, 682–697 (2023).
Yu, Z. et al. Rational solvent molecule tuning for high-performance lithium metal battery electrolytes. Nat. Energy 7, 94–106 (2022).
Kim, M. S. et al. Suspension electrolyte with modified Li+ solvation environment for lithium metal batteries. Nat. Mater. 21, 445–454 (2022).
Liu, Y. et al. Solubility-mediated sustained release enabling nitrate additive in carbonate electrolytes for stable lithium metal anode. Nat. Commun. 9, 3656 (2018).
Deng, W. et al. Quantification of reversible and irreversible lithium in practical lithium-metal batteries. Nat. Energy 7, 1031–1041 (2022).
McShane, E. J. et al. Quantifying graphite solid–electrolyte interphase chemistry and its impact on fast charging. ACS Energy Lett. 7, 2734–2744 (2022).
Xiang, Y. et al. Quantitatively analyzing the failure processes of rechargeable Li metal batteries. Sci. Adv. 7, eabj3423 (2021).
von Holtum, B. et al. Accessing the primary solid–electrolyte interphase on lithium metal: a method for low-concentration compound analysis. ChemSusChem 16, e202201912 (2023).
Jaworowski, R. J., Potts, J. R. & Hobart, E. W. The determination of oxygen in lithium. Anal. Chem. 35, 1275–1279 (1963).
Sax, H. I. & Steinmetz, H. Determination of Oxygen in Lithium Metal (Oak Ridge National Laboratory, 1958).
Lu, J. et al. A lithium–oxygen battery based on lithium superoxide. Nature 529, 377–382 (2016).
Steinberg, K. et al. Imaging of nitrogen fixation at lithium solid electrolyte interphases via cryo-electron microscopy. Nat. Energy 8, 138–148 (2022).
Cao, X. et al. Effects of fluorinated solvents on electrolyte solvation structures and electrode/electrolyte interphases for lithium metal batteries. Proc. Natl Acad. Sci. USA 118, e2020357118 (2021).
Ren, X. et al. Role of inner solvation sheath within salt-solvent complexes in tailoring electrode/electrolyte interphases for lithium metal batteries. Proc. Natl Acad. Sci. USA 117, 28603–28613 (2020).
Fiedler, C., Luerssen, B., Rohnke, M., Sann, J. & Janek, J. XPS and SIMS analysis of solid electrolyte interphases on lithium formed by ether-based electrolytes. J. Electrochem. Soc. 164, A3742–A3749 (2017).
Aurbach, D., Ein‐Ely, Y. & Zaban, A. The surface chemistry of lithium electrodes in alkyl carbonate solutions. J. Electrochem. Soc. 141, L1–L3 (2019).
Leung, K., Soto, F., Hankins, K., Balbuena, P. B. & Harrison, K. L. Stability of solid electrolyte interphase components on lithium metal and reactive anode material surfaces. J. Phys. Chem. C. 120, 6302–6313 (2016).
Michan, A. L. et al. Fluoroethylene carbonate and vinylene carbonate reduction: understanding lithium-ion battery electrolyte additives and solid electrolyte interphase formation. Chem. Mater. 28, 8149–8159 (2016).
Jaumann, T. et al. Role of 1,3-dioxolane and LiNO3 addition on the long term stability of nanostructured silicon/carbon anodes for rechargeable lithium batteries. J. Electrochem. Soc. 163, A557–A564 (2016).
Corder, G. W. & Foreman, D. I. in Nonparametric Statistics for Non-Statisticians 122–154 (John Wiley & Sons, 2011).
Huang, Y. et al. Eco-friendly electrolytes via a robust bond design for high-energy Li metal batteries. Energy Environ. Sci. 15, 4349–4361 (2022).
Liu, S. et al. An inorganic-rich solid electrolyte interphase for advanced lithium-metal batteries in carbonate electrolytes. Angew. Chem. 60, 3661–3671 (2021).
Qiu, F. et al. A concentrated ternary‐salts electrolyte for high reversible Li metal battery with slight excess Li. Adv. Energy Mater. 9, 1803372 (2018).
Wang, Q. et al. Interface chemistry of an amide electrolyte for highly reversible lithium metal batteries. Nat. Commun. 11, 4188 (2020).
Yuan, X., Liu, B., Mecklenburg, M. & Li, Y. Ultrafast deposition of faceted lithium polyhedra by outpacing SEI formation. Nature 620, 86–91 (2023).
Peled, E., Golodnitsky, D. & Ardel, G. Advanced model for solid electrolyte interphase electrodes in liquid and polymer electrolytes. J. Electrochem. Soc. 144, L208 (1997).
Li, Y. et al. Correlating structure and function of battery interphases at atomic resolution using cryoelectron microscopy. Joule 2, 2167–2177 (2018).
Cao, X., Jia, H., Xu, W. & Zhang, J.-G. Review—localized high-concentration electrolytes for lithium batteries. J. Electrochem. Soc. 168, 010522 (2021).
Chen, S. et al. High-voltage lithium-metal batteries enabled by localized high-concentration electrolytes. Adv. Mater. 30, 1706102 (2018).
Shkrob, I. A., Marin, T. W., Zhu, Y. & Abraham, D. P. Why bis(fluorosulfonyl)imide is a ‘magic anion’ for electrochemistry. J. Phys. Chem. C. 118, 19661–19671 (2014).
Moon, J. et al. Non-fluorinated non-solvating cosolvent enabling superior performance of lithium metal negative electrode battery. Nat. Commun. 13, 4538 (2022).
Ko, S. et al. Electrode potential influences the reversibility of lithium-metal anodes. Nat. Energy 7, 1217–1224 (2022).
Yu, Z. et al. Molecular design for electrolyte solvents enabling energy-dense and long-cycling lithium metal batteries. Nat. Energy 5, 526–533 (2020).
Kim, S. C. et al. Data-driven electrolyte design for lithium metal anodes. Proc. Natl Acad. Sci. USA 120, e2214357120 (2023).
Fan, X. et al. Highly fluorinated interphases enable high-voltage Li-metal batteries. Chem 4, 174–185 (2018).
Sayahpour, B. et al. Quantitative analysis of sodium metal deposition and interphase in Na metal batteries. Energy Environ. Sci. 17, 1216–1228 (2024).
Adams, B. D., Zheng, J., Ren, X., Xu, W. & Zhang, J. G. Accurate determination of Coulombic efficiency for lithium metal anodes and lithium metal batteries. Adv. Energy Mater. 8, 1702097 (2017).
Acknowledgements
This work made use of the Massachusetts Institute of Technology (MIT) Materials Research Science and Engineering Center Shared Experimental Facilities, supported by the National Science Foundation under award number DMR-14-19807. This work also made use of the MIT Department of Chemistry Instrumentation Facility. Titration method development was supported in part by an Electrochemical Society–Toyota Young Investigator Award (B.M.G. and K.S.). The Cryo-HRTEM work was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under award number DE-SC0022955 (Y.L. and C.W.). We also thank T. Buonassisi, MIT, for helpful discussions regarding the statistical analysis presented herein.
Author information
Authors and Affiliations
Contributions
G.M.H. and B.M.G. conceived the idea and designed the experiments. G.M.H. carried out experiments and measurements. C.W. and Y.L. co-designed Cryo-HRTEM experiments and performed imaging. K.S. assisted with method development. G.M.H. and B.M.G. analysed the data and prepared the initial draft of the paper. All authors discussed the results and contributed to the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Energy thanks Xinyong Tao, Atsuo Yamada and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Methods, Notes 1–7, Figs. 1–46 and Tables 1–11.
Source data
Source Data Fig. 1
Source data of all samples used for statistical analysis.
Source Data Fig. 2
Source data of all samples used for statistical analysis.
Source Data Fig. 3
Source data of all samples used for statistical analysis.
Source Data Fig. 4
Integrated NMR data of all samples.
Source Data Fig. 5
Source data of all samples used for statistical analysis.
Source Data Fig. 6
Source data of all samples used for statistical analysis.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hobold, G.M., Wang, C., Steinberg, K. et al. High lithium oxide prevalence in the lithium solid–electrolyte interphase for high Coulombic efficiency. Nat Energy (2024). https://doi.org/10.1038/s41560-024-01494-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41560-024-01494-x
This article is cited by
-
Hidden potential of lithium oxide
Nature Energy (2024)
