
Abstract
The evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has highlighted the need for versatile diagnostic assays that can discriminate among emerging variants of the virus. Here we report the development and performance benchmarking of an inexpensive (approximately US$0.30 per test) assay for the rapid (sample-to-answer time within 30 min) colorimetric detection of SARS-CoV-2 variants. The assay, which we integrated into foldable paper strips, leverages nucleic acid strand-displacement reactions, the thermodynamic energy penalty associated with single-base-pair mismatches and the metal-ion-controlled enzymatic cleavage of urea to amplify the recognition of viral RNAs for the colorimetric readout of changes in pH via a smartphone. For 50 throat swab samples, the assay simultaneously detected the presence of SARS-CoV-2 and mutations specific to the SARS-CoV-2 variants Alpha, Beta and Gamma, with 100% concordance with real-time quantitative polymerase chain reaction and RNA sequencing. Customizable and inexpensive paper-based assays for the detection of viruses and their variants may facilitate viral surveillance.
Similar content being viewed by others


Main
The rapid evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) challenges current strategies for combating the ongoing pandemic, such as vaccination1,2, and has become a most urgent concern in the pandemic. However, there is no affordable and scalable diagnostic tool for the detection of SARS-CoV-2 variants, which impedes their diagnosis and epidemiological surveillance. Emerging variants raise uncertainties concerning viral transmission, pathogenicity and vaccine effectiveness3,4,5. The Alpha (also called B.1.1.7 or 501Y.V1) variant, for instance, has 43% to 90% higher transmissibility6 and 61% higher mortality risk7 than the predecessor lineage. The Beta (B.1.351 or 501Y.V2) variant contributes to a reduction of the clinical trial efficacy of AZD1222 vaccine from 70% to 22% and of NVX-CoV237 vaccine from 89% to 49% (refs. 8,9,10). A SARS-CoV-2 variant with E484K and D614G mutations has been reported to enable vaccine breakthrough infection11. The Delta (B.1.617.2) variant drove a second wave of SARS-CoV-2 infection in India that caused over 300,000 new cases per day (ref. 12) and has spread rapidly all over the world. In the face of such threats, tools that allow a scalable test of SARS-CoV-2 variants will help us to precisely assess the infection and lethality risk of SARS-CoV-2, quickly segregate high-risk or vaccine-deficient infectors, epidemiologically evaluate the evolution of SARS-CoV-2 and efficiently guide the use and iterative development of vaccines.
Currently, however, tools allowing us to rapidly resolve mutations in SARS-CoV-2 for identifying its variants are not readily available. Tools for scalable screening of SARS-CoV-2 variants should ideally (i) allow detection of single-nucleotide mutations in viral RNAs, (ii) confer a short sample-to-result turnaround time and (iii) provide a multiplexing capability to identify multiple variants. Reverse transcription quantitative polymerase chain reaction (RT–qPCR) is the gold-standard nucleic acid test and dominates SARS-CoV-2 diagnosis13. Advances in the replacement of PCR by isothermal amplification strategies such as loop-mediated isothermal amplification (LAMP)14,15,16, recombinase polymerase amplification17 and nucleic acid sequence-based amplification15, or the integration of CRISPR-Cas systems17,18,19, mitigate the need for bulky temperature-control instruments and shorten the assay time, thus facilitating SARS-CoV-2 diagnosis in resource-limited regions. Point-of-care nucleic acid tests using the Abbott ID Now and Cepheid GeneXpert have already been authorized by the US Food and Drug Administration. However, the aforementioned methods usually require nucleic acid amplification, entailing increased complexity and reagent costs for SARS-CoV-2 tests. Moreover, the identification of single-nucleotide changes in viral RNAs, for example by hybridization discrimination20,21 or enzyme-based recognition22,23, can be challenging and these methods have not yet been further developed to resolve SARS-CoV-2 variants. CRISPR-Cas-based nucleic acid tests hold promise for detecting genetic variation by exploiting the sequence discrimination capacity of Cas proteins16,24,25. Yet, limited nucleotide mutation-sensitive regions of 3–6 nt close to fixed sequences, such as a protospacer adjacent motif20,26, may prevent CRISPR-Cas-based diagnostics from covering some nucleotide mutations of interest in SARS-CoV-2, and its potential has so far not been sufficiently explored for detecting SARS-CoV-2 variants in clinical samples. Thus far, the only available strategy for profiling SARS-CoV-2 variants involves sequencing13,15,27, whose use is hindered, particularly in resource-limited regions, by the shortage of infrastructure as well as its high labour-intensiveness and costs.
The need for scalable SARS-CoV-2 tests that allow variants to be resolved has motivated our efforts to explore single-nucleotide-resolved viral RNA detection strategies. The assay reported here, termed MARVE (for multiplexed, nucleic-acid-amplification-free, single-nucleotide-resolved viral evolution), allows nucleic-acid-amplification-free visual detection of viral RNAs with single-nucleotide resolution. We employ a basic nucleic acid strand displacement process to sustain a specific recognition of input viral RNAs, which enables us to discriminate viral RNA mutations. MARVE uses an enzyme-induced colorimetric process to amplify the recognition events of viral RNAs; this eliminates the need for a reverse transcription and nucleic acid amplification process, thus dramatically reducing the test’s cost and the sample-to-result turnaround time. To demonstrate the simplicity, portability and multiplexing capability of MARVE, we measured SARS-CoV-2 and its five key variants in an integrated testing paper using a smartphone. We also developed a smartphone application (app) to guide the diagnosis, and to visualize and record the test results for clinical and cold-chain food samples, facilitating on-site profiling of SARS-CoV-2 variants by minimally trained personnel. MARVE has potential as a streamlined technology for the diagnosis and screening of SARS-CoV-2 variants without the need for complex laboratory settings.
Results
Working principle of MARVE
MARVE achieves nucleic-acid-amplification-free detection of viral RNA mutations by coupling a highly programmable nucleic acid strand displacement process with a metal ion-controlled enzymatic reaction using a double-stranded DNA probe (Fig. 1). The probe, termed the toehold exchange DNA probe (TEprobe), is designed to recognize mutation markers in the SARS-CoV-2 variants Alpha, Beta, Gamma (also called P.1) and Delta. The TEprobe contains a pair of terminal single-stranded overhangs, shown in blue and red in Fig. 1, referred to as the forward and reverse toehold, respectively. The reverse toehold domain includes a cytosine–cytosine (C:C) mismatch, allowing selective incorporation of the metal ion Ag(I), and forms a metallo-mediated C-Ag(I)-C artificial base pair by base–metal cation interactions28,29,30. Competitive binding of viral RNAs with the Rec strand in TEprobes, facilitated by the forward toehold, disrupts the reverse toehold and releases the clamped Ag(I) ion. The net effect of the strand displacement process is determined by the formation of new base pairs within the forward toehold and the disruption of the former base pairs within the reverse toehold. TEprobes are programmable and can be designed with a forward and reverse toehold that offer a slightly negative free energy of the strand displacement reaction induced by the mutated viral RNAs, while a biased positive free energy is generated when wild viral RNA binds due to the thermodynamic energy penalty derived from single-base-pair mismatch. On the basis of this principle, only mutated viral RNAs can efficiently initiate the strand displacement reaction, and mutations in the SARS-CoV-2 genome can therefore be specifically recognized. The recognition event leads to the release of Ag(I) ion from the TEprobe, the ion serving as an inhibitor of urease31, thus yielding an amplified effect arising from the production of NH4+ by the enzymatic cleavage of urea. The level of NH4+ product determines the solution pH. Therefore, SARS-CoV-2 mutations and variants can be identified and visualized by the naked eye or a smartphone using pH indicators.

A double-stranded DNA probe, the TEprobe, is designed to precisely control the net thermodynamic energy of the strand displacement reaction induced by input RNAs by tuning its terminal forward toehold and reverse toehold sequences. Mutated and wild viral RNAs can be distinguished by the strand displacement reaction on the basis of the thermodynamic energy penalty derived from a single-base-pair mismatch. Recognition of viral RNAs leads to the release of a urease inhibitor metal ion, Ag(I), and thus controls the cleavage of urea and the level of NH4+. Key mutation markers of SARS-CoV-2 variants such as Alpha, Beta, Gamma and Delta are identified and visualized using pH indicators. The diagnosis results are determined and presented to users by smartphone.
We further develop a paper-folding ‘origami’ strategy to integrate the viral detection steps, a smartphone imaging procedure to achieve portable detection, and a smartphone app to guide the operation and interpret the diagnostic results, facilitating on-site diagnosis of SARS-CoV-2 variants by users with minimal training.
We used a chemically synthesized short RNA sequence (22 nucleotides, nt) as a model target RNA to validate each reaction step that constitutes MARVE. Phenol red—frequently used in colorimetric LAMP—is a highly sensitive pH indicator32,33, serving to monitor the presence of NH4+. The specific binding of Ag(I) ion to a C:C base mismatch was verified by measuring the melting temperature of TEprobes with different base pairs or mismatches incorporated (Supplementary Fig. 1). Urease catalysis, Ag(I) inhibition of urease and target RNA-mediated strand displacement were demonstrated by the absorbance change of phenol red in each reaction step (Fig. 2a, and Supplementary Figs. 2 and 3). Target RNA-induced strand displacement and subsequent release of free Ag(I) ion were further confirmed by gel electrophoresis and the measurement of free Ag(I) in solution (Fig. 2b,c). A 20 min reaction time yielded a maximal colour change induced by the strand displacement reaction (Fig. 2a), promising a rapid assay for detecting RNAs. We found that throat swab samples had a negligible effect on the pH of the detection solution and the activity of urease (Supplementary Fig. 4). The presence of up to 50 mM electrolyte species typically found in clinical samples for SARS-CoV-2 tests34 did not change the urease activity (Supplementary Fig. 5).

a, Kinetic measurements of the MARVE reaction by measuring the absorbance at 560 nm. The absorbance measurement started upon the addition of phenol red and urea. Photographs of the product after a 20 min reaction (marked by a dashed line) are placed at the end of each curve. b, Electrophoretic analysis of the target RNA-mediated strand displacement reaction. Concentrations of Rec strand, Block strand and target RNA were all 1.5 μM. To facilitate the separation between the band of Block-Rec hybridization and the band of target RNA-Rec hybridization, the Block strand was added with 15 nt poly T at the 3′ terminus. c, Determination of the level of free Ag(I) ion in each step of MARVE using iodometric titration. d, Identification of single-nucleotide mutations using MARVE. Shown are the tested RNA sequences (left), the absorbance at 560 nm and the discrimination factor obtained for each RNA sequence (middle), and the free energy of the strand displacement reaction with an input of each RNA sequence (right). Discrimination factor, DF = Anon-target/Atarget_ave, where Anon-target is the absorbance at 560 nm of mutated RNA-present sample, and Atarget_ave is the average absorbance at 560 nm of target RNA-present sample (n = 3). Concentrations of phenol red, urea, urease, TEprobe and target RNA (or other RNA sequences in d) used in a, c and d were 250 μM, 500 mM, 1 nM, 3 nM and 3 nM, respectively. The urea cleavage reaction proceeded for 20 min in d. The TEprobe with an 8 nt forward toehold and a 7 nt reverse toehold was used in a–d. Data in a, c and d are mean ± s.d. (n = 3).
We next explored the capacity of MARVE to identify mutations in RNA sequences. We designed TEprobes with a tunable reverse toehold (1–9 nt) and a fixed forward toehold (8 nt) (sequences are listed in Supplementary Table 1). Among these, the TEprobe with a 7 nt reverse toehold contributed a highly efficient strand displacement reaction and a red-to-yellow colour change in the presence of the target RNA but hindered an obvious colour change with the mutated RNAs (Supplementary Fig. 6 and Extended Data Fig. 1). To systematically estimate the capacity of MARVE to detect single-nucleotide mutations, we synthesized a series of input RNA sequences modified from the target sequence with single-nucleotide substitution, insertion or deletion (Fig. 2d). Substitutions were located throughout the sequence with a 1 nt interval, while at the 12th nt (from the 5′ terminus), all possible nucleotide mutations including substitution, insertion and deletion were designed. To assess the discriminative ability of MARVE, we defined the discrimination factor as the ratio of the absorbance in the presence of a mutated input RNA to that in the presence of the target RNA. Using the TEprobe with a 7 nt reverse toehold, MARVE yielded discrimination factor values of over 10 for most of the mutant RNAs. To further explain the results of mutation discrimination, we measured the free energy of the C-Ag(I)-C artificial base pair using isothermal titration calorimetry (Supplementary Fig. 7), and calculated the free energy of the strand displacement reaction upon addition of different input RNAs (Supplementary Note 1). The mutations in the input RNAs resulted in a positive reaction free energy, with the exceptions of nucleotide substitution at position 22, and C or G base insertions at position 12 (Supplementary Tables 3 and 4). The mismatch resulting from the terminal mutation (position 22) only contributed to the loss of one neighbour base stacking35, thus yielding a lower discrimination factor compared with that of the internal mutation. The negative reaction free energy derived from C and G base insertions at position 12 may result from underestimating the energy penalty caused by nucleotide insertion. Except for the two cases of nucleotide insertion, thermodynamics-based prediction can facilitate the design of specific TEprobes to detect mutations in RNA sequences.
Visual detection of respiratory RNA viruses using MARVE
We tested MARVE to detect a series of human-associated respiratory viruses, including three coronaviruses (severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus (MERS-CoV) and SARS-CoV-2 and seven influenza virus subtypes.
TEprobes for the three coronaviruses were designed to target the US CDC-recommended sequences for RT–qPCR tests on the N gene. Because SARS-CoV-2 and SARS-CoV have similar N gene sequences (Fig. 3a), distinction of the two coronaviruses would indicate the specificity of MARVE. Pseudotyped viruses (or pseudoviruses), which are safe surrogate viruses carrying the N gene, the E gene and open reading frames 1a and 1b (ORF1ab) of SARS-CoV, MERS-CoV and SARS-CoV-2, were constructed using lentiviruses as vectors and served as positive controls (Supplementary Fig. 8). An 8 nt forward toehold could not support efficient hybridization between TEprobes and viral RNAs, as the presence of SARS-CoV-2 failed to sufficiently turn the solution colour to yellow (Extended Data Fig. 2). We reasoned that the long viral RNA sequence formed secondary structures (Supplementary Fig. 9) that might have hindered its hybridization to the forward toehold and thus reduced the colour change. Extension of the forward toehold length to 15 nt dramatically increased the response of TEprobes towards SARS-CoV-2 while discriminating SARS-CoV-2 from a stoichiometric amount of SARS-CoV or MERS-CoV (Extended Data Fig. 2d). Length-optimized TEprobes enabled visual detection of SARS-CoV, MERS-CoV and SARS-CoV-2, and negligible cross-reaction was observed among the three coronaviruses using their cognate TEprobes (Fig. 3a). The discrimination factors for the three coronaviruses were all greater than 20 (Extended Data Fig. 2g), indicating a highly specific detection of coronaviruses by MARVE. A test of MARVE with a dilution series of SARS-CoV-2 pseudovirus indicated that 8,000 copies per μl of SARS-CoV-2 pseudovirus can be identified by the naked eye, and 800 copies per μl can be distinguished from the background by absorbance measurement (Welch’s t-test: *P < 0.05) (Supplementary Fig. 10).

a, Cross-reaction test of SARS-CoV-2, SARS-CoV and MERS-CoV. Left: sequences of coronavirus N genes targeted by MARVE. Middle: heat map recording the absorbance at 560 nm using each TEprobe for profiling the three coronaviruses. Right: the corresponding visual detection panel for the three coronaviruses. b, Visual detection of seven influenza virus subtypes. c, Working principle of the origami paper. TEprobes, urease and urea/phenol red are loaded on page 2, 3 and 4, respectively. MARVE reactions are initiated by sequential paper-folding. d, Paper-based detection of respiratory viral RNAs. GAR is the proportion of regions with a green component in the detection sites. e–g, Sensitivity estimation of paper-based detection by targeting the N gene (e,g) or the E gene (f) of SARS-CoV-2. The red line in g indicates the threshold for classification. MARVE yielded positive results for all 21 replicates of samples, with 400 copies per μl of SARS-CoV-2 pseudovirus. h, Detection of respiratory RNA viruses spiked in throat swab samples. Photographs (left) and GAR values (right) of origami papers testing samples with SARS-CoV-2, SARS-CoV or MERS-CoV. SARS-CoV and MERS-CoV were detected by targeting their N genes. The presence of the human RNase P gene indicates successful sampling and preservation of throat swab samples. Urea cleavage reaction proceeded for 20 min in a and b. The concentration of each virus was 30,000 copies per μl in a and b, and 2,400 copies per μl in d and h. Data in b and d are mean ± s.d. (n = 3). Data in e and f are from 6 replicates. Data in g are mean ± s.d. (n = 21). Statistical significance in e, f and g was determined by a two-tailed Welch’s t-test: **P < 0.01, ***P < 0.001, ****P < 0.0001.
Influenza viruses cause symptoms similar to SARS-CoV-2 in humans and their infection can be confused with SARS-CoV-2 on the basis of clinical symptoms alone. We designed TEprobes to detect H1N1, H1N2, H3N2, H5N1, H7N9, H9N2 and H10N8 by targeting gene sites according to World Health Organization (WHO) recommendations. The presence of 30,000 copies per μl of each influenza virus subtype yielded a saturated colour change of phenol red using its cognate TEprobe (Fig. 3b), indicating that MARVE can also be readily applied for the detection of influenza viruses.
To facilitate out-of-laboratory detection, we used the origami strategy to integrate each reaction of MARVE. The origami papers were fabricated from Whatman No. 1 filter papers using a hot wax printing strategy according to a published method (Fig. 3c and Supplementary Fig. 11)36. We added pullulan to provide an oxygen-impermeable thin film to entrap urease37. The protection strategy maintained urease activity above 70% for 28 d at 4 °C (Supplementary Fig. 12). Sample loading, strand displacement-mediated Ag(I) release and Ag(I)-controlled pH visualization were carried out sequentially by folding the origami paper. The papers were finally imaged using a smartphone and the images were processed by Circle Hough Transform and K-means clustering (Supplementary Fig. 13). The proportion of regions with a green component in the circular detection sites (green area ratio (GAR), %) was used to quantify the colour change, which corresponds to the presence of viral RNAs. A concentration of 800 Ag(I) ions per μl induced a GAR value that is distinguishable from background (Supplementary Fig. 14) (Welch’s t-test: ****P < 0.0001). We estimated analytical variance among five batches of origami papers and among three different operators, and found that MARVE yielded a stable result for detecting the N gene of SARS-CoV-2 in the tested condition (Supplementary Fig. 15).
Origami papers designed with multiple loading sites permitted a multiplexed detection of viral RNAs of interest, exemplified here by coronaviruses and influenza viruses. We found that TEprobes matrixed in filter papers worked robustly to sense the presence of SARS-CoV-2 (by targeting either the N gene or the E gene), SARS-CoV, MERS-CoV, H1N1, H7N9 or H9N2 (Fig. 3d). MARVE was able to reliably detect as few as 400 copies per μl of SARS-CoV-2 by targeting either the N gene or the E gene of SARS-CoV-2 on the basis of smartphone imaging (Fig. 3e–g) (Welch’s t-test: **P < 0.01, ***P < 0.001, ****P < 0.0001).
We next tested the capacity of MARVE to detect respiratory RNA viruses in clinical and cold-chain food samples. Throat swab samples are most commonly taken for the diagnosis of SARS-CoV-2 infection38. The risk of transmission of SARS-CoV-2 on cold-chain food and environment underscores the need for monitoring SARS-CoV-2 residues on inanimate surfaces39,40. SARS-CoV-2, SARS-CoV and MERS-CoV pseudoviruses, and H1N1, H7N9 and H9N2 viruses, were spiked into healthy throat swabs to serve as infected samples or smeared on the surface of frozen belt fish and food packaging to mimic virus-contaminated samples. The presence of 2,400 copies per μl of SARS-CoV-2 yielded an obvious colour change using TEprobes targeting the E gene and the N gene (Fig. 3h, Supplementary Fig. 16 and Extended Data Fig. 3). Each coronavirus in throat swab, frozen belt fish and food packaging samples could be distinguished by its corresponding TEprobe. These results showed the robustness of MARVE for sensing RNA viruses in clinical and food samples.
Multiplexed detection of SARS-CoV-2 variants by MARVE
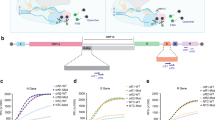
We next challenged MARVE to profile mutations in the SARS-CoV-2 genome. WHO has recommended several of the most critical SARS-CoV-2 variants as variants of concern (VOCs), including the Alpha, Beta, Gamma and Delta variants. The Alpha variant spread to 94 countries within 4 months (ref. 41) and causes sharply increased infectivity, while the Beta and Gamma variants confer a dramatic decrease in vaccine efficacy2. Delta is currently the most widespread variant. Key mutations in VOCs include the single-nucleotide substitutions D614G, N501Y, K417N, K417T, E484K, L452R, T478K and P681R, and the nucleotide deletion Δ69/70, in the S gene (Fig. 4a,b). We synthesized pseudoviruses of wild SARS-CoV-2 and variants with a single mutation (D614G, V367F and R408I), as well as Alpha, Beta, Gamma and Delta variants (Supplementary Fig. 8).

a, V367F, R408I and mutations in VOCs. b, Mutation information of VOCs including Alpha, Beta, Gamma and Delta variants. c, Design and validation pipeline for TEprobes targeting mutations of interest in the SARS-CoV-2 genome. d, Visual detection of mutations listed in a. e, MARVE can distinguish the SARS-CoV-2 D614G variant from wild SARS-CoV-2 at up to a 50-fold molar excess of the variant. The green shadow indicates the range of discrimination factors. The concentration of the SARS-CoV-2 D614G variant was 30,000 copies per μl. The concentration of SARS-CoV-2 pseudovirus was 30,000 copies per μl in d. Data in d and e are mean ± s.d. (n = 3).
We propose a pipeline to guide the design of TEprobes for identifying mutations of interest in the SARS-CoV-2 genome (Fig. 4c). First, a ~30–40 nt domain spanning the mutation of interest is chosen as the target sequence and the TEprobe is designed with toeholds to yield a slightly negative free energy of the strand displacement reaction with an input of mutation-present target sequence (−1 to −2 kcal mol−1) and a positive reaction energy with an input of mutation-absent target sequence20. Next, to facilitate TEprobe binding with the SARS-CoV-2 genome, which forms the secondary structure, 10–15 TEprobes derived from the thermodynamically predicted TEprobe by extending the forward toehold were synthesized (Supplementary Table 5). The optimized TEprobes were experimentally screened by estimating the discrimination factor against the mutation. On the basis of this pipeline, we obtained TEprobes targeting 11 mutations including key mutations in VOCs, as well as V367F and R408I in the S gene (Supplementary Figs. 17–20). The discrimination factor values of each TEprobe against its target mutation were all above 15 (Fig. 4d) and no crosstalk occurred in a test of the D614G mutation (Supplementary Fig. 21) using the optimized TEprobe. This result indicated the feasibility of the pipeline for obtaining specific TEprobes to detect mutations in the SARS-CoV-2 genome. Notably, MARVE retained its high discriminative capacity (a discrimination factor of 11.2) with up to a 50-fold molar excess of wild SARS-CoV-2 (Fig. 4e). Visual detection of the D614G-, V367F- or R408I-mutated SARS-CoV-2 pseudoviruses was further demonstrated using the origami paper (Supplementary Fig. 22).
We envisaged that a streamlined detection of SARS-CoV-2 variants would need to encompass (i) successful sampling, (ii) diagnosis of SARS-CoV-2 infection and (iii) identification of the mutation(s) in SARS-CoV-2. Thus, we designed an origami paper with three functional zones: the Control zone was designed to detect the human RNase P gene, ensuring successful sampling and the validation of reactive detection; the NE zone allowed detection of the N and E genes of SARS-CoV-2; and the Mutation zone was aimed at identifying mutations in SARS-CoV-2. We challenged MARVE to visualize SARS-CoV-2 and its variants (D614G-, V367F- or R408I-mutated) in throat swab, frozen fish and food packaging samples. The results in Extended Data Fig. 4 showed that the presence of SARS-CoV-2 or its variants was detectable via targeting the N gene or the E gene at the NE zone. With a positive result for SARS-CoV-2, variants containing D614G, V367F or R408I mutation were identified. These results demonstrate that MARVE can achieve a specific and multiplexed detection of SARS-CoV-2 variants.
Rapid survey of VOCs will be critical for efficient identification of high-risk and vaccine-ineffective infectors. We therefore quickly established an additional MARVE assay for covering the test of Alpha, Beta, Gamma and Delta variants by expanding the Mutation zone of the origami paper to detect nine mutations in VOCs. The obtained TEprobe for each mutation was used to discriminate wild SARS-CoV-2 and the tested variants (Supplementary Fig. 19). Wild SARS-CoV-2 and the D614G-mutated, Alpha, Beta, Gamma and Delta variants could be identified by the naked eye or by setting a threshold GAR value via smartphone imaging (Fig. 5a). We established the MARVE assay for VOCs within 18 d, demonstrating the power of MARVE to be adapted to the challenge of SARS-CoV-2 evolution (Fig. 5b). Customized optimization of MARVE for emerging SARS-CoV-2 variants can be further shortened to 4 d excluding the synthesis of pseudoviruses. Such short turnaround time and customizability for emerging variants are crucial for supervising SARS-CoV-2 evolution and adapted diagnosis.

a, Detection of and discrimination among Alpha, Beta, Gamma and Delta variants. Design of origami papers (top left), photographs and GAR values of origami papers with the addition of wild SARS-CoV-2 or SARS-CoV-2 variants (bottom), and the identification of mutations in each sample (top right). A GAR value above the red dashed line indicates a positive result. b, MARVE can be customized to detect emerging SARS-CoV-2 variants within 18 d. c, Streamlined detection of SARS-CoV-2 variants. d, Equipment and consumables needed for performing MARVE. e, Streamlined detection protocols achieved detection of 400 copies per μl of SARS-CoV-2 pseudovirus. Data in e are from 2 replicates. The concentration of SARS-CoV-2 pseudovirus was 2,400 copies per μl in a.
To demonstrate the feasibility of screening for SARS-CoV-2 and its variants outside a laboratory setting, we used smartphones to serve as the detector and a thermal lysis strategy24 to obtain viral RNAs (Fig. 5c). Nucleic acid extraction is the bottleneck of nucleic acid tests for point-of-care use. Thermal lysis is one of the most promising strategies for rapid nucleic acid extraction. We optimized the current thermal lysis protocol24 by adding RNA carrier (provided in the E.Z.N.A. viral RNA kit) to release RNAs from virions more efficiently. The process can be performed using a standard heat block within 5 min, eliminating the need for instruments such as centrifuges. We devised an attachment for smartphones that helps to image origami papers using a stable light source (Supplementary Fig. 23) and a smartphone app to guide users to complete the testing of different samples (Supplementary Figs. 24 and 25). The app also permitted the visualization of results, with geographic information supported by the phone’s GPS. The whole out-of-laboratory detection process could be completed within 30 min and requires only a standard heat block, a testing paper, a smartphone and a smartphone attachment (Fig. 5d), while providing the sensitivity to detect 400 copies per μl of SARS-CoV-2 pseudovirus (Fig. 5e and Supplementary Fig. 26). To further ease manual operation and reduce the risk of contamination during detection, we modified the attachment and the origami paper so that paper-folding and the MARVE reaction can be conducted in a closed chamber (Supplementary Fig. 27).
Detection of SARS-CoV-2 and its variants in clinical throat swab samples
Finally, we tested the feasibility of MARVE for detecting SARS-CoV-2 and its variants in clinical throat swab samples. Extracted RNAs from 50 clinical throat swab samples were provided by West China Hospital (Chengdu, China). These samples were collected from persons with suspected SARS-CoV-2 infection in Sichuan Province between 4 April and 23 June 2020. Thirty-five samples were confirmed positive and 15 samples were confirmed negative for SARS-CoV-2 by the hospital on the basis of RT–qPCR results.
The samples were analysed in parallel by an in-house RT–qPCR assay and the MARVE assay. RT–qPCR was performed using US CDC-approved primers for the SARS-CoV-2 N gene and E gene. RT–qPCR measurements resulted in cycle threshold (Ct) values of 25.06–33.37 by targeting the E gene of the positive samples confirmed by West China Hospital, corresponding to concentrations of SARS-CoV-2 ranging from 348 to 156,036 copies per μl (Supplementary Figs. 28 and 29). During this test of MARVE, because the Delta variant was not prevalent, we only made origami papers covering detection of the human RNase P gene, the N gene and the E gene, as well as mutations D614G, N501Y, K417N, K417T, E484K and Δ69/70, aiming to screen wild SARS-CoV-2 and the D614G-mutated, Alpha, Beta and Gamma variants (Fig. 6a and Supplementary Fig. 30). As the volume of RNA samples was limited, paper-based detection of each gene or mutation of SARS-CoV-2 was performed only once. We specified a normalized GAR to quantify the MARVE results, defined as (GARgene – GARnegative)/(GARRNase P – GARnegative), where GARgene, GARnegative and GARRNase P are GAR values of the detection sites for target viral genes or mutations, the negative control and the human RNase P gene, respectively. On the basis of the normalized GAR signal, MARVE showed 100% (35 out of 35) positive predictive agreement and 100% (15 out of 15) negative predictive agreement relative to the RT–qPCR results based on the N gene test, and 88.6% (31 out of 35) positive predictive agreement and 100% (15 out of 15) negative predictive agreement relative to the RT–qPCR results based on the E gene test (Fig. 6b,c and Supplementary Table 6). MARVE failed to positively detect clinical samples 16, 19, 21 and 41 by targeting the E gene. Nevertheless, the simultaneous detection of the N gene and the E gene increased the accuracy of the diagnosis of SARS-CoV-2 infection.

a, Accumulated GARs of tested genes and mutations for each throat swab sample. D614G-mutated, Alpha, Beta and Gamma variants were covered in the detection using MARVE. b, Comparison of detection results by MARVE and RT–qPCR. Ct value of RT–qPCR is indicated as grey circles. Normalized GAR is shown in histogram. Normalized GAR is defined as (GARgene – GARnegative)/(GARRNase P – GARnegative), where GARgene, GARnegative and GARRNase P are GARs of the detection sites for target viral genes or mutations, the negative control and the human RNase P gene, respectively. Thresholds for Ct and normalized GAR values for positive and negative tests (red dashed lines) were obtained on the basis of ROC curves. Samples 1–15 were PCR-negative and 16–50 were PCR-positive. c, ROC accuracy of MARVE for SARS-CoV-2 diagnosis by targeting the N gene or the E gene. d, Detection results of SARS-CoV-2 and its mutations in 50 throat swab samples using MARVE.
We found that clinical samples 25, 30 and 32 carried SARS-CoV-2 with a D614G mutation (Fig. 6d). We sequenced the extracted RNAs from the 3 throat swab samples and validated the presence of the D614G mutation (Supplementary Fig. 31). We did not find N501Y, K417N/T, E484K or Δ69/70 mutations among the tested samples. The samples we tested were obtained before July 2021, during which period the Alpha, Beta and Gamma variants were not frequent. Collectively, we have demonstrated a rapid workflow for two-gene marker (N and E genes) simultaneous determination of SARS-CoV-2 and a screen for multiple SARS-CoV-2 variants with a short sample-to-result time and a low cost (US$0.30 per test) (Supplementary Table 7) using a testing paper and a smartphone, which is highly promising for scalable screening of SARS-CoV-2 variants.
Discussion
Rapid and efficient monitoring of the mutational evolution of SARS-CoV-2 is central to ending the COVID-19 pandemic. Inadequate access to sequencing infrastructures has caused failure of the regular diagnosis of SARS-CoV-2 with mutation information, and also slowed the tracking of viral variants and the acquisition of abundant epidemiological data on the evolution of SARS-CoV-2. Here we show that an assay, termed MARVE, promises to fulfill the need for scalable screening of SARS-CoV-2 variants. MARVE renders the identification of SARS-CoV-2 variants as easy as tests for SARS-CoV-2 infection by moving SARS-CoV-2 variant diagnosis and screening from sequencing centres to out-of-laboratory settings. Thus, it makes abundant testing for SARS-CoV-2 variants possible in both highly developed and resource-limited global regions.
Importantly, MARVE offers short-turnaround-time customization of SARS-CoV-2 variant detection, which is enabled by MARVE’s general and simple probe design, as well as its multiplexing capability. We have demonstrated the establishment of a MARVE assay for the multiplexed profiling of VOCs (Alpha, Beta, Gamma and Delta variants) within 18 d, and the turnaround time can be further shortened by accelerating the synthesis of pseudotyped virus. Shaped by their fitness for replication, transmission and immune escape, SARS-CoV-2 variants will continuously emerge and impact the spread of the virus, and may further compromise the efficacy of existing vaccines27. A short turnaround time for developing customized assays for SARS-CoV-2 variants is imperative to offer an efficient follow-up diagnostic tool for the monitoring of viral evolution and prevalence.
In addition, MARVE features nucleic-acid-amplification-free, extremely low-cost and mobile-device-dependent detection, which, collectively, facilitates population-based mass screening. MARVE dispenses with reverse transcription and nucleic acid amplification processes as it directly detects viral RNA via a urease-based colorimetric readout. The nucleic-acid-amplification-free assaying strategy dramatically reduces the sample-to-result turnaround time (down to 25–30 min) and eliminates the risk of amplicon contamination, which is a serious issue in SARS-CoV-2 diagnosis42. The cheap reagents required, such as DNA probes and urease, make the estimated cost as little as US$0.30 for a single test that covers the N gene, the E gene and five variants: the D614G-mutated, Alpha, Beta, Gamma and Delta variants (Supplementary Table 7). It has been reported that 48.33% of the world’s population currently owns a smartphone43. The wide availability of smartphones is key to the abundant, regular and on-site diagnosis of SARS-CoV-2 and its variants. MARVE, which uses smartphones for sensing, allows for the detection of as few as 400 copies per μl of SARS-CoV-2 RNA. Since the viral load of SARS-CoV-2 in positive swab samples reportedly ranges from 641 copies per ml to 1.34 × 1011 copies per ml (ref. 44), our out-of-laboratory detection protocol based on smartphones can reliably diagnose SARS-CoV-2 infection. Moreover, detection results can be visualized and interpreted by a smartphone app, which could also provide recommendations about quarantine and care procedures according to the diagnostic results.
We note, however, that MARVE cannot find unknown mutations in the SARS-CoV-2 genome. MARVE will probably serve as a complement to sequencing techniques. We envisage that virologists will initially use sequencing and epidemiological data for the detection and confirmation of key mutations and variants that need to be monitored for viral control, followed by the rapid customization of MARVE to fulfill the need to identify these key variants.
This proof-of-concept study for the rapid and multiplexed detection of SARS-CoV-2 variants uses test papers and smartphones. Additional efforts will be necessary to translate this work into a routine tool for testing SARS-CoV-2 variants. First, although the detection of variants has been achieved with the use of pseudoviruses, MARVE has only confirmed positive clinical test samples for D614G-mutated SARS-CoV-2 infection due to the inaccessibility in Sichuan province of clinical samples that are positive for infection with other variants, such as Delta. Cases in which MARVE is used to diagnose other key SARS-CoV-2 variants in clinical samples will further support its applicability as a routine test for SARS-CoV-2 variant diagnosis. Second, the nucleic acid strand-displacement reaction can be predicted, and therefore, in principle, optimized TEprobes can be obtained on the basis of thermodynamic prediction. However, long SARS-CoV-2 RNA of up to ~30,000 bases will result in complex secondary structures, dramatically increasing the difficulty in precisely predicting the strand displacement-based recognition process. Thus, further experimental optimization by tuning the forward toehold of TEprobes may often be needed to obtain a specific probe for identifying SARS-CoV-2 mutations. In addition, MARVE is less sensitive than RT–qPCR partly due to the lack of nucleic acid amplification. The effect of increasing the number of C-Ag(I)-C artificial base pairs in TEprobes showed potential to improve the response and sensitivity of MARVE via introducing an amplification effect in the strand displacement reaction (Extended Data Fig. 5 and Supplementary Note 2). Using urease with an enhanced enzyme activity may also contribute to an improved inhibitory effect derived from Ag(I) ion, in turn improving the response of MARVE towards low-concentration virus.
In the future, cheap, portable and user-friendly viral screening strategies such as MARVE could be quickly modified to target other threatening pathogens such as the dengue and Zika viruses, which mainly afflict resource-constrained developing countries.
Methods
Oligonucleotides
DNA oligonucleotides were purchased from Sangon and purified using PAGE. RNA oligonucleotides were synthesized by Takara and purified by HPLC. Information on DNA and RNA oligonucleotides is given in Supplementary Tables 1 and 2.
Pseudovirus preparation
Pseudoviruses of SARS-CoV-2, MERS-CoV, SARS-CoV and SARS-CoV-2 variants (V367F-mutated, R408I-mutated, D614G-mutated, Alpha, Beta, Gamma and Delta variants) were generated using the lentiviral vector system. SARS-CoV-2, SARS-COV and MERS-CoV contain a truncated fragment of the N gene, the E gene and ORF1ab. SARS-CoV-2 variants contain a truncated fragment of the N gene, the E gene and the S gene. For ORF1ab, the E gene and the N gene, WHO-recommended sequences targeted by RT–qPCR were included in the gene fragment. For the S gene, sequences spanning mutations of interest (that is, to be detected) were contained in the gene fragment (Supplementary Table 8). Pseudoviruses were produced according to a published method with several modifications45. Briefly, the N gene, the E gene and ORF1ab (or the S gene) were cloned into CMV/R vector. psPAX2 vector (5 μg), pMD2.G vector (5 μg) and CMV/R vector carrying SARS-CoV-2, SARS-CoV or MERS-CoV genes (10 μg) were transfected into 3–8 × 106 293T cells using a calcium phosphate transfection kit (Thermo Fisher, K278001). Pseudoviruses were obtained by collecting the supernatants 48 h after transfection and filtering them using a 0.22 μm syringe filter, and removing residual vector DNA with DNase I. The copy number of pseudoviruses was estimated using RT–qPCR and reference plasmids containing the N gene of SARS-CoV-2, SARS-CoV or MERS-CoV. Pseudovirus samples were aliquoted and kept at –80 °C before use. Considering the differences in sample transfer during the detection processes, the concentration of viruses stated in the Article was estimated on the basis of the copy number of viruses and the sample volume used (after RNA extraction) for solution-based or origami paper-based detection (Supplementary Fig. 32).
TEprobe preparation
TEprobes consisted of two single-stranded DNAs, Rec strand and Block strand. They were prepared by incubating 5 μl Rec strand (300 nM), 5 μl Block strand (300 nM), 5 μl AgNO3 (300 nM), 15 μl NaNO3 (0.6 M, pH 6.8) and 20 μl H2O at room temperature for 20 min, and kept at 4 °C.
Measurement of melting temperatures (T m) of TEprobes
Tm of TEprobes was determined using QuantStudio 3 (ABI). EvaGreen (1×) (Biotium, 31000) was added as an indicator of hybridization of TEprobes. The temperature was raised from 35 °C to 95 °C at a rate of 6 °C min−1. Excitation wavelength was 494 nm. Fluorescence intensity was recorded at 521 nm at 5 s intervals.
Analysis of the C-Ag(I)-C artificial base pair using isothermal titration calorimetry (ITC)
ITC experiments were performed at 25 °C using Nano ITC (TA Instruments). TEprobes and AgNO3 were dissolved in 150 mM NaNO3. AgNO3 (1 mM) was titrated with 20 consecutive 5 μl additions into the cell containing 40 μM TEprobes. The titration data were analysed by ITC data processing software (Nano Analyze).
Viral RNA detection in solution
Viral RNAs were detected by mixing 2 μl RNA samples with 10 μl TEprobes for 20 min at room temperature in 96-well plates, followed by the addition of 10 μl urease (10 nM; TCI, U0017, EC 3.5.1.5) and 78 μl colour indicator mixture (10 μl urea (5 M), 10 μl phenol red (2.5 mM) and 58 μl H2O). The enzyme activity of the urease used was 203.13 U mg−1, measured using a urease activity assay kit (Merck, MAK120-1KT). The absorbance of the solution was measured using a Synergy H1 microplate reader (BioTek) and images of the solution were obtained using a HUAWEI P40 smartphone.
Gel electrophoresis was carried out to analyse the DNA products in each step of MARVE, using 5% agarose with Tris-acetate-EDTA buffer and Gelred dye (Biotium, 41001). The final sample volume was 6 μl, containing 5 μl of reaction solution and 1 μl 6× gel loading buffer. Gel electrophoresis was carried out at 120 V for 40 min and the DNA was imaged using UV light with a Gel Doc XR+ system (Bio-Rad).
Viral RNA extraction
Negative throat swab specimens were acquired from healthy donors in the Deng laboratory and deposited in 200 μl 1 M NaNO3 medium. Pseudoviruses or influenza viruses were spiked into throat swab specimens or smeared onto the surface of food packaging and frozen belt fish. Washing buffer (200 μl 1 M NaNO3) was used to collect the virus on food packaging and frozen belt fish. RNA was extracted from the 200 μl samples using the E.Z.N.A. viral RNA kit (Omega, R6874-02). Briefly, 500 μl QVL lysis buffer was added to the sample and incubated at room temperature for 10 min to lyse cells, followed by the addition of 350 μl 100% ethanol. The solutions were transferred into a HiBind RNA mini column and centrifuged at 13,000 g for 15 s. Next, 500 μl VHB buffer was added to the column, which was then centrifuged at 13,000 g for 15 s. Finally, the column was washed twice using Wash Buffer II and the RNA was eluted using 20 μl molecular biology grade H2O (Corning, 46-000-CM).
The thermal lysis strategy used to obtain viral RNAs was conducted following the reported HUDSON assay with a few modifications24. A 20 μl sample was transferred into a centrifuge tube containing 1 μl tris(2-carboxyethyl)phosphine (100 mM), 1 μl EDTA (0.5 mM) and 1 μl RNA carrier (provided in the E.Z.N.A. viral RNA kit). The mixture was heated at 95 °C for 5 min and was then ready for the subsequent MARVE assay.
Preparation of origami paper
Whatman no. 1 filter paper was printed using a Xerox ColorQube 8580/8880N colour printer. The printed origami paper was then placed on a heat block for 10 s at 170 °C, allowing the wax to melt and spread through the paper to form hydrophobic barriers. TEprobes (5 μl, 3 nM), 5 μl urease (1 nM urease in 6 nM pullulan solution) and 5 μl colour indicator mixture (1 μl urea (2.5 M), 1 μl phenol red (1.25 mM) and 3 μl H2O) were separately loaded on page 2, 3 and 4, respectively, of the origami paper. The number of detection sites was designed on demand. Prepared origami papers were kept at 4 °C and used within 1 week.
Paper-based viral RNA detection
Viral RNA was detected on a prepared origami paper. Viral RNA samples (5 μl) were added to page 1 of the origami paper, which was then folded to page 2 (containing TEprobes) to enable contact between RNA samples and TEprobes. After a 20 min incubation at room temperature, page 2 was folded to page 3 (loaded with urease), initiating the binding of the released Ag(I) with urease. Finally, page 3 was folded to contact page 4 (containing urea/phenol red) to indicate the pH change.
Origami papers were imaged using a smartphone and a 3D-printed attachment (30 × 18 × 20 cm), which provided a stable light source for imaging the papers. The detection sites were identified and analysed on the basis of Circle Hough Transform and K-means clustering. A smartphone app on Android 11 was designed to guide the detection process and visualize the diagnostic results.
RT–qPCR testing
Viral RNAs extracted from clinical samples were reverse-transcribed into complementary DNA in a total volume of 20 μl using a RevertAid First Strand cDNA synthesis kit (Thermo Fisher, K1622). First, primers were hybridized by mixing 5 μl RNA sample, 1 μl random hexamer primer and 6 μl H2O and incubating at 65 °C for 5 min. Next, 2 μl deoxynucleoside triphosphates (10 mM), 1 μl RiboLock RNase inhibitor (20 U μl−1), 1 μl RevertAid RT (200 U μl−1) and 4 μl 5× reaction buffer were added and the mixture was incubated sequentially for 5 min at 25 °C, 60 min at 42 °C and 5 min at 70 °C to terminate the reaction. The products were used directly for qPCR procedures with the Platinum SYBR Green qPCR SuperMix-UDG (Thermo Fisher, 11744100). qPCR was carried out on the CFX96 Touch RT-PCR system (Bio-Rad) with the following protocol: 50 °C for 5 min, 95 °C for 5 min, and then 45 cycles of 95 °C for 15 s, 60 °C for 15 s and 72 °C for 45 s. The 20 μl reaction system contained 2 μl of products obtained in the previous step, 10 μl of SYBR Green qPCR Supermix, 1 μl each of forward primer and reverse primers (10 μM) and 6 μl H2O. US CDC-approved primers for the SARS-CoV-2 N gene and E gene were used. The forward and reverse primers for the N gene amplification were 5′-AGTCAAGCCTCTTCTCGTTCCTC-3′ and 5′-CAGCAGCAGATTTCTTAGTGACAGTTT-3′, respectively; those for the E gene amplification were 5′-CTTGCTTTCGTGGTATTCTTGCT-3′ and 5′-TAGACCAGAAGATCAGGAACTCT-3′, respectively.
Clinical samples and ethics statement
Clinical throat swab samples were collected from the West China Hospital of Sichuan University (Ethical Approval no. 2020(100)) and frozen extracted RNAs from these samples were provided by the hospital for MARVE and in-house RT–qPCR testing. RNA samples were thawed on ice, divided into 5 μl aliquots and stored at −80 °C until use. Negative throat swab specimens were acquired from healthy donors in the Deng laboratory. Influenza viruses were kindly provided by Prof. Yi Shi (Institute of Microbiology, Chinese Academy of Sciences, Beijing, China).
Statistics
Data analysis and illustrations were performed using Primer5, SnapGene 3.2.1 and Origin 2019b. Image processing was performed using Matlab 2020b. Welch’s t-test was conducted using SPSS 25.0. Receiver operating characteristic (ROC) curves to assess the diagnostic accuracy of the MARVE assay were produced using GraphPad Prism 8.0.1.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data supporting the results of this study are available within the paper and its Supplementary Information. The raw datasets generated and analysed during the study are available from the corresponding authors on reasonable request. Source data are provided with this paper.
Code availability
The code for image processing is available on GitHub at https://github.com/Nelson233/MARVEL.
References
Garcia-Beltran, W. F. et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 184, 2372–2383 (2021).
Fontanet, A. et al. SARS-CoV-2 variants and ending the COVID-19 pandemic. Lancet 397, 952–954 (2021).
Gupta, R. K. Will SARS-CoV-2 variants of concern affect the promise of vaccines? Nat. Rev. Immunol. 21, 340–341 (2021).
Altmann, D. M., Boyton, R. J. & Beale, R. Immunity to SARS-CoV-2 variants of concern. Science 371, 1103–1104 (2021).
Abdool Karim, S. S. & de Oliveira, T. New SARS-CoV-2 variants-clinical, public health, and vaccine implications. N. Engl. J. Med. 384, 1866–1868 (2021).
Davies, N. G. et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 372, eabg3055 (2021).
Davies, N. G. et al. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature 593, 270–274 (2021).
Madhi, S. A. et al. Efficacy of the ChAdOx1 nCoV-19 Covid-19 vaccine against the B.1.351 variant. N. Engl. J. Med. 384, 1885–1898 (2021).
Cohen J. South Africa suspends use of AstraZeneca’s COVID-19 vaccine after it fails to clearly stop virus variant. Science https://doi.org/10.1126/science.abg9559 (2021).
Wadman M., Cohen J. Novavax vaccine delivers 89% efficacy against COVID-19 in U.K.–but is less potent in South Africa. Science https://doi.org/10.1126/science.abg8101 (2021).
Hacisuleyman, E. et al. Vaccine breakthrough infections with SARS-CoV-2 variants. N. Engl. J. Med. 384, 2212–2218 (2021).
Vaidyanathan, G. Coronavirus variants are spreading in India–what scientists know so far. Nature 593, 321–322 (2021).
Carter, L. J. et al. Assay techniques and test development for COVID-19 diagnosis. ACS Cent. Sci. 6, 591–605 (2020).
Zhang, C. et al. Rapid one-pot detection of SARS-CoV-2 based on a lateral flow assay in clinical samples. Anal. Chem. 93, 3325–3330 (2021).
Wu, Q. et al. INSIGHT: a population-scale COVID-19 testing strategy combining point-of-care diagnosis with centralized high-throughput sequencing. Sci. Adv. 7, eabe5054 (2021).
Broughton, J. P. et al. CRISPR–Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 38, 870–874 (2020).
Joung, J. et al. Detection of SARS-CoV-2 with SHERLOCK one-pot testing. N. Engl. J. Med. 383, 1492–1494 (2020).
Fozouni, P. et al. Amplification-free detection of SARS-CoV-2 with CRISPR-Cas13a and mobile phone microscopy. Cell 184, 323–333 (2021).
Ackerman, C. M. et al. Massively multiplexed nucleic acid detection with Cas13. Nature 582, 277–282 (2020).
Hong, F. et al. Precise and programmable detection of mutations using ultraspecific riboregulators. Cell 180, 1018–1032 (2020).
Wang, J. S. & Zhang, D. Y. Simulation-guided DNA probe design for consistently ultraspecific hybridization. Nat. Chem. 7, 545–553 (2015).
Larsson, C., Grundberg, I., Söderberg, O. & Nilsson, M. In situ detection and genotyping of individual mRNA molecules. Nat. Methods 7, 395–397 (2010).
Abi, A. & Safavi, A. Targeted detection of single-nucleotide variations: progress and promise. ACS Sens. 4, 792–807 (2019).
Myhrvold, C. et al. Field-deployable viral diagnostics using CRISPR-Cas13. Science 360, 444–448 (2018).
de Puig, H. et al. Minimally instrumented SHERLOCK (miSHERLOCK) for CRISPR-based point-of-care diagnosis of SARS-CoV-2 and emerging variants. Sci. Adv. 7, eabh2944 (2021).
Freije, C. A. & Sabeti, P. C. Detect and destroy: CRISPR-based technologies for the response against viruses. Cell Host Microbe 29, 689–703 (2021).
Plante, J. A. et al. The variant gambit: COVID-19’s next move. Cell Host Microbe 29, 508–515 (2021).
Johannsen, S. et al. Solution structure of a DNA double helix with consecutive metal-mediated base pairs. Nat. Chem. 2, 229–234 (2010).
Kondo, J. et al. High-resolution crystal structure of a silver(I)–RNA hybrid duplex containing Watson–Crick-like C–silver(I)–C metallo-base pairs. Angew. Chem. Int. Ed. 54, 13323–13326 (2015).
Ono, A. et al. Specific interactions between silver(I) ions and cytosine–cytosine pairs in DNA duplexes. Chem. Commun. 39, 4825–4827 (2008).
Ambrose, J. F., Kistiakowsky, G. B. & Kridl, A. G. Inhibition of urease by silver ions. J. Am. Chem. Soc. 73, 1232–1236 (1951).
Dao Thi, V. L. et al. A colorimetric RT-LAMP assay and LAMP-sequencing for detecting SARS-CoV-2 RNA in clinical samples. Sci. Transl. Med. 12, eabc7075 (2020).
Tanner, N. A., Zhang, Y. & Evans, T. C. Jr Visual detection of isothermal nucleic acid amplification using pH-sensitive dyes. BioTechniques 58, 59–68 (2015).
Jenkins, G. N. The Physiology and Biochemistry of the Mouth (Blackwell Scientific Publications,1978).
Serra, M. J. & Turner, D. H. Predicting thermodynamic properties of RNA. Methods Enzymol. 259, 242–261 (1995).
Reboud, J. et al. Paper-based microfluidics for DNA diagnostics of malaria in low resource underserved rural communities. Proc. Natl Acad. Sci. USA 116, 4834–4842 (2019).
Jahanshahi-Anbuhi, S. et al. Pullulan encapsulation of labile biomolecules to give stable bioassay tablets. Angew. Chem. Int. Ed. 53, 6155–6158 (2014).
Weissleder, R., Lee, H., Ko, J. & Pittet, M. J. COVID-19 diagnostics in context. Sci. Transl. Med. 12, eabc1931 (2020).
Larsen, D. A. & Wigginton, K. R. Tracking COVID-19 with wastewater. Nat. Biotechnol. 38, 1151–1153 (2020).
Yao, L. et al. Detection of coronavirus in environmental surveillance and risk monitoring for pandemic control. Chem. Soc. Rev. 50, 3656–3676 (2021).
Collier, D. A. et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature 593, 136–141 (2021).
Robinson-McCarthy, L. R. et al. Anomalous COVID-19 tests hinder researchers. Science 371, 244–245 (2021).
How Many Smartphones are in the World? (BankMyCell); https://www.bankmycell.com/blog/how-many-phones-are-in-the-world
Pan, Y. et al. Viral load of SARS-CoV-2 in clinical samples. Lancet Infect. Dis. 20, 411–412 (2020).
Yang, Z.-y et al. Evasion of antibody neutralization in emerging severe acute respiratory syndrome coronaviruses. Proc. Natl Acad. Sci. USA 102, 797–801 (2005).
Acknowledgements
This work was funded by the National Natural Science Foundation of China (No. 22034004, No. 21621003, No. 22074100), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB36000000), Tsinghua University Spring Breeze Fund (No. 2020Z99CFZ019) and the Sichuan Science and Technology Program (2021YFS0403, 2019YFS0003).
Author information
Authors and Affiliations
Contributions
R.D., J.L. and W.L. conceived the project. R.D., T.Z., J.L., Y.W. and W.L. designed the experiments. T.Z., R.D., Y. W., C. Wu, T.X., Y.Z. and Y.H. performed the experiments. R.D., T.Z., K.Z., X.T., C. Wang, C. Wu, R.L.-A., C.Y., N.G. and J.L. analysed the results. J.L., R.D., T.Z., K.Z., R.L.-A. and Q.H. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Biomedical Engineering thanks Feng Xu, Yurong Yan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Electrophoretic analysis of the strand displacement reaction using TEprobes with different reverse toeholds.
a, Schematic illustration of the strand displacement reaction using TEprobes with different reverse toeholds. b, Sequences of TEprobes. c, Electrophoretic images of the products of the strand displacement reaction. Concentrations of TEprobe and the target RNA were both 500 nM. The strand displacement reaction was proceeded for 20 min. The strand displacement reaction was hindered by increasing the reverse toehold length up to 9 nt.
Extended Data Fig. 2 Design of TEprobes for discriminating SARS-CoV-2, SARS-CoV and MERS-CoV.
a, Target sequences on the N gene of SARS-CoV-2, SARS-CoV and MERS-CoV. Nucleotides in SARS-CoV and MERS-CoV differing from SARS-CoV-2 are marked in red. b, The TEprobe with a 7-nt reverse toehold and an 8-nt forward toehold cannot sufficiently respond to the target virus, SASR-CoV-2. c, TEprobes with extended forward toehold lengths up to 12 nt, 15 nt, 18 nt, 20 nt and 25 nt. d, Absorbance at 560 nm and visual result using TEprobes targeting SARS-CoV-2 in the presence of SARS-CoV-2, SARS-CoV or MERS-CoV. e, Discrimination factors to distinguish SARS-CoV-2 from SARS-CoV and MERS-CoV using TEprobes in c. Forward toehold up to 15 nt can support a sufficient color change response to the target virus, SARS-CoV-2. And a further increase of forward toehold reduced the discrimination factors due to the elevated color change induced by the non-target virus, SARS-CoV. f, Detection of SARS-CoV-2, SARS-CoV or MERS-CoV using its cognate TEprobe. g, Discrimination factor to distinguish SARS-CoV-2, SARS-CoV and MERS-CoV. Concentrations of urea, phenol red, urease and TEprobes were 500 mM, 250 μM, 1 nM and 3 nM, respectively. The measurement of absorbance was proceeded after 20-min urea cleavage reaction. The concentration of viruses was 30,000 copies/μl. Date in b and f are mean ± s.d. (n = 3).
Extended Data Fig. 3 Visual detection of influenza viruses in throat swab, food packaging and frozen belt fish samples using MARVE.
a, Origami papers designed to detect SARS-CoV-2, H1N1, H9N2 and H7N9. b, Photographs of origami papers detecting SARS-CoV-2 and influenza viruses in throat swab, food packaging, and frozen belt fish samples. c, GAR values of images in b. 5 μl SARS-CoV-2 (2,400 copies/μl) was dropped on page 1 of the origami paper. 5 μl TEprobes (3 nM), 5 μl urease (1 nM urease in 6 nM pullulan solution) and 5 μl colour indicator mixture (1 μl urea (2.5 M), 1 μl phenol red (1.25 mM) and 3 μl H2O) were separately loaded on page 2, 3 and 4, respectively, of the origami paper.
Extended Data Fig. 4 Origami papers for multiplexed detection of R408I-, D614G- or V367F-mutated SARS-CoV-2 variants in throat swab, food packaging and frozen belt fish samples.
a, Design of origami papers. b, Photographs of origami papers testing different samples. c, GAR value of each gene or mutation tested in b. The concentrations of R408I-muated, D614G-muated, and V367F-muated SARS-CoV-2 variants were all 2,400 copies/μl. 5 μl extracted viral RNA was added on the origami paper. 5 μl TEprobes (3 nM), 5 μl urease (1 nM urease in 6 nM pullulan solution) and 5 μl colour indicator mixture (1 μl urea (2.5 M), 1 μl phenol red (1.25 mM) and 3 μl H2O) were used for the colorimetric detection.
Extended Data Fig. 5 Investigation of the effect of the number of C-Ag(I)-C artificial base pairs in TEprobes on the response of MARVE towards target RNA.
a, Absorbance at 560 nm in the presence of 1 nM target RNA and absence of target RNA (upper), and the ratio of background (in the absence of target RNA) to signal (in the presence of 1 nM target RNA) using TEprobes with 1, 2 and 3 C-Ag(I)-C artificial base pairs (bottom). b, Sequences of TEprobes with 1, 2 or 3 C-Ag(I)-C artificial base pairs. c, Evaluation of the discrimination capacity of TEprobes that anchored multiple C-Ag(I) ion-C artificial base pairs. The TEprobes were designed with forward toehold length ranged from 9 nt to 15 nt. The reverse toehold length of all TEprobes was 7 nt. Data in a and c are mean ± s.d. (n = 3). Concentrations of urea, phenol red, urease and TEprobes were 500 mM, 250 μM, 1 nM and 3 nM, respectively. The measurement of absorbance was proceeded after 20-min urea cleavage reaction.
Supplementary information
Supplementary Information
Supplementary notes, figures, tables and references.
Source data
Source Data for Fig. 2
Source data and unprocessed gel.
Source Data for Fig. 3
Source data.
Source Data for Fig. 4
Source data.
Source Data for Fig. 5
Source data.
Source Data for Fig. 6
Source data.
Source Data for Extended Data Fig. 1
Unprocessed gel.
Source Data for Extended Data Fig. 2
Source data.
Source Data for Extended Data Fig. 3
Source data.
Source Data for Extended Data Fig. 4
Source data.
Source Data for Extended Data Fig. 5
Source data.
Rights and permissions
About this article

Cite this article
Zhang, T., Deng, R., Wang, Y. et al. A paper-based assay for the colorimetric detection of SARS-CoV-2 variants at single-nucleotide resolution. Nat. Biomed. Eng 6, 957–967 (2022). https://doi.org/10.1038/s41551-022-00907-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41551-022-00907-0
This article is cited by
-
Long-term monitoring of ultratrace nucleic acids using tetrahedral nanostructure-based NgAgo on wearable microneedles
Nature Communications (2024)
-
Rapid and on-site wireless immunoassay of respiratory virus aerosols via hydrogel-modulated resonators
Nature Communications (2024)
-
Biosensors based on potent miniprotein binder for sensitive testing of SARS-CoV‑2 variants of concern
Microchimica Acta (2024)
-
A one-pot isothermal Cas12-based assay for the sensitive detection of microRNAs
Nature Biomedical Engineering (2023)
-
Multiplexed discrimination of SARS-CoV-2 variants via plasmonic-enhanced fluorescence in a portable and automated device
Nature Biomedical Engineering (2023)
