
Abstract
Staphylococcus aureus is a high-priority pathogen causing severe infections with high morbidity and mortality worldwide. Many S. aureus strains are methicillin-resistant (MRSA) or even multi-drug resistant. It is one of the most successful and prominent modern pathogens. An effective fight against S. aureus infections requires novel targets for antimicrobial and antistaphylococcal therapies. Recent advances in whole-genome sequencing and high-throughput techniques facilitate the generation of genome-scale metabolic models (GEMs). Among the multiple applications of GEMs is drug-targeting in pathogens. Hence, comprehensive and predictive metabolic reconstructions of S. aureus could facilitate the identification of novel targets for antimicrobial therapies. This review aims at giving an overview of all available GEMs of multiple S. aureus strains. We downloaded all 114 available GEMs of S. aureus for further analysis. The scope of each model was evaluated, including the number of reactions, metabolites, and genes. Furthermore, all models were quality-controlled using MEMOTE, an open-source application with standardized metabolic tests. Growth capabilities and model similarities were examined. This review should lead as a guide for choosing the appropriate GEM for a given research question. With the information about the availability, the format, and the strengths and potentials of each model, one can either choose an existing model or combine several models to create models with even higher predictive values. This facilitates model-driven discoveries of novel antimicrobial targets to fight multi-drug resistant S. aureus strains.
Similar content being viewed by others

Introduction
Staphylococcus aureus is an opportunistic pathogen that asymptomatically and permanently colonizes the nose of up to one third of the human population1. It is a commensal of the mucosae and the human skin, but can also cause severe infections with high morbidity, mortality, and healthcare-associated costs2. Methicillin-resistant S. aureus (MRSA) is one of the most successful modern pathogens3. In 2017, the WHO published a priority pathogens list for the research and development of new antibiotics. Among the clarithromycin-resistant Helicobater pylori and the vancomycin-resistant Enterococcus faecium, S. aureus, especially the methicillin-resistant S. aureus (MRSA), vancomycin intermediate (VISA), and vancomycin resistant strains (VRSA), are high priority pathogens4.
Staphylococcus aureus bacteremia (SAB) is a common infection5. The incidence rate ranges from approximately 20 cases per 100,000 persons per year in Canada6 to approximately 50 cases per 100,000 persons, inferred from the United States surveillance data7. The higher incidence rate might be due to the greater burden of MRSA5. SAB can be classified into three categories: (1) Hospital onset of health-care associated infections, e.g., nosocomial; (2) Community onset of health-care associated infections, and (3) community acquired infections8. Besides SAB, S. aureus, and especially MRSA, is the leading cause of endocarditis, bone and joint infections, skin and soft tissue infections, and further hospital-acquired infections3. A study from 2013 revealed over 80,000 invasive infections and 11,000 deaths per year due to MRSA in the United States. Compared to the previous years, the number of invasive MRSA infections declined slightly9. Unfortunately, the rate decline of MRSA infections has recently slowed down according to the “Morbidity and Mortality Weekly Report” of the Centers for Disease Control and Prevention10, while the number of methicillin-susceptible S. aureus (MSSA) bloodstream infections even slightly increased. In 2017, nearly 120,000 S. aureus bloodstream infections and 20,000 associated deaths occurred in the United States10. Hence, strategies for preventing infections inside and outside acute care settings are required to further reduce the amount of invasive MRSA infections.
The transmission of S. aureus in general, and MRSA in particular, is facilitate by the long persistence time of S. aureus colonization. Nearly any item with skin contact can serve as fomes. In a hospital setting, this can include coats and clothes from doctors and nursing staff, pens, and mobile devices, such as cell phones3. Studies also suggest that infecting S. aureus isolates also persist in households three months after skin infections11. Even across and within athletic fitness facilities, S. aureus is found on different surfaces, including weight plates and treadmill handles12.
Besides the challenge of controlling S. aureus colonization in multiple environments, S. aureus strains evolve and adapt to different environments due to variability in diversity, mobile genetic elements (MGEs), and accumulation of mutations13,14,15. Mediators of virulence, immune evasion, and antibiotic resistance are commonly found within the accessory components of the S. aureus genomes, consisting of MGEs with pathogenicity islands, chromosomal cassettes, transposons, plasmids, and bacteriophages. Compared to the core genome, the accessory genome is more variable and also often more strain-specific3. MGEs in S. aureus can carry antibiotic resistance genes for resistances against penicillin, trimethoprim, erythromycin, clindamycin, and tetracyclines15. However, strains not only evolve and develop antibiotic resistances, they even replace each other within the same host14.
To fight S. aureus infections, several new antimicrobial and antistaphylococcal drugs have been developed recently3,13, including oritavancin and ceftaroline16,17. Despite the development of new antibiotics, S. aureus in general, and MRSA in particular, remains a prominent pathogen with persisting high mortality3. Since S. aureus will continue to evolve and develop new resistances13, the research on S. aureus and the development of new antimicrobials is of urgency to fight S. aureus infections.
One possibility for the identification of novel targets for antimicrobial therapies is the use of genome-scale metabolic models (GEMs). Advances in high-throughput techniques and whole-genome sequencing facilitate the construction of GEMs18,19. They are reconstructed based on information from genome sequences and experimentally obtained biochemistry19,20. With this information, stoichiometry-based and mass-balanced metabolic reactions can be formulated using gene-protein-reaction associations (GPRs). These stoichiometry-based GEMs can predict metabolic flux values within the constructed network21 and optimization techniques. Optimization techniques, such as flux balance analysis (FBA), use linear programming20. Recent advances in the reconstruction of GEMs and the fast analysis and integration of omics data enabled metabolic studies with model-driven hypotheses and context-specific simulations22,23. Among the multiple applications of GEMs is the drug targeting in pathogens and the modeling of interactions among multiple cells or organisms20. These approaches could be used to investigate and develop novel antimicrobials or antistaphylococcals. However, depending on the pathogen and strain, various models of S. aureus strains might be required to investigate the best antistaphylococcal target for a certain S. aureus strain.
In this review, we present all currently available GEMs of S. aureus from various databases. The available models were compared regarding their scope, their availability, their format, and their immediate usability. For various reasons, some of the models required revisions, such as converting spreadsheet file formats to SBML24 or ensuring the syntactic validity of SBML files. After having all models available as syntactically valid SBML files, their growth-capabilities, their predictive value, and the similarities between the various models were investigated. This review gives an overview of the available models and their properties to identify the appropriate model for a specific research question.
Model overview
Introduction of the models
Databases such as BiGG25 or BioModels26 comprise a variety of genome-scale metabolic models. Together with models from other databases and supplementary information from scientific publications, a large number of genome-scale metabolic models of S. aureus is available: The BioModels database contains two models of S. aureus by Becker et al.27 and Heinemann et al.28, both build in 2005. The BioModels database also harbors the models created within the Path2Models project29. In this project, 33 whole genome metabolism models of S. aureus were automatically created and curated between 2012 and 201329. The BiGG Models Database contains two GEMs of S. aureus: the already mentioned model by Becker et al.27 and a recently published model by Seif et al.30 from 2019. Lee et al. published thirteen genome-scale metabolic reconstructions of multiple Staphylococcus aureus strains in 200931. In 2016, Bosi et al.32 curated and published 64 genome-scale metabolic models of various S. aureus strains. Together with the S. aureus model published within the gut microbiota resource of the Virtual Metabolic Human (VMH) Database33,34, a total number of 114 genome-scale metabolic models of Staphylococcus aureus exists today.
All models were downloaded, tested, and evaluated using COBRApy35 and MEMOTE36. MEMOTE is an open-source software that contains a standardized and community-maintained set of metabolic model tests36. The overall MEMOTE score comprises information about annotations of metabolites, reactions, and genes, the inclusion of Systems Biology Ontology (SBO) terms, and the model’s consistency. Within the annotations sections, the presence and conformity of different database identifiers is evaluated. In the SBO term section, the annotation of model instances with appropriate SBO terms is assessed. The model consistency check comprises tests to evaluate the stoichiometric consistency, mass and charge balances, metabolite connectivity, and unbounded fluxes in default medium36. However, the MEMOTE score currently does not consider information about e.g., realistic growth rates, orphan or dead-end metabolites, stoichiometrically balanced cycles, or duplicated reactions. MEMOTE includes this information in its report but does not incorporate it into the calculated score. The number of model instances and their MEMOTE score are indicated in Fig. 1.

For all models, the number of reactions, metabolites, and genes in the model is illustrated. MEMOTE conducts standardized and community-maintained metabolic tests for quality control and quality assurance of genome-scale metabolic models (GEMs) and assigns the tested model a score ranging from 0 to 100%. Lee et al.31, Bosi et al.32, and the Path2Models Project29 published a collection of different S. aureus models. For the collections, the mean number of model instances is shown and the error bar indicates the standard deviation (s.d.).
iSB619—GEM by Becker et al.
The first, initial draft of an S. aureus genome-scale reconstruction was curated by Becker and Palsson in 2005. They reconstructed the S. aureus strain N315 with 619 genes, 743 reactions, and 655 metabolites. The GEM was curated based on the key metabolic pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database37. Subsequently, The Institute for Genomic Research (TIGR) website38 was browsed for additional reactions. 91% of all reactions are linked with genes or open reading frames in so-called gene-protein-reaction associations (GPRs). This first-draft GEM is almost completely elementally and charge balanced. The biomass objective function was formulated based on the biomass data from Bacillus subtilis39 and substituted where necessary. It contains metabolites, such as amino acids, nucleotides, lipids, and cell wall constituents27. The first S. aureus GEM reached a MEMOTE score of 67% and is available as a file in SBML Level 3 Version 140 format with flux balance constraints (fbc) extension41 and BiGG identifiers.
iMH551—GEM by Heinemann et al.
In the same year, the second genome-scale reconstruction of S. aureus was published by Heinemann et al. Both research groups curated the S. aureus strain N315 and used the KEGG37 and TIGR database38, together with literature for genome regions with limited sequence homology for gene function assignments. A new biomass objective function was specifically defined for S. aureus based on integration of literature data from a variety of different S. aureus strains. The biomass objective function was build upon the five polymer categories DNA, RNA, proteins, lipids, and cell wall components, and extended by pool solutes. The reconstruction includes 801 metabolites and 860 reactions that are based on 551 genes and simulates aerobic and anaerobic growth28. This S. aureus GEM reached a MEMOTE score of 35% and is also available as SBML Level 3 file with fbc extension. The genes are not included in the SBML file.
GEMs by Lee et al.
Lee et al. utilized the ERGOTM bioinformatics suite42 and the KEGG ligand/reaction database37 to curate metabolic reconstructions of multiple S. aureus genomes. The DNA sequence and associated open reading frames (ORFs) or protein sequences were integrated into the ERGO genome database. ORFs were called via a combination of programs and annotated automatically or manually. BLAST was used to compute the protein similarities. Functional assignments, relationship computation, and pathway analyses based on existence of ortholog and protein family clusters led to automated metabolic reconstructions. Manual steps included the review of every gene in the genome, pathway curations, and the consideration and reconciliation of motif/domain database results for functional assignments. For identified missing steps within a certain pathway, Lee et al. searched for orthologs or published biochemical activities. For all complete, incomplete, or partial EC number annotations, associated reactions were identified via the ERGO pathway collections and KEGG database. Lee et al. used both biomass compositions from Becker et al.27 and Heinemann et al.28 for their analyses. On average, the thirteen S. aureus reconstructions included 1476 ± 14 reactions and 1406 ± 11 metabolites. All models are available as Excel spreadsheet files with KEGG identifiers.
GEMs from Path2Models
More than 140,000 freely available and automatically generated mathematical models from pathway representations are available through the Path2Models project. KEGG37, BioCharta43, MetaCyc44, and SABIO-RK45 served as databases to generate three types of models, including genome-scale metabolic reconstructions. The pipeline for generating GEMs starts with the extraction of pathway data from KEGG37 and MetaCyc44. To reconcile the different metabolite and reaction identifiers, MNXref was used46. MNXref was further used to define default metabolite formulas and charge states. It allowed the mapping to different databases for a semantical annotation in accordance with the Minimal Information Required In the Annotation of Models (MIRIAM) guidelines47. To all GEMs, a default biomass objective function containing all 20 amino acids, RNA and DNA nucleotide precursors, glycogen, and ATP was added. Between 2012 and 2013, 33 S. aureus GEMs were curated with the help of this pipeline, including one bovine strain. This strain had 6110 reactions, 4416 metabolites, and 1198 genes. The other S. aureus GEMs have on average 3064 ± 103 reactions, 2186 ± 75 metabolites, and 519 ± 12 genes. All models have a MEMOTE score of 48% and are available at the BioModels database as SBML Level 2 files48 with mixed nomenclature.
GEMs by Bosi et al.
In 2016, Bosi et al. constructed 64 GEMs of different S. aureus strains. They started by extending and adding content from KEGG37, Model SEED49, and MetaCyc44 to the S. aureus N315 model iSB619 by Becker et al. This manually curated model was used as reference for other S. aureus strains. Shared genes and reactions were identified and subsequently, strain-specific metabolic content available from KEGG37, Model SEED49, and BioCyc50 was manually added to the strain-specific GEMs. Since an S. aureus biomass composition was not available, the biomass objective functions from Becker et al.27 and Heinemann et al.28 were combined and S. aureus-specific data regarding the fatty acid composition in the biomass were used to adjust the biomass objective function. A gap-filling step further refined the models. On average, the models have 1460 ± 94 reactions, 1446 ± 47 metabolites, and 788 ± 116 genes with an average MEMOTE score of 36 ± 1%. All models are available as SBML Level 3 files51 with fbc extension and BiGG nomenclature.
GEM by Magnúsdóttir et al.
To elucidate the role of microbial communities in human metabolism and health, Magnúsdóttir et al. semi-automatically generated genome-scale metabolic reconstructions of 773 human gut bacteria, including S. aureus USA300-FPR375733. By using a comparative metabolic reconstruction method that propagates refinements from one metabolic reconstruction to others, the model quality of all 773 models was improved. The basis for each reconstruction were draft GEMs from Model SEED49 and KBase52 including gap-filling, refinement via rBioNet53, and quality control and quality assurance testing. Further refinement steps included the verification of reaction directionalities as well as mass and charge imbalances. The reconstructions were extended by gut-microbiota specific subsystems and central metabolic subsystems, and anaerobic growth was enabled. Leak tests and the removal of infeasible flux loops further refined the model. The S. aureus model contains 1403 reactions, 1193 metabolites, and 859 genes, and reached a MEMOTE score of 45%. It is available as SBML Level 3 file with fbc extension and VMH nomenclature.
iYS854—GEM by Seif et al.
Seif et al. manually reconstructed a comprehensive genome-scale metabolic model of S. aureus USA300 str. JE2 containing 886 genes, 1455 reactions, 1335 metabolites, and 673 three-dimensional protein structures. The GEM was build upon one of the reconstructions of Bosi et al.32. Extensive and detailed manual curation was supported by literature reviews and network evaluations. The initial model was extended by an updated biomass objective function. Model instances, such as genes, reactions, and metabolites, were enriched with cross-references and metadata. More than 50 metabolic sub-modules were examined, curated, and added to the GEM, together with over 200 confidence scores and 300 references. By this manual curation, 569 new metabolic processes, 214 new ORF assignments and 207 new metabolites were added. Experimental validation of the model revealed an 85% agreement with gene essentiality data and 68% agreement with experimental physiological data30. A model evaluation with MEMOTE revealed with 81% the highest MEMOTE score of all tested models. The model is available as SBML Level 3 file with fbc extension and BiGG identifiers.
Presence of strains
The 114 currently available GEMs divide into 65 different S. aureus strains. In Fig. 2, the diverse S. aureus strains and their occurrence in the different publications is illustrated. Some strains, such as USA300-FPR3757 or N315 occur several times in different databases. Others, like the GEM for S. aureus strain JE2 occur only once in literature so far. The colors indicate the metabolite and reaction identifier in the respective model. Among the five models of the strain N315, two models exist that both carry BiGG identifiers. Models with same identifiers can be compared more easily than models with discriminating identifiers. Thirteen S. aureus strains occur at least in three different databases or publications with varying identifiers.

Sixty-five different strains of S. aureus are available over the seven publications. Some S. aureus strains, such as the S. aureus strain USA300-FPR3757, occur in several publications, for other strains, only one publication is available. The colors indicate the utilized metabolite and reaction identifiers in the respective models. Models with similar or same identifiers can be compared more easily.
Due to the vast amount of different S. aureus strains, we elucidate only the strains that are shared over multiple databases. As already mentioned, the GEMs of the S. aureus strain N315 are the most prevalent. This strain was isolated from the pharyngeal smear of a Japanese patient in 198254. It is a methicillin-resistant S. aureus (MRSA). The only effective antibiotic against it was vancomycin. However, in 1997, a vancomycin-resistant MRSA strain, Mu50, was discovered in a Japanese infant with a surgical wound infection54. The closely related strain Mu3 is a hetero vancomycin-intermediate MRSA strain. Strains with heterogeneous vancomycin resistance can spontaneously produce cells with increasing resistance against vancomycin55,56.
The isolates JH1 and JH9 stem from a series of MRSA isolates obtained from a patient receiving extensive therapy. These strains are also vancomycin-intermediate S. aureus. The first isolate, JH1, was taken before the chemotherapy and was fully susceptible to vancomycin. The last isolate, JH9, from the end of the therapy showed decreased susceptibility to vancomycin57.
The S. aureus strains of type USA300 are clones of the community-acquired MRSA58,59. It causes invasive infections in children and adults in the USA58, but also in Canada and Europe59. It is suggested that USA300 is more virulent than other community-acquired MRSA strains58. FPR3757 is a multidrug-resistant USA300 strain with acquired mobile genetic elements (MGEs) encoding resistance and virulence determinant that probably lead to enhanced pathogenicity59. The other USA300 isolate, TCH1516, also named USA300-HOU-MR, was isolated at the Texas Children’s Hospital in 2007. Significant differences to other MRSA strains lie within the plasmid content and the antibiotic susceptibility profiles58.
MW2 is another community-acquired MRSA isolate. It carries a wide range of virulence and resistance genes60. At the moment, more than fifteen different pathogenicity islands are identified in S. aureus. Interestingly, MW2 contains almost the same complement of pathogenicity islands as USA300-TCH151658. In contrast, the S. aureus strain COL contains six pathogenicity islands, such as Mu50, but in different combinations58. COL is one of the first MRSA isolates from the early 1960s. It is a penicillinase-negative strain61,62. In contrast to the highly virulent MW2 strain, where virulence factors are found outside of prophages, fewer virulence factors are found outside of prophages in S. aureus strain Newman. This strain carries four integrated prophages and two large pathogenicity islands with important contributions for the pathogenesis. This S. aureus strain is susceptible to methicillin63.
As the Newman strain, the S. aureus isolate MSSA476 is a methicillin-susceptible clone. It is a community-acquired strain, such as MW2. It was isolated in 1998 and susceptible to most commonly used antibiotics, excluding penicillin and fusidic acid64. In contrast MRSA252 is a clinically important hospital-acquired MRSA lineage. It is genetically diverse to other S. aureus strains64.
S. aureus does not only infect humans, it is also the cause of a mastitis in cattle. Strain RF122 contains genomic features that distinguish the human and the bovine pathogens65.
Eight different S. aureus isolates belong to the South German clone lineage ST228. This clone spread over 10 years in a hospital in Switzerland. The isolates were collected between 2001 and 2008. The eight isolates represent the evolutionary history of the clone. As many others, it is an MRSA66.
Model improvements
A variety of different S. aureus models from various strains is available. However, not all 114 downloaded S. aureus models were of the same quality: Some SBML files were syntactically invalid, others utilized an older SBML format, or were not available as SBML file at all. To provide a collection of usable and updated SBML models, we performed debugging and/or improvement steps on some of the models. Models with valid SBML files of the latest level were not improved. All debugging and improvement steps served the purpose of standardizing and annotating the models. No content changes were performed that affect model calculations.
GEMs by Bosi et al.
The 64 S. aureus models by Bosi et al.32 were downloaded and evaluated using COBRApy35. The built-in validity check for SBML files returned a number of errors. In a first step, a pipeline for debugging the errors was created. All files lacked the XML declaration, which was added together with the XML version number and the encoding attribute. According to the SBML language specifications, metabolite, reaction, and model identifier need to fulfill certain properties67, e.g., model identifiers cannot start with a number. The identifiers were adapted according to the guidelines. The downloaded SBML file contained an empty compartment list, which was filled with the compartments during the debugging. As the compartment list comprises all cellular compartments in which metabolites and reactions occur, the different compartments were extracted from the metabolites’ information and subsequently incorporated into the compartment list. The charges, chemical formulas, and compartments of the models’ metabolites were adapted or added, where necessary. After these debugging steps, the models were exported as valid SBML files and evaluated with MEMOTE. The MEMOTE score of 36% in Table 1 is the score after these debugging steps, since MEMOTE requires a syntactically valid SBML file as input.
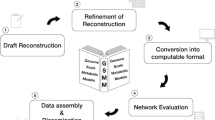
Since a pipeline for altering all 64 S. aureus GEMs already existed, we added further steps to the pipeline to extend the models with respect to their annotations. With the use of the Systems Biology Ontology (SBO), semantic information about model components can be provided. This information allows an explicit and unambiguous understanding of the components’ meaning68. For the model genes and metabolites, appropriate SBO terms were defined. Reactions were divided into metabolic and transport reactions, each receiving different SBO terms. Transport reactions were even further refined to active, passive, or co-transport with antiporters or symporters. After the assignment of appropriate SBO terms, further annotations were added using ModelPolisher69. ModelPolisher accesses the BiGG Models Database for the annotation and autocompletion of SBML models69. With the help of the ModelPolisher, additional metadata was incorporated for the different model instances. After those extensions, the MEMOTE score of the 64 GEMs increased on average to 83 ± 1%, which is an average improvement of 47%. The complete pipeline for debugging and extending all 64 models and saving them as valid SBML files is summarized in Fig. 3.

Not all the 64 SBML files downloaded from the supplement of Bosi et al. did directly pass the syntactic validation. In seven steps, the errors reported in the validity check were solved to receive valid SBML files. The valid files were then further extended with appropriate SBO terms for genes, species, and reactions. In a final step, annotations were added to the model using ModelPolisher69.
All debugging and extension steps served the purpose of making the models simulatable. Since reaction-sets, metabolite-sets, or gene-sets were not altered, the models’ simulation behavior is not affected. However, the models can now directly be used, as they are now all available as valid SBML files.
GEMs by Lee et al.
The thirteen GEMs by Lee et al. were available as Excel spreadsheet. For all reactions and metabolites in the list, the respective information, such as reaction or metabolite name, or chemical formula was extracted from the KEGG database37, where available. Based on the information from the KEGG database and the Excel spreadsheet, a consensus model including all reactions was created. Both biomass objective functions from Becker et al.27 and Heinemann et al.28 were added to the consensus model, as well as exchange reactions for all extracellular metabolites. ModelPolisher69 was used for annotating the model. Based on this consensus model, the individual models of the thirteen S. aureus strains were curated: The strain-specific reactions listed in the Excel spreadsheet were added to the respective models, and the biomass objective function from Becker et al. was adapted strain-specifically. The KEGG database was browsed for the strain-specific gene identifiers. The models now include on average 491 ± 8 genes, except for S. aureus strain RF122, where no strain-specific KEGG gene identifier was available. Further annotations, such as KEGG annotations and EC-codes were added to the models. Despite manual effort, all thirteen models do not show growth for neither of of the biomass objective functions. The MEMOTE score for all models excluding the model for the S. aureus strain RF122 reached 66%. Since the GEM for the RF122 strain does not contain any genes, its MEMOTE score only adds up to 57%. Comparing the originally published models concerning model simulations and growth predictions is not possible, because only Excel spreadsheets with reactions and metabolites were available.
GEMs from Path2Models
The 33 models from the Path2Models project are the only models of S. aureus that are still SBML Level 2 Version 470. Since the fbc package is officially only available from Level 3, it is not yet integrated in the files. We updated all models to SBML Level 3 Version 140 with the fbc package enabled using libSBML71. However, the original chemical formulas did not match the scheme that the official fbc package72 requires. In order to avoid creating syntactically invalid SBML files, all chemical formulas needed to be adapted according to the fbc specification72. The original chemical formulas can still be found in the notes field. This notes field further contained a variety of annotations from different databases, including BRENDA73, KEGG37, MetaCyc44, MetaNetX46, Rhea74, BiGG25, Reactome75, Model SEED49, Unipathway76, the Human Metabolome Database (HMDB)77, ChEBI78, and InChI79. All database annotations that can be found in the identifiers.org47 registry were transferred to the annotations, using identifiers.org uniform Resource Identifiers (URIs). The service identifiers.org provides directly resolvable identifiers from a multitude of different databases. The final and valid SBML files were evaluated using MEMOTE. The total score for the GEMs from the Path2Models project increased from 48 to 59% and all models are now available as SBML Level 3 files. Again, no changes on the reaction, metabolite, or gene content were performed, which would affect the model simulations.
Model analysis
In the following section, we examined the available models for their predictive value and their similarity. As the growth behavior of S. aureus is reported in various defined media, the models’ capability of reflecting growth under these conditions indicates the predictive value of the model. Subsequently, the publications were checked for the inclusion of experimental data in the models or the verification of model-driven hypothesis. Additionally, the predictions of gene essentialities using different models are compared. In the last step, the models’ similarities were examined concerning their reaction and gene content.
Growth capabilities
The growth of genome-scale metabolic models on different media is an important characteristic of a model’s capabilities and flexibility to reflect the organisms behavior in different environments. Since S. aureus is known to grow in a variety of different environments, its growth was simulated in chemically defined environments to investigate the model’s capabilities.
Chemically defined medium (CDM)
The CDM is a complete defined medium with 18 amino acids, two purines, and six vitamins and initially developed to study the slime production by coagulase-negative staphylococci80. It was used by Halsey et al. to study the amino acid catabolism in S. aureus81. Either no carbon source was added (CDM), or glucose (CDM_glc) or galactose (CDM_gal) was added to the medium. The growth of S. aureus strain JE2 is already computationally and experimentally validated and verified on CDM and its variants30.
Synthetic nasal medium (SNM)
The primary ecological niche of S. aureus is the human nose82,83. Krismer et al. developed a defined synthetic nasal medium (SNM) based on the composition of nasal fluid components determined by metabolomics84,85. This medium was initially developed to monitor the growth of S. aureus under similar physiological conditions as in the nose. Growth in this medium is experimentally verified for the S. aureus strains USA300 LAC and Newman. Since the medium is chemically defined, it can also be established in growth simulations in systems biology.
Gut medium
Already in the 1950s and 1960s, the intestinal colonization of S. aureus was reported82. Recent interest in the gut microbiome revealed and enlightened the relevant role and influence of S. aureus on the intestinal microbial ecology and diversity83,86,87,88,89. Intestinal colonization by S. aureus is, e.g., assumed to induce pseudo-membranous colitis and to change the gut microbial ecology89. Alterations in the composition of the gut microbiota can result in the development of chronic diseases, such as type 2 diabetes, colorectal cancer, and obesity90. Hence, studying the role of S. aureus in the context of the gut microbiome is of high relevance. Magnúsdóttir et al. generated 773 genome-scale metabolic reconstructions for 773 members of the human gut microbiome, including S. aureus. To simulate the growth in the gut, they chemically defined a medium according to experimental data. The medium definition was extracted from the S. aureus model created by Magnúsdóttir et al.33. Magnúsdóttir et al. validated two of the 773 genome-scale reconstructions experimentally, where S. aureus was not included. However, as their model grew in the defined medium, and S. aureus is reported to colonize the intestine, we inferred that growth should be possible.
SCFM
S. aureus does not only occur on the human skin, in the human nose82,83, or the nasopharyngeal tract91,92. It is furthermore observed in patients, especially in children, with cystic fibrosis (CF)93, an autosomal recessive disease. As one of the earliest and also most prevalent pathogens, S. aureus causes chronic airway infections in patients with CF94. To investigate the role of S. aureus and other associated pathogens, such as Pseudomonas aeruginosa93, Palmer et al. developed a synthetic cystic fibrosis medium (SCFM), mimicking the nutritional composition of the sputum of patients with CF by chromatographic and enzymatic analyses of the CF sputum. This medium was initially created to analyze the nutritional behavior of Pseudomonas aeruginosa in CF sputum95. Clinical isolates of S. aureus are reported to grow in SCFM96.
Since the thirteen models by lee et al. and the 33 models from the path2models project did not exhibit any growth in full medium, these models were not included in the analysis of growth capabilities. During the analysis, three of the models by Bosi et al. reported a low growth rate of 0.00186 mmol/(gDW × h) without any active exchange reactions (models sa_118, sa_gr1, and sa_lct). A positive growth rate without active exchange reactions can be an indicator for futile cycles and a necessity for manual verification and refinements.
Not all models by Bosi et al. were capable of growing on any of the tested media. In total, 33 out of the 61 remaining models were not able to grow on any of the tested media. This might be explained by the auxotrophies for amino acids and vitamins in several S. aureus strains observed by Bosi et al.32. None of the models by Bosi et al. grew on the SCFM or the gut medium. For SNM and the CDM compositions, different patterns emerged: ten strains, including N315, only grew on SNM, while six strains only grew on the CDM with galactose. Seven strains grew on all three variants of the CDM and the remaining five strain models grew on both the SNM and all CDM. The model iSB619 by Becker et al. only grew on the gut medium, while the model iMH551 by Heinemann et al. returned a positive growth rate for all tested media types. The model iYS854 exhibits growth on almost all tested media, except for the SCFM and the gut medium. It is comparable to the models by Bosi et al., with the difference of a higher growth rate. The model by Magnúsdóttir only grew on its own gut medium. In Fig. 4, the growth capabilities of the various S. aureus GEMs under different environmental conditions is illustrated.

All models with initial growth (see Table 1) were tested on the different media. For the GEMs by Bosi et al.32, the prefix “Bosi” was added to the model strain name. The other models are named according to their published model ID or, in case of the model from the VMH database, by the author. Media types are the synthetic nasal medium (SNM), synthetic cystic fibrosis medium (SCFM), gut medium, chemically defined medium (CDM), CDM with glucose (CDM_glc), and CDM with galactose (CDM_gal). Models that did not show growth in any of the tested media were excluded. The color-bar indicates the growth rate: the darker the color, the higher the growth rate of the model organism on the given medium.
Presence of experimental data
Besides the correct prediction of growth in a defined environment, a model’s predictive value also increases when laboratory data is included or in silico observations are verified in laboratory experiments.
Automatically curated GEMs
The models from the Path2Models project were automatically constructed. Within automated reconstruction processes, the inclusion of experimental data for individual models is complicated. For this reason, the GEMs from the Path2Models project do not contain experimental data29. Moreover, the models are not simulatable and, thus, can also not predict any growth. Verification of model predictions is hence not possible.
Semi-automatically curated GEMs
Curating a collection of multiple GEMs is time and labor intense. Manual reconstruction would take a significant amount of time. Thus, the models from Lee et al., Bosi et al., and Magnúsdóttir et al. were constructed semi-automatically.
Lee et al. verified their models using gene essentially analysis and growth experiments of two models. They found literature evidence and experimental verification for six of the 44 identified genes that were essential in all strains in silico. The growth experiments supported their minimal-medium predictions31.
The models from Bosi et al. were examined for the correct simulation of already known auxotrophies. Furthermore, the predictions of the growth capability in the presence of spermidine, and the growth on chemically defined media were verified in laboratory experiments for several strains32.
The model from Magnúsdóttir et al. was curated based on literature-derived experimental data. However, it is not specified which experimental data is used exactly. Metabolic predictions of two of the 773 reconstructions were validated against experimental data33.
Manually curated GEMs
Becker et al., Heinemann et al., and Seif et al. manually curated their strain-specific GEMs. The in silico growth predictions of the model iSB619 in a minimal medium were compared to laboratory experiments. Becker et al. additionally predicted essential genes. As this was the first available GEM of S. aureus, no experimental data was available to compare the predicted essential genes with27. The model iMH551 was compared to available knowledge about auxotrophies in S. aureus. The model’s growth predictions under aerobic and anaerobic conditions were validated against available experimental evidence28.
The model iYS854 underwent the most experimental verifications compared to all other models. Its predictions are in 85% agreement with gene essentiality experiments. The in silico predictions of the catabolism of carbon sources are in 68% agreement with experimental physiological data. They compared the models’ growth predictions on various media with laboratory experiments, and performed extensive condition-specific GEM validation and evaluation in the presence and absence of glucose.
Prediction of gene essentialities
Another indicator for the predictive value of a model is the correctness of predicted gene essentialities. The essentiality of a gene depends on the environment and the availability of nutrients. To identify essential genes in silico, each gene is individually knocked out in a so-called single gene deletion analysis and its effect on the growth rate is evaluated. This analysis, however, requires a model’s capacity to simulate growth in the investigated environment. As the models from the Path2Models project and Lee et al. did not show any initial growth (see Table 1), these models were excluded from the single gene deletion analysis. Additionally, this review aims to compare models from different sources. Since the models from the Path2Models project and Lee et al. were already excluded from this analysis, only two strains remain with more than one model: S. aureus USA300-FPR3757 and S. aureus N315. Two models from Bosi et al. and Magnúsdóttir et al. are available for the strain USA300-FPR3757, which can simulate growth. The model from Magnúsdóttir et al. contains gene identifiers that cannot be resolved within the PATRIC database97, leading to its exclusion from this analysis. With only one remaining model from Bosi et al., a comparison of predicted gene essentialities for the strain USA300-FPR3757 is not possible anymore.
Becker et al., Heinemann et al., and Bosi et al. curated models for the strain N315 simulating growth. The model from Heinemann et al., however, had to be excluded from the single-gene-deletion analysis as the model did not contain any GPRs and, thus, no genes. We downloaded the list of 302 essential genes for N315 from the Database of Essential Genes (DEG)98 and mapped all genes to the respective KEGG gene identifier. The medium is indicated as a rich medium in the DEG, but no further description of the chemical definition is given. Therefore, all exchange reactions were opened for the single gene deletion analysis.
The model from Bosi et al. predicted 117 essential genes, while the model from Becker et al. predicted 80. Of the 302 essential genes from the DEG, only 176 and 107 genes were present in the models from Bosi et al. and Becker et al., respectively. From the 117 predicted essential genes by Bosi et al., 27 (23.1%) were predicted correctly, while 90 (76.9%) of the predicted essential genes are not in accordance with the experimentally derived essential genes. Similarly, from the 80 predicted essential genes by Becker et al., 18 (22.5%) were predicted correctly, while 62 (77.5%) of the predicted essential genes are not listed in the DEG. One possible explanation for the similar predictions of essential genes is that the models from Bosi et al. are based on the model from Becker et al. The low number of true positive predicted essential genes could indicate further refinement potential of the two models.
Similarities between models
The analysis of the growth capabilities implied a clustering of models with similar growth behavior, especially for the models by Bosi et al. To identify further similarities between the models, the reaction sets were compared. Mapping identifiers between different databases induces a bias, since a complete mapping is currently not feasible. Tools, such as ModelPolisher69, can be helpful for annotating and comparing models. However, these tools rely on cross-references in various databases, which holds some challenges: The tools can only search with the correct identifier; if a model, however, has identifiers not included in the database, the tools will not find any annotations for that model instance. One other challenge lies within the administration and topicality of the databases. Changes in one database might not be reported or updated in the cross-references of other databases, leading to erroneous allocations that would bias the result of the comparison.
Heat maps of reaction similarity
Since the models have diverging identifiers, we divided them into three different groups. The first group comprises the 33 models from the Path2Models project with consistently mixed identifiers The second group includes all thirteen models by Lee et al. with KEGG IDs. The third group includes all models with BiGG identifiers, namely all models by Bosi et al., as well as the models iSB619 and iYS854. Furthermore, this third group contains the model created by Magnúsdóttir et al. This model possesses VMH identifiers, however, those identifiers can easily be converted to BiGG identifiers since they bear a resemblance to the BiGG IDs. Within these groups, all reactions were listed and checked for their occurrence in the models. With this table of reaction occurrences, the Jaccard distance was calculated between all pairwise combinations of the models.
With this distance matrix, the heat-map in Fig. 5 was created. The models iSB619, Magnúsdóttir, and iYS854 vary widely between each other and the models by Bosi et al. Within the Bosi models, clusters of more and less similar models can be identified (Fig. 5c). Such clusters are expected, as we assumed that genetically similar strains also lead to more similar GEMs, due to the gene–protein-reaction associations (GPRs). For example, the two closely related USA300 strains TCH1516 and FPR3757 have a distance value of 0.015, while the distance to one of the isolates of the ST228 lineage (ST228-16035) is 0.160. Strain MRSA252 is reported to be genetically diverse compared to other S. aureus strains. Its distance, however, to the USA300-TCH1516 strain is smaller (0.06) than the distance to the isolates of the ST228 lineage. Hence, the genetic differences between the different strains are not necessarily reflected in their respective GEMs so far.

The models were divided into three groups based on their metabolite and reaction identifiers: (a) has all models of the Path2Models project with consistently mixed identifiers, (b) has all models with KEGG identifiers (hence, all GEMs by Lee et al.), and (c) contains all models with BiGG identifiers. Within the three groups, all pairwise Jaccard distances were calculated based on the models’ reaction sets. The distances are displayed in the heat map. The color bar range is equal for (a) and (c) for better comparison. As the distances in (b) are much smaller, the color bar’s range was adapted.
The distances between the models with BiGG IDs (group three) ranged from 0 to 0.8, with the maximal distances between the models iSB619, Magnúsdóttir, and iYS854. The models by Lee et al., however, are more similar, indicated by the scaling of the color-bar that ranges from 0 to 0.05. The model of the S. aureus strain TCH1516 differs the most from all other models (Fig. 5b). Unlike the models from Bosi et al., the two USA300 strains (TCH1516 and USA300) do not cluster. They have a distance of 0.037. In contrast to the models of Lee et al., the strain TCH1516 does not stand out in the groups with BiGG IDs and the Path2Models models.
Most distances between the models from the Path2Models project (group one), ranged from 0.25 to 0.35. However, the model of strain RF122 protrudes with a mean distance of 0.62. This trend can also be observed in the heat-map of the models by Lee et al., but not as prominent as in Fig. 5a. One possible explanation is given in the taxonomy for the S. aureus strain RF122, which is an bovine mastitis-associated isolate with notable differences to human clones of S. aureus99. This difference is, however, not as obvious in the Models of Bosi et al. compared to the models of Lee et al. and the Path2Models project.
Venn diagrams of gene similarity
Despite significant effort to standardize and consistently annotate all models using different annotating tools, such as the ModelPolisher, or database requests for aliases from databases like BiGG or ModelSEED, a satisfying comparison of the reaction sets between different identifiers is still not possible. For example, for the models with KEGG identifiers from Lee et al., we could not use the ModelPolisher, as this annotation tool currently requires BiGG identifiers. For that reason, we browsed the BiGG Models Database locally for cross-references to KEGG identifiers. Unfortunately, 842 out of 1486 KEGG reaction identifier were not referenced at all in BiGG, 359 KEGG identifiers were not uniquely mapped to a BiGG identifier, and only 285 identifiers were uniquely mapped. We checked some of the non-referenced KEGG identifiers in the ModelSEED database for aliases but could not determine the respective identifiers.
For that reason, we looked at the gene content of the models. Most models used KEGG gene identifiers, regardless of the identifier database of the reactions and metabolites. As the different strains have strain-specific gene identifiers, the following analysis was conducted strain-wise. Strains with at least three models from various resources were taken into account (see also Fig. 2): For eleven strains, three models are available, for the strain USA300-FPR3757, four models are present in this collection, and for the strain N315, five models are available. However, the SBML file of the N315 model by Heinemann et al. does not include any genes. Thus, the model was excluded from the comparison. Same accounts for the RF122 strain-specific model by Lee et al., which also does not contain any genes. For this reason, the model was also excluded from the analysis. By that, the strain RF122 did no longer fulfill the criterion of at least three available models.
The gene sets from the remaining models were compared. As indicated, most models used KEGG gene identifiers, but not all. The model by Magnúsdóttir et al. included strain-specific and unspecific PATRIC identifiers97. With the help of the PATRIC ID mapping service, the respective KEGG gene identifiers were extracted. However, this was only feasible for the strain-specific identifiers. Despite significant effort, the unspecific identifiers could not be resolved, as no mapping scheme could be identified. Thus, from the 859 genes included in the Magnúsdóttir model, only 192 could be resolved to KEGG identifiers.
Model iSB619 contained new locus tags, whereas the KEGG identifiers correspond to the old locus tags. With the GenBank flat file (gbff)100 of S. aureus strain N315, the locus tags were mapped. For the 619 new locus tags 611 respective old locus tags, and thus KEGG identifiers, were extracted.
The models by Bosi et al. included mostly KEGG gene identifiers. Within the strains JH1 and JH9, the gene identifiers were truncated by the included word “DRAFT” to make them consistent with the actual KEGG identifiers. For example, the initial identifier SaurJH1DRAFT_0595 was truncated to the correct KEGG identifier SaurJH1_0595.
After these mapping and adapting steps, the gene sets within the different strains from the different resources were compared, and Venn diagrams were created as shown in Fig. 6. Across all twelve comparisons, the models by Bosi et al. have the largest portion of genes that are solely reflected in these models. This number varies between 20.1% in the N315 strain and 59% in the Newman strain. As these models have the highest gene content on average with approximately 788 ± 116 genes per model, this seems apparent. The models from the Path2Models project have an average gene content of 519 ± 12 genes per model, and the models by Lee et al. contain 488 ± 149 genes on average. It was already mentioned that the gene identifiers from the JH1 and JH9 models by Bosi needed to be adapted. Despite this adaption, only half of the gene content is present in the other models as well. For the Newman, MW2, and Mu3 strains, we further analyzed the gene identifiers after these observed discrepancies between the gene contents with the models from the other two databases. These three strain-specific models from Bosi include non-strain-specific gene identifiers, which could not be mapped to the corresponding strain-specific gene identifier.

For all models occurring in at least three different resources, the gene content was compared strain-specifically. After unifying the gene identifiers to KEGG IDs, Venn diagrams were created comparing the gene content. The models from Bosi et al. have, on average, the highest gene content, explaining the large fraction of genes occurring only in these models. The models by Lee et al. and the Path2Models project seem more similar, which could be explained by the fact that both are curated based on the KEGG database. Although all models in one Venn diagram (and thus, one comparison) represent the same strain, the models have differences, indicating the influence of the reconstruction method on the final model content.
The models from Lee et al. and the Path2Models project are relatively similar concerning their gene content. Since both models are curated based on the KEGG database, this similarity is evident. The four models of the S. aureus USA300-FPR3757 strain have a gene content overlap of 15.7%. The model by Magnúsdóttir et al. has only 0.3% gene content that is not reflected in the other three models. However, one needs to keep in mind that many genes in the model are not strain-specific and could not be mapped and compared.
With these twelve gene content comparisons, we again calculated the Jaccard distance between the models from Bosi et al., Lee et al., and the Path2Models project. As already visible from the Venn diagrams, the models from Lee and the Path2Models project are most similar with respect to their gene content. They have a mean Jaccard distance of 0.288 ± 0.004. However, one might have speculated that the models are more similar based on the Venn diagrams. It needs to be highlighted that the Venn diagrams are calculated based on the gene content of all compared models. In contrast, the Jaccard distance calculates pairwise distances and, thus, only considers two models at once. For that reason, the models from Lee et al. and the Path2Models project are still the most similar ones, but their identity might not be as large as first expected when looking at the Venn diagrams. The Bosi models have a mean distance to the Lee models of 0.666 ± 0.179 and to the Path2Models project models a mean distance of 0.616 ± 0.203.
Although the different models from the various databases reflect the same strain, the models have distinct diversities. This can be explained by the differences in the reconstruction process. How the model is curated seems to play a pivotal role for the final model and its model instances. Thus, the reconstruction method needs to be chosen carefully, and manual or semi-automated additions might be required.
Decision guidance
With the vast amount of different strain-specific S. aureus models, the identification of the suitable GEM for a specific research question or purpose might become difficult. Table 2 gives an overview about the main features of the S. aureus GEMs. The features were assigned based on the strengths of the different models or model collections after the model improvement steps. If one is interested in simulatable models, the table guides the reader to the corresponding models. By combining different required features, the selection can be tailored. If one needs, e.g., a model with BiGG IDs that grows on different media, the models by Bosi et al. or the model iYS854 are suggested, depending on the desired strain. High MEMOTE scores indicate a high degree of annotations, which facilitates the re-usability and comparability of a model.
A predictive value score was calculated based on the model analysis regarding their growth capabilities and the presence of experimental data. If a model was not simulatable, it received a predictive value score of 0. Otherwise, a score of 1 was added. For growth capabilities in one environment, a score of 1 was added; for growth in multiple environments, 2 was added. For every experimental verification procedure, such as growth verifications, auxotrophies, compliance with physiological data, or other experiments, a score of 1 was added. The prediction of essential genes was not included in this score, as this analysis was only conducted for two models. By this scheme, the model iYS854 had the highest predictive value score of 7, followed by iMH551 and some models by Bosi et al. The models by Bosi et al. received a score between 3 and 5, as some models do not predict growth in any tested environment, while others do. As the models from Lee et al. and the Path2Models project are not simulatable, they received a predictive value score of 0. Models with high predictive value score and high MEMOTE score are recommended for further use, while models with low predictive value score might need further refinement and experimental verification before usage.
This table does not contain strain-specific information. Including the information from Figs. 2 and 4 will further guide the decision for a suitable model.
Discussion
The analyses show that despite genomic and genetic similarities, GEMs of related strains are not necessarily similar to each other. This accounts for both models of the same strain curated by different research groups and to related strains curated by the same group. One example is the model from Magnusdóttir et al. with the S. aureus strain USA300-FPR3757 and the corresponding model from Bosi et al. Despite it is the same strain, the GEMs are quite different in their reaction content. In contrast, the two strain-specific models of the strains MRSA252 and USA300-TCH1516 by Bosi et al. are quite similar despite the genetic diversity of the strain MRSA252. This observation might have several reasons. The first, and probably most striking, reason is the incompleteness of the models. As high-quality genome-scale metabolic reconstructions require manual curation and evaluation101, and many models introduced in this review were created automatically or semi-automatically, some models might lack general or strain-specific reactions. This lack of required reactions is also visible when optimizing the flux distributions of the models. For multiple models, no growth could be simulated in FBA, not even in full medium. This was especially the case for the automatically curated models from the Path2Models project and the semi-automatically curated models from Lee et al. But also some of the semi-automatically curated models from Bosi et al. did not show any growth. Thus, a connection between automated or semi-automated curation and the functionality of the models seems to exist. However, automated or semi-automated curation does not necessarily result in poor growth prediction, especially when the basis for the (semi-) automated processes underwent significant manual curation. The other models from Bosi et al. showed growth on up to four different media. The semi-automatically constructed model by Magnusdóttir et al. could be simulated on one medium, which is also the case for the manually curated model iSB619. Furthermore, some of the S. aureus strains have plasmids carrying additional genes. For a strain-specific model, these additional genes need to be incorporated into the GEM as well. Especially the metabolic and transporter genes are relevant for the strain-specific model. The plasmid of the S. aureus strain N315, e.g., carries a gene for the cadmium resistance transporter CadD, which facilitates the export of cadmium ions and other cationic compounds102. Besides further proteinogenic genes, the plasmid of strain N315 also carries a gene for the penicillin-hydrolyzing class A β-lactamase enzyme. These two genes are, e.g., also present on the plasmid of the S. aureus strain USA300-TCH1516.
As explained previously, the challenge lies within the different reaction and metabolite identifiers. In this review, we additionally tried to annotate the GEMs further to simplify the comparison of models with differing identifiers. However, only approximately one third of all reactions and metabolites are annotated with identifiers of external databases. It is still challenging to find all cross-references for a particular metabolite or reaction in a specific database. For that reason, we additionally evaluated the gene content of the strain-specific models, as most models contained identifiers from the KEGG database. The gene identifiers from other databases were mapped to the KEGG identifiers. Again, a bias is introduced when identifiers are mapped between databases: On the one hand, not all identifiers can be resolved in the other database. On the other hand, some identifiers do not comply with the databases’ identifiers scheme and do not have annotations. This makes an automated mapping of several hundred identifiers infeasible. Extensive manual labor would be necessary to map these identifiers. The usage of consistent identifiers that comply with the database scheme and additional annotations is highly recommended and would simplify the re-usability, translatability, and comparability of models103. The comparison of the strain-specific models’ gene content confirmed that GEMs from different resources could vary, despite their genetic equality, highlighting the relevance of the curation process on the resulting GEM. This observation is even more explicit when comparing the models by Lee et al. and from the Path2Models project: both rely on the KEGG database. However, the models are not equal, as the two groups used different approaches for the curation of the models.
Missing reactions and strain-specific genes might also affect the growth behavior of a strain-specific model on a given medium. Only the model iMH551 showed growth on all tested media. Additional growth experiments for specific S. aureus strains can help to identify the missing growth capabilities of the model. The model’s ability to adapt to different environmental conditions is crucial to simulate an organism in silico. This is also reflected in the predictive value score, which was assigned to the models. Especially for models with a low predictive value score, additional experiments would help determine and also increase the predictive value of the model.
The models from Lee et al., the Path2Models project, Bosi et al., and Magnúsdóttir et al. are curated automatically or semi-automatically. Except for the models from Bosi et al., all models have a comparatively low predictive value score than the manually curated models. The models from the Path2Models project and Lee et al. have a score of 0. The low score from the Path2Models projects’ models might go back to the lack of experimental data in both the curation and verification process, thus highlighting its importance for predictive genome-scale metabolic reconstructions. The low score for the models from Lee et al. accentuates the importance of standardized GEMs, which allow re-usability. Although the models from Bosi et al. are curated semi-automatically, their predictive value scores are comparable high. They based their pipeline on a manually refined model and verified their predictions with experimental data. More experimental data accompany more knowledge. The latest model, iYS854 has the highest predictive value score, was manually curated, and extensively experimentally validated. The result of such a time- and labor-intensive work is a GEM with a high predictive value and a strong recommendation for future usage.
Conclusion and outlook
In this review, all 114 currently available genome-scale metabolic models (GEMs) of Staphylococcus aureus were presented and evaluated. It serves as guide for the different available reconstructions in various databases, using differing metabolite and reaction identifiers. Some models originally comprise a large number of reactions, metabolites, and genes, after undergoing several manual curation steps and extensive annotating. Such models have a high MEMOTE score. The model with the highest MEMOTE score is the iYS854 model by Seif et al. Other models have a vast amount of reactions and metabolites, such as the reconstructions of the Path2Models project. Such models could, e.g., serve as information sources for the reconstruction or refinement of already existing strain-specific models. Based on the information regarding availability, model format, MEMOTE score, growth behavior, used database identifiers, predictive value, and similarities between models, together with a previously defined research question, the appropriate genome-scale reconstruction can be chosen from the vast amount of available GEMs. Another approach would be to use the strengths of every reconstruction and incorporate it into merged or combined models, which increase the correctness and the predictive value of a strain-specific model. Despite the vast amount of presented models in this review, there is no suitable model for every S. aureus strain available. Furthermore, missing annotations or identifiers that do not comply with the database identifier scheme impede the models’ re-usability and comparability. Standardization of all models would be desirable but is currently not feasible with the available tools without extensive manual labor for hundreds of identifiers. No omics data was incorporated into many of the published GEMs so far. Information about transcription profiles, for example, can help to refine metabolic reconstructions to better reflect the metabolic state of an organism in a defined environment. The incorporation of omics data can thus increase the predictive value of genome-based metabolic reconstructions104.
However, with the help of the already available reconstructions and further information, strain-specific models could be created or extended. Information from literature, merging of strain-specific models, and manual curation steps could further improve the predictive value of simulations and analyses of metabolic features of S. aureus. Having predictive GEMs can eventually lead to the identification of novel targets for antimicrobial therapies in the fight against antibiotic resistant strains of S. aureus.
Data availability
The availability of all models, including the improved models, is listed in the supplementary Table S1. The model collection was deposited in BioModels105 within COMBINE archive files (in OMEX format)106 and assigned the identifiers (1) MODEL2007110001, (2) MODEL2007150001, and (3) MODEL2007150002.
Code availability
All the necessary scripts and resources for model modifications and improvements are available in a git repository at github.com/draeger-lab/S_aureus_GEMs_Collection.
References
Sakr, A., Brégeon, F., Mège, J. L., Rolain, J. M. & Blin, O. Staphylococcus aureus nasal colonization: an update on mechanisms, epidemiology, risk factors, and subsequent infections. Front. Microbiol. 9, 2419 (2018).
Schmidt, A., Bénard, S. & Cyr, S. Hospital cost of staphylococcal infection after cardiothoracic or orthopedic operations in France: a retrospective database analysis. Surg. Infect. 16, 428–435 (2015).
Turner, N. A. et al. Methicillin-resistant Staphylococcus aureus: an overview of basic and clinical research. Nat. Rev. Microbiol. 17, 203–218 (2019).
Tacconelli, E. et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327 (2018).
van Hal, S. J. et al. Predictors of mortality in Staphylococcus aureus bacteremia. Clin. Microbiol. Rev. 25, 362–386 (2012).
Laupland, K., Ross, T. & Gregson, D. Staphylococcus aureus bloodstream infections: risk factors, outcomes, and the influence of methicillin resistance in calgary, Canada, 2000–2006. J. Infect. Dis. 198, 336–343 (2008).
Klevens, R. M. et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. J. Am. Med. Assoc. 298, 1763–1771 (2007).
Friedman, N. D. et al. Health care-associated bloodstream infections in adults: a reason to change the accepted definition of community-acquired infections. Ann. Intern. Med. 137, 791–797 (2002).
Dantes, R. et al. National burden of invasive methicillin-resistant Staphylococcus aureus infections, United States, 2011. JAMA Intern. Med. 173, 1970–1979 (2013).
Kourtis, A. P. et al. Vital signs: epidemiology and recent trends in methicillin-resistant and in methicillin-susceptible Staphylococcus aureus bloodstream infections—United States. MMWR 68, 214–219 (2019).
Eells, S. J. et al. Persistent environmental contamination with USA300 methicillin-resistant Staphylococcus aureus and other pathogenic strain types in households with S. aureus skin infections. Infect. Control Hosp. Epidemiol. 35, 1373–1382 (2014).
Dalman, M. et al. Characterizing the molecular epidemiology of Staphylococcus aureus across and within fitness facility types. BMC Infect. Dis. 19, 69 (2019).
Monaco, M., Pimentel de Araujo, F., Cruciani, M., Coccia, E. M. & Pantosti, A. In Current Topics in Microbiology and Immunology, vol. 409, 21–56 (Springer, 2017).
Azarian, T. et al. Intrahost evolution of methicillin-resistant Staphylococcus aureus USA300 among individuals with reoccurring skin and soft-tissue infections. J. Infect. Dis. 214, 895–905 (2016).
Malachowa, N. & Deleo, F. R. Mobile genetic elements of Staphylococcus aureus. Cell. Mol. Life Sci. 67, 3057–3071 (2010).
Corey, G. R. et al. Pooled analysis of single-dose oritavancin in the treatment of acute bacterial skin and skin-structure infections caused by Gram-positive pathogens, including a large patient subset with methicillin-resistant Staphylococcus aureus. Int. J. Antimicrob. Agents 48, 528–534 (2016).
Arshad, S. et al. Ceftaroline fosamil monotherapy for methicillin-resistant Staphylococcus aureus bacteremia: a comparative clinical outcomes study. Int. J. Infect. Dis. 57, 27–31 (2017).
Mienda, B. S., Salihu, R., Adamu, A. & Idris, S. Genome-scale metabolic models as platforms for identification of novel genes as antimicrobial drug targets. Future Microbiol. 13, 455–467 (2018).
Liu, L., Agren, R., Bordel, S. & Nielsen, J. Use of genome-scale metabolic models for understanding microbial physiology. FEBS Lett. 584, 2556–2564 (2010).
Gu, C., Kim, G. B., Kim, W. J., Kim, H. U. & Lee, S. Y. Current status and applications of genome-scale metabolic models. Genome Biol. 20, 1–18 (2019).
Dräger, A. & Planatscher, H. in Encyclopedia of Systems Biology (eds. Dubitzky, W. et al.) 1249–1251 (Springer, 2013).
Lewis, N. E., Nagarajan, H. & Palsson, B. O. Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methods. Nat. Rev. Microbiol. 10, 291–305 (2012).
O’Brien, E., Monk, J. & Palsson, B. Using genome-scale models to predict biological capabilities. Cell 161, 971–987 (2015).
Renz, A., Mostolizadeh, R. & Dräger, A. In Systems Medicine (ed. Wolkenhauer, O.) vol. 3, 362–371 (Academic Press, 2020). https://www.sciencedirect.com/science/article/pii/B9780128012383115247.
Norsigian, C. J. et al. BiGG Models 2020: multi-strain genome-scale models and expansion across the phylogenetic tree. Nucleic Acids Res. https://doi.org/10.1093/nar/gkz1054/5614178 (2020)
Glont, M. et al. BioModels: expanding horizons to include more modelling approaches and formats. Nucleic Acids Res. 46, D1248–D1253 (2018).
Becker, S. A. & Palsson, B. Ø. Genome-scale reconstruction of the metabolic network in Staphylococcus aureus N315: an initial draft to the two-dimensional annotation. BMC Microbiol. 5, 8 (2005).
Heinemann, M., Kümmel, A., Ruinatscha, R. & Panke, S. In silico genome-scale reconstruction and validation of the Staphylococcus aureus metabolic network. Biotechnol. Bioeng. 92, 850–864 (2005).
Büchel, F. et al. Path2Models: large-scale generation of computational models from biochemical pathway maps. BMC Syst. Biol. 7, 116 (2013).
Seif, Y. et al. A computational knowledge-base elucidates the response of Staphylococcus aureus to different media types. PLoS Comput. Biol. 15, e1006644 (2019).
Lee, D.-S. et al. Comparative genome-scale metabolic reconstruction and flux balance analysis of multiple Staphylococcus aureus genomes identify novel antimicrobial drug targets. J. Bacteriol. 191, 4015–4024 (2009).
Bosi, E. et al. Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc. Natl Acad. Sci. USA 113, E3801–9 (2016).
Magnúsdóttir, S. et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 35, 81–89 (2016).
Noronha, A. et al. The virtual metabolic human database: integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res. 47, D614–D624 (2019).
Ebrahim, A., Lerman, J. A., Palsson, B. O. & Hyduke, D. R. COBRApy: COnstraints-based reconstruction and analysis for python. BMC Syst. Biol. 7, 74 (2013).
Lieven, C. et al. MEMOTE for standardized genome-scale metabolic model testing. Nat. Biotechnol. 38, 272–276 (2020).
Kanehisa, M., Sato, Y., Furumichi, M., Morishima, K. & Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 47, D590–D595 (2019).
Peterson, J. D. The comprehensive microbial resource. Nucleic Acids Res. 29, 123–125 (2001).
Dauner M, S. U. Stoichiometric growth model for riboflavin-producing Bacillus subtilis. Biotechnol. Bioeng. 76, 132–143 (2001).
Hucka, M. et al. Systems biology markup language (SBML) level 3 version 1 core. J. Integr. Bioinform. 15, 1 (2018).
Olivier, B. G. & Bergmann, F. T. SBML level 3 package: flux balance constraints version 2. J. Integr. Bioinform. 15, 20170082 (2018).
Overbeek, R. et al. The ErgoTM genome analysis and discovery system. Nucleic Acids Res. 31, 164–171 (2003).
Schaefer, C. F. et al. PID: The pathway interaction database. Nucleic Acids Res 37, D674–D679 (2009).
Caspi, R. et al. The MetaCyc database of metabolic pathways and enzymes—a 2019 update. Nucleic Acids Res. 48, D445–D453 (2019).
Wittig, U., Rey, M., Weidemann, A., Kania, R. & Müller, W. SABIO-RK: an updated resource for manually curated biochemical reaction kinetics. Nucleic Acids Res. 46, D656–D660 (2018).
Moretti, S., Tran, V. D. T., Mehl, F., Ibberson, M. & Pagni, M. MetaNetX/MNXref: unified namespace for metabolites and biochemical reactions in the context of metabolic models. Nucleic Acids Res. 49, D570–D574 (2021).
Juty, N., Le Novere, N. & Laibe, C. Identifiers.org and MIRIAM Registry: community resources to provide persistent identification. Nucleic Acids Res. 40, D580–D586 (2012).
Finney, A. & Hucka, M. Systems biology markup language: level 2 and beyond. Biochem. Soc. Trans. 31, 1472–1473 (2003).
Henry, C. S. et al. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 28, 977–982 (2010).
Karp, P. D. et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 20, 1085–1093 (2019).
Keating, S. M. et al. SBML level 3: an extensible format for the exchange and reuse of biological models. Mol. Syst. Biol. 16, e9110 (2020).
Arkin, A. P. et al. KBase: The United States Department of Energy systems biology knowledgebase. Nat. Biotechnol. 36, 566–569 (2018).
Thorleifsson, S. G. & Thiele, I. rBioNet: a COBRA toolbox extension for reconstructing high-quality biochemical networks. Bioinformatics 27, 2009–2010 (2011).
Kuroda, M. et al. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357, 1225–1240 (2001).
Hiramatsu, K. et al. Dissemination in Japanese hospitals of strains of Staphylococcus aureus heterogeneously resistant to vancomycin. Lancet 350, 1670–1673 (1997).
Neoh, H. M. et al. Mutated response regulator graR is responsible for phenotypic conversion of Staphylococcus aureus from heterogeneous vancomycin-intermediate resistance to vancomycin-intermediate resistance. Antimicrob. Agents Chemother. 52, 45–53 (2008).
Mwangi, M. M. et al. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc. Natl Acad. Sci. USA 104, 9451–9456 (2007).
Highlander, S. K. et al. Subtle genetic changes enhance virulence of methicillin resistant and sensitive Staphylococcus aureus. BMC Microbiol. 7, 99 (2007).
Diep, B. A. et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367, 731–739 (2006).
Baba, T. et al. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet 359, 1819–1827 (2002).
Gill, S. R. et al. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 187, 2426–2438 (2005).
Dyke, K. G., Jevons, M. P. & Parker, M. T. Penicillinase production and intrinsic resistance to penicillins in Staphylococcus aureus. Lancet 1, 835–838 (1966).
Baba, T., Bae, T., Schneewind, O., Takeuchi, F. & Hiramatsu, K. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J. Bacteriol. 190, 300–310 (2008).
Holden, M. T. et al. Complete genomes of two clinical Staphylococcus aureus strains: evidence for the evolution of virulence and drug resistance. Proc. Natl Acad. Sci. USA 101, 9786–9791 (2004).
Herron-Olson, L., Fitzgerald, J. R., Musser, J. M. & Kapur, V. Molecular correlates of host specialization in Staphylococcus aureus. PLoS ONE 2, e1120 (2007).
Vogel, V., Falquet, L., Calderon-Copete, S. P., Basset, P. & Blanc, D. S. Short term evolution of a highly transmissible methicillin-resistant Staphylococcus aureus clone (ST228) in a Tertiary care hospital. PLoS ONE 7, e38969 (2012).
Hucka, M. et al. Systems Biology Markup Language (SBML) level 3 version 2 core release 2. J. Integr. Bioinform. 16, 1 (2019).
Courtot, M. et al. Controlled vocabularies and semantics in systems biology. Mol. Syst. Biol. 7, 543–543 (2014).
Römer, M. et al. ZBIT Bioinformatics Toolbox: a web-platform for systems biology and expression data analysis. PLOS ONE 11, e0149263 (2016).
Hucka, M., Finney, A., Hoops, S., Keating, S. M. & Le Novère, N. Systems biology markup language (SBML) Level 2: structures and facilities for model definitions. Tech. Rep. Nat. Preced. https://doi.org/10.1038/npre.2008.2715.1 (2008).
Bornstein, B. J., Keating, S. M., Jouraku, A. & Hucka, M. LibSBML: an API Library for SBML. Bioinformatics 24, 880–881 (2008).
Olivier, B. G. & Bergmann, F. T. The systems biology markup language (SBML) level 3 package: flux balance constraints. J. Integr. Bioinform. 2, 660–690 (2015).
Chang, A. et al. BRENDA, the ELIXIR core data resource in 2021: new developments and updates. Nucleic Acids Res. 49, D498–D508 (2021).
Lombardot, T. et al. Updates in Rhea: SPARQLing biochemical reaction data. Nucleic Acids Res. 47, D596–D600 (2019).
Jassal, B. et al. The reactome pathway knowledgebase. Nucleic Acids Res. 48, D498–D503 (2020).
Morgat, A. et al. UniPathway: a resource for the exploration and annotation of metabolic pathways. Nucleic Acids Res. 40, D761–D769 (2012).
Wishart, D. S. et al. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 46, D608–D617 (2018).
Hastings, J. et al. ChEBI in 2016: improved services and an expanding collection of metabolites. Nucleic Acids Res. 44, D1214–D1219 (2016).
Heller, S. R., McNaught, A., Pletnev, I., Stein, S. & Tchekhovskoi, D. InChI, the IUPAC International Chemical Identifier. J. Cheminform. 7, 23 (2015).
Hussain, M., Hastings, J. G. M. & White, P. J. A chemically defined medium for slime production by coagulase-negative staphylococci. J. Med. Microbiol. 34, 143–147 (1991).
Halsey, C. R. et al. Amino acid catabolism in Staphylococcus aureus and the function of carbon catabolite repression. mBio 8, e01434–16 (2017).
Williams, R. E. Healthy carriage of Staphylococcus aureus: its prevalence and importance. Bacteriol. Rev. 27, 56–71 (1963).
Acton, D. S., Tempelmans Plat-Sinnige, M. J., Van Wamel, W., De Groot, N. & Van Belkum, A. Intestinal carriage of Staphylococcus aureus: How does its frequency compare with that of nasal carriage and what is its clinical impact? Eur. J. Clin. Microbiol. Infect. Dis. 28, 115–127 (2009).
Krismer, B. et al. Nutrient limitation governs Staphylococcus aureus metabolism and niche adaptation in the human nose. PLoS Pathog. 10, e1003862 (2014).
Krismer, B., Weidenmaier, C., Zipperer, A. & Peschel, A. The commensal lifestyle of Staphylococcus aureus and its interactions with the nasal microbiota. Nat. Rev. Microbiol. 15, 675–687 (2017).
Piewngam, P. et al. Pathogen elimination by probiotic Bacillus via signalling interference. Nature 562, 532–537 (2018).
Ren, D. et al. Effects of mixed lactic acid bacteria on intestinal microbiota of mice infected with Staphylococcus aureus. BMC Microbiol. 18, 109 (2018).
Dong, D. et al. Effects of intestinal colonization by Clostridium difficile and Staphylococcus aureus on microbiota diversity in healthy individuals in China. BMC Infect. Dis. 18, 1–8 (2018).
Sannasiddappa, T. H., Costabile, A., Gibson, G. R. & Clarke, S. R. The influence of Staphylococcus aureus on gut microbial ecology in an in vitro continuous culture human colonic model system. PLoS ONE 6, e23227 (2011).
Clemente, J. C., Ursell, L. K., Parfrey, L. W. & Knight, R. The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270 (2012).
Bogaert, D. et al. Colonisation by Streptococcus pneumoniae and Staphylococcus aureus in healthy children. Lancet 363, 1871–1872 (2004).
Rosenfeld, M. et al. Prevalence of cystic fibrosis pathogens in the oropharynx of healthy children and implications for cystic fibrosis care. J. Cyst. Fibros. 11, 456–457 (2012).
Hurley, M. N. Staphylococcus aureus in cystic fibrosis: Problem bug or an innocent bystander? Breathe 14, 87–90 (2018).
Schwerdt, M. et al. Staphylococcus aureus in the airways of cystic fibrosis patients—a retrospective long-term study. Int. J. Med. Microbiol. 308, 631–639 (2018).
Palmer, K. L., Aye, L. M. & Whiteley, M. Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. J. Bacteriol. 189, 8079–8087 (2007).
Sweeney, E. et al. An ex vivo cystic fibrosis model recapitulates key clinical aspects of chronic Staphylococcus aureus infection. Microbiology 167, 000987 (2021).
Davis, J. J. et al. The PATRIC Bioinformatics Resource Center: expanding data and analysis capabilities. Nucleic Acids Res. 48, D606–D612 (2020).
Luo, H., Lin, Y., Gao, F., Zhang, C. T. & Zhang, R. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 42, D574 (2014).
Herron, L. L. et al. Genome sequence survey identifies unique sequences and key virulence genes with unusual rates of amino acid substitution in bovine Staphylococcus aureus. Infect. Immunity 70, 3978–3981 (2002).
Clark, K., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J. & Sayers, E. W. GenBank. Nucleic Acids Res. 44, D67–D72 (2016).
Thiele, I. & Palsson, B. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 5, 93–121 (2010).
Crupper, S. S., Worrell, V., Stewart, G. C. & Iandolo, J. J. Cloning and expression of cadD, a new cadmium resistance gene of Staphylococcus aureus. J. Bacteriol. 181, 4071–4075 (1999).
Carey, M. A., Dräger, A., Beber, M. E., Papin, J. A. & Yurkovich, J. T. Community standards to facilitate development and address challenges in metabolic modeling. Mol. Syst. Biol. 16, e9235 (2020).
Ramon, C., Gollub, M. G. & Stelling, J. Integrating -omics data into genome-scale metabolic network models: principles and challenges. Essays Biochem. 62, 563–574 (2018).
Chelliah, V. et al. BioModels: ten-year anniversary. Nucleic Acids Res. 43, D542–D548 (2015).
Bergmann, F. T. et al. COMBINE archive and OMEX format: one file to share all information to reproduce a modeling project. BMC Bioinform. 15, 369 (2014).
Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy—EXC 2124—390838134 and supported by the Cluster of Excellence “Controlling Microbes to Fight Infections” (CMFI). A.D. is supported by the German Center for Infection Research (DZIF, doi: 10.13039/100009139) within the Deutsche Zentren der Gesundheitsforschung (BMBF-DZG, German Centers for Health Research of the Federal Ministery of Education and Research), grant № 8020708703.
Author information
Authors and Affiliations
Contributions
A.R. conceived the study and improved the models. A.D. supervised the study. A.R. and A.D. wrote the manuscript. Both authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Renz, A., Dräger, A. Curating and comparing 114 strain-specific genome-scale metabolic models of Staphylococcus aureus. npj Syst Biol Appl 7, 30 (2021). https://doi.org/10.1038/s41540-021-00188-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41540-021-00188-4
This article is cited by
-
Logic programming-based Minimal Cut Sets reveal consortium-level therapeutic targets for chronic wound infections
npj Systems Biology and Applications (2024)
-
Integration of graph neural networks and genome-scale metabolic models for predicting gene essentiality
npj Systems Biology and Applications (2024)
