
Abstract
Parkinson’s disease (PD) is pathologically characterized by the abnormal accumulation of α-synuclein fibrils (Lewy bodies) in the substantia nigra and other brain regions, although the role of Lewy bodies remains elusive. Constipation usually precedes the motor symptoms in PD, which is in accordance with the notion that α-synuclein fibrils start from the intestinal neural plexus and ascend to the brain in at least half of PD patients. The gut microbiota is likely to be involved in intestinal and brain pathologies. Analyses of the gut microbiota in PD, rapid-eye-movement sleep behavior disorder, and dementia with Lewy bodies suggest three pathological pathways. First, Akkermansia, which is increased in PD, degrades the intestinal mucus layer and increases intestinal permeability, which triggers inflammation and oxidative stress in the intestinal neural plexus. Second, decreased short-chain fatty acids (SCFAs)-producing bacteria in PD reduce the number of regulatory T cells. Third, SCFAs also aggravate microglial activation with an unelucidated pathway. In addition, in dementia with Lewy bodies (DLB), which is another form of α-synucleinopathies, increased genera, Ruminococcus torques and Collinsella, may mitigate neuroinflammation in the substantia nigra by increasing secondary bile acids. Interventions for the gut microbiota and their metabolites may potentially delay or mitigate the development and progression of PD and other Lewy body diseases.
Similar content being viewed by others


Introduction
Parkinson’s disease (PD) is pathologically characterized by abnormal accumulation of α-synuclein fibers (Lewy bodies) in the substantia nigra and other brain regions. However, the role of Lewy bodies has not been fully elucidated. PD is a movement disorder characterized by rigidity (muscle stiffness), postural instability, gait disturbance, bradykinesia (slowness of muscle movement), and resting tremor. However, PD patients often present with non-motor symptoms, including mild to severe cognitive impairment. Gastrointestinal symptoms of PD constitute the most common non-motor symptoms, with constipation being present in ~80% of PD patients1. Constipation usually precedes the motor symptoms of PD2 and worsens with disease progression. Similarly, the transit times of the small and large intestines are prolonged in PD3. In addition to constipation, non-motor symptoms such as rapid-eye-movement (REM) sleep behavior disorder (RBD), excessive daytime sleepiness, anosmia/hyposmia, and depression occur before the onset of the motor symptoms of PD.
Braak et al. examined the appearance of Lewy bodies in PD patients without cognitive symptoms and healthy participants and observed that the distribution of Lewy bodies ascended from the dorsal vagal nucleus to the locus coeruleus and substantia nigra4. The dorsal vagal nucleus innervates the gastrointestinal tract, and its distribution coincides with the manifestation of constipation as a prodromal PD symptom. In RBD, the sub-laterodorsal tegmental nucleus (SLD), located from the medulla oblongata to the bridge, is affected. SLD has a direct or indirect input to the spinal motor nuclei and inhibits skeletal muscle contraction during REM sleep, and its impairment may cause abnormal REM sleep behavior5. The locus coeruleus may also be involved in hypersomnolence in PD patients.
Two pathways have been proposed for the entry of a neurotropic pathogen into the nervous system in PD6. One pathway is from the olfactory bulb, and the other is from the oral and nasal cavities to the gastrointestinal tract and enteric nervous system (ENS)6. Although the olfactory pathway still remains controversial, the presence of the enteric pathway is supported by the ascending ɑ-synuclein pathology from the brainstem to the substantia nigra along with the ascendance of the disease loci for constipation, RBD, and depression prior to the onset of motor symptoms7. As discussed below, an approximately 50% reduction in the prevalence of PD after truncal vagotomy also supports the enteric pathway of PD8,9.
Radiological analysis has indicated the following two types of PD: body-first and brain-first PD10. PD patients with RBD, as well as idiopathic RBD patients, show the body-first trajectory, which represents an ascending progression from the intestinal tract and the cardiac sympathetic nervous system to the striatum. In contrast, PD patients without RBD show the brain-first trajectory, which represents descending progression from the amygdala to the intestinal tract and cardiac sympathetic nervous system. Gut microbiota is likely to be involved in the body-first PD.
In this article, we review the involvement of the gut–brain axis in the development and progression of PD from the perspective of the gut microbiota.
Parkinson’s disease and gastrointestinal disorders
Inflammatory bowel diseases (IBD) include ulcerative colitis (UC) and Crohn’s disease (CD). A meta-analysis revealed that UC and CD increase the risk ratio of PD 1.28- and 1.30-fold, respectively11. Interestingly, anti-tumor necrosis factor (TNF) therapy for IBD reduces the risk of PD12,13. The increased risk of PD in IBD patients is consistent with the fact that increased intestinal permeability is causally related to PD. However, only one in 23 colectomized patients with UC (average age, 52.0 years) has shown the presence of Lewy bodies in the ENS14, which indicates a low prevalence of abnormal α-synuclein fibrils in the ENS in UC. A genome-wide association study has shown that the leucine-rich repeat kinase 2 (LRRK2) gene is a major locus associated with increased susceptibility to CD15. Pathogenic variants of LRRK2 are the most common cause of familial PD and also account for ~1% of sporadic PD16. As high LRRK2 levels are observed in the inflamed colon in CD patients and in peripheral immune cells in sporadic PD patients, high levels and/or activities of LRRK2 may aggravate inflammatory processes in both CD and PD17,18. Thus, high LRRK2 levels in CD may be a potential biomarker for PD development and a potential therapeutic target to reduce the risk of PD in CD patients17.
Epidemiological studies show that total vagotomy reduces the hazard ratio of PD by approximately 50%8,9. The prion-like transmission of α-synuclein fibrils via the vagal nerve has also been observed in animal models19. Furthermore, stimulation of the vagal nerve in mice after administering lipopolysaccharides (LPS) suppresses the inflammation-induced production of TNF-α and interleukin (IL)-6 by microglia, which is not observed in vagotomized mice20. In addition, appendectomy may21 or may not22 reduce the risk of PD, which is in agreement with the observation that α-synuclein pathology begins in the vermiform appendix23. The vagal nerve is likely to play an essential role in the transmission of α-synuclein fibrils from the gut to the brain24.
Norovirus infection increases ɑ-synuclein production in the ENS, and ɑ-synuclein accelerates intestinal inflammation, suggesting an association between microbial infection and ɑ-synuclein pathology25. However, these observations are limited to PGP9.5-positive neurites, and the plexuses of Meissner and Auerbach have not been evaluated for the presence of α-synuclein-positive inclusions. Intestinal bacteria may be involved in gastrointestinal inflammation and promote the formation of abnormal protein aggregates in the intestine. Oral administration of the bacterial amyloid protein, curli, increases the levels of inflammatory markers and the aggregation of α-synuclein in the rat brain26. The intraperitoneal administration of LPS, which is a cell wall component of gram-negative bacteria, such as Escherichia coli and Salmonella spp, increases intestinal permeability and causes the accumulation of phosphorylated α-synuclein fibrils in the intestinal mucosa and dorsal vagal nuclei27. LPS also generates a self-renewable and structurally distinct strain of α-synuclein fibrils in vitro28.
PD is a multifactorial disease which is considerably influenced by environmental factors, and inheritance accounts for less than 10% of PD patients. Environmental exposure to herbicides and pesticides increases the risk of developing PD29. The inhibition of mitochondrial function or the induction of oxidative stress is mainly attributed to the toxicity of herbicides and pesticides30. Studies in mice have revealed that the pesticide rotenone, which inhibits mitochondrial electron transport complex I, causes the abnormal aggregation of α-synuclein fibrils in the ENS and subsequently in the substantia nigra pars compacta31,32. Vagotomy prevents the rotenone-mediated retrograde propagation of α-synuclein fibrils to the dorsal motor nucleus of the vagal nerve33.
Pathology of Lewy bodies in the gastrointestinal tract
The presence of Lewy bodies in the gastrointestinal plexus was first reported by Qualman et al. in 198434. Subsequently, Lewy bodies have been found in the vasoactive intestinal peptide-positive plexus but not in the tyrosine hydroxylase-positive plexus, which are both present in the myenteric plexus (Auerbach’s plexus)35. Some enteroendocrine cells (EECs), named neuropods, directly synapse to the vagal afferents36, which have a subsequent synaptic pathway to the substantia nigra and striatum37. In addition to the ENS, EECs also express α-synuclein38, which raises the possibility that α-synuclein fibrils originate in EECs and propagate to Auerbach’s neural plexus. Colon biopsies have revealed that Lewy bodies are already present in the intestinal perikarya and neurites at 2–5 years39 or up to 8 years40 prior to the onset of PD.
In PD, α-synuclein fibrils accumulate more frequently in the upper gastrointestinal tract than those in the lower gastrointestinal tract41,42. However, this observation remains controversial. First, the prevalence of α-synuclein fibrils in biopsies of the upper and lower gastrointestinal tracts remains undetermined40. Second, as the cells bearing Lewy bodies are vulnerable and decrease in abundance with the progression of PD, the upper gastrointestinal tract may have more viable cells with a small number of Lewy bodies. Indeed, 11C-donepezil positron emission tomography (PET) has revealed a sequential decrease in the parasympathetic innervations of the small intestine, colon, and kidney in PD43,44, although the presence of Lewy bodies in parasympathetic neurons has not been reported to the best of our knowledge. Therefore, the colon might have already lost cells with α-synuclein fibrils at the time of autopsy. Third, during an autopsy, the intestinal mucosa rapidly loses its structure, making histopathological evaluation difficult. Fourth, the intraperitoneal administration of LPS in mice induces α-synuclein fibrils more remarkably in the large intestine than that in the small intestine and increases intestinal permeability only in the large intestine27. Similar to the gastrointestinal tract, the abnormal accumulation of α-synuclein fibrils in the cutaneous sympathetic nerves has been found in the thoracic skin in two of 20 patients (10%), but none in the legs45. As the impairment of cutaneous sympathetic nerves is more remarkable in the extremities than that in the thorax with PD progression45, the lack of α-synuclein fibrils in the legs is likely to represent the loss of α-synuclein fibril-bearing sympathetic nerves. These observations may justify the apparent decrease in the number of α-synuclein fibrils in the lower gastrointestinal tract.
Smoking, coffee, and gut microbiota in PD
Both genetic and environmental factors influence the development of PD, and genetic factors have higher effects on PD, whereas environmental factors have higher numbers of affected individuals, compared to the other factors46. Smoking has decreased PD development by ~50% in 44 retrospective and four prospective studies47. Twin studies have shown that the risk of PD is inversely correlated with the dose of cigarette smoking48. Nicotine in cigarettes may exert protective effects against PD49. Gut microbiota may also mediate protective effects. Smoking reinforces the barrier of the large intestine50,51, which is potentially achieved by the increased abundance of Bacteroides and Prevotella and decreased abundance of Firmicutes and Actinobacteria, which are induced by smoking52,53. In contrast, smoking enhances oncogenic MAPK/ERK signaling and impairs the gut barrier function in mice54.
Coffee consumption also reduces PD development by ~30%55 and is also negatively correlated with the prevalence of constipation56. The polyphenols in coffee may exert protective effects against PD57. Coffee activates gallbladder contraction and colonic motility, increases the abundance of Bifidobacterium, and decreases the abundance of Clostridium58.
Constipation in PD
Constipation is observed in 54% of the patients with PD59. Constipation itself alters intestinal microbiota and increases mucosal permeability and inflammation. Patients with idiopathic constipation show increased serum antibody titers against Staphylococcus aureus and E. coli, suggesting the invasion of these bacteria across the orogastrointestinal barrier60. In addition, longer intestinal transit time is associated with the increased abundance of Akkermansia muciniphila, Bacteroides spp, and Alistipes61. The effects of confounding factors on gut microbiota in PD have shown that constipation increases the abundance of two genera (Hungatella and Lactobacillus) and decreases the abundance of three genera (Faecalibacterium, Lachnospiraceae ND3007 group, and Lachnospiraceae UCG-004)62. Our study63, along with a previous study64, showed that the degree of constipation in PD patients correlates with a decrease in the serum levels of LPS-binding protein (LBP). Chronic invasion of LPS from the intestinal tract may potentially reduce serum LBP levels. In PD, intestinal permeability is increased, and E. coli and nitrotyrosine, markers for protein oxidization, are abnormally stained in the intestinal mucosa64.
Gut microbiota in PD
Gut microbiota in PD has been analyzed using quantitative PCR, 16S rRNA sequencing (16S rRNA-seq), and shotgun metagenomic analysis (shotgun-seq), wherein qPCR has been performed in three studies63,65,66. Although qPCR can analyze a limited number of bacteria, it determines the absolute number of each bacterium, whereas 16S rRNA-seq and shotgun-seq determine the relative abundance of each bacterium. In our qPCR analysis, the sum of the absolute number of 19 representative intestinal bacteria, which account for 71.3% of the total intestinal bacteria, in PD was ~80% of that of controls63. This suggests that metabolites generated by the gut microbiota are likely to be reduced in PD.
The relative abundance of intestinal bacteria has been determined either using 16S rRNA-seq or shotgun-seq, and the results vary considerably among studies67,68,69,70,71,72. In addition, bacterial classifications and names vary considerably among reference databases. SILVA is the most commonly used taxonomic database, along with three other widely used databases. The difference in reference databases makes the direct comparison of different reports difficult. Thus, we established a method to meta-analyze non-parametric datasets and analyzed our own 16 S rRNA-seq dataset along with four other 16S rRNA-seq datasets62. Meta-analysis of gut microbiota in PD and controls across Japan, the United States, Finland, Russia, and Germany has shown an increased abundance of the mucin-degrading genus Akkermansia and decreased abundance of Roseburia, Faecalibacterium, and Lachnospiraceae ND3007, all of which produce short-chain fatty acids (SCFAs). In our dataset, the abundance of Akkermansia increased while that of Roseburia and Faecalibacterium decreased with PD progression. PICRUSt2 enables the prediction of functional effects from the 16 S rRNA-seq dataset. PICRUSt2 analysis has shown that butyrate and propionate metabolisms are altered in the gut microbiota in PD. This is consistent with a decrease in the abundance of SCFA-producing bacteria in PD. Metagenomic shotgun analyses68,72,73 also show a decrease in the abundance of SCFA-producing bacteria and an increase in that of Akkermansia, which is in accordance with 16S rRNA-seq analyses.
We66,74 and another group70 have reported longitudinal studies of the gut microbiota in PD. All three studies indicate that the gut microbiota is essentially unchanged over 2 years in both controls and PD. Our report shows that the increased abundance of Bifidobacterium at year 0 is associated with worsening in two years of the Universal Parkinson’s Disease Rating Scale (UPDRS) Part I, representing the non-motor experiences of daily living66. In contrast, Aho et al. reported that decreased Prevotella abundance at year 0 is associated with the rapid progression of PD70. Random forest models to differentiate PD patients who were at the same or better Hoehn & Yahr (HY) stage in 2 years (the stable group) and who moved to the advanced HY stage in 2 years (the deteriorated group) using gut microbiota at year 0 revealed that the abundance of only two SCFA-producing genera, Fusicatenibacter and Faecalibacterium, at year 0 were able to predict the progression of PD patients at HY stage 1 in 2 years with an area-under-the-curve of receiver operating characteristics (AUROC) of 0.86874. PD patients with low Fusicatenibacter and Faecalibacterium levels deteriorate faster than those with high levels of these bacteria, indicating that low SCFAs may accelerate the progression of PD.
Gut microbiota in RBD
RBD is prodromal to PD, and ~90% of RBD patients develop other Lewy body diseases, predominantly PD. Non-negative matrix factorization of gut microbiota using Liger, a tool for single-cell RNA sequencing analysis, in controls, RBD, and PD revealed four enterotypes. The proportion of controls decreased in the order of enterotypes A to D, whereas the proportion of severe PD increased in the same order, indicating that patients with severe PD tend to have specific enterotype(s)75. Akkermansia abundance is significantly increased in RBD patients compared with that in controls in Japan and Germany75,76. In contrast, the abundance of SCFA-producing genera Faecalibacterium, Roseburia, and Lachnospiraceae ND3007 group, all of which are decreased in PD, were not decreased in RBD. A low abundance of SCFA-producing bacteria may accelerate the transition of α-synuclein pathologies from RBD to PD.
Gut microbiota in dementia with Lewy bodies (DLB)
PD patients develop motor symptoms at first, and later develop dementia, which is called PD dementia (PDD), in advanced stages. In contrast, the onset of dementia in dementia with Lewy bodies (DLB) is before or less than one year after the onset of motor symptoms77. DLB is characterized by visual hallucinations, fluctuating cognitive impairment, sleep disturbance, movement disorders (Parkinsonism), and autonomic dysfunctions77,78. The clinical symptoms, cognitive profiles, and brain pathologies are similar between PDD and DLB79. The factors differentiating PDD and DLB remain unknown, and the gut microbiota is a potentially responsible factor.
Our recent study has shown that the abundance of SCFA-producing bacteria is decreased, and that of Akkermansia is increased in DLB80, as has been observed in PD62. In addition, the abundance of Ruminococcus torques and Collinsella is increased in DLB, but not in PD. The overall gut microbiota profiles are similar in patients with DLB and PD at HY stages 3 and 4 (HY3&4), which represent moderate to severe cases of PD. A random forest model to differentiate DLB from HY3&4 shows that high Ruminococcus torques, high Collinsella, and low Bifidobacterium abundances are predictive of DLB with an AUROC of 0.825. Low Bifidobacterium abundance is also observed in Alzheimer’s disease81,82, and supplementation with Bifidobacterium improves Alzheimer’s disease, presumably by inducing the brain-derived neurotrophic factor83. The increased abundance of Ruminococcus torques and Collinsella are likely to cause abnormal intestinal permeability84,85. In addition, Ruminococcus torques and Collinsella are major secondary bile acid-producing bacteria86. Indeed, the measurement of fecal secondary bile acids has revealed that the production of ursodeoxycholate (UDCA), a major secondary bile acid, is increased in DLB80. The anti-inflammatory87 and antioxidant effects88 of UDCA have been previously reported. The effects of UDCA on PD have also been reported89,90. Although not directly related to Lewy body diseases, countries where healthy people have a high abundance of Collinsella, have low mortality rates due to COVID-19, which may also be attributed to the anti-inflammatory effects of UDCA91. Thus, the anti-inflammatory effects of Ruminococcus torques and Collinsella are likely to be protective against neuroinflammation in the substantia nigra and retards the development of motor symptoms in DLB.
Intestinal bacterial metabolites in PD
Intestinal bacteria produce SCFA, vitamins, polyamines, secondary bile acids, branched-chain amino acids, trimethylamine N-oxide, tryptophan, and indole derivatives91,92. Shotgun metagenomic analyses have shown that biotin metabolism, glycan degradation, and the biosynthesis of phenylalanine, tyrosine, and tryptophan are decreased in PD68,73. High fecal concentrations of branched-chain and aromatic amino acids in PD patients are associated with an increased abundance of Alistipes, Rikenellaceae_RC9_gut_group, Bifidobacterium, and Parabacteroides, as well as a decreased abundance of Faecalibacterium93. A similar analysis has shown high levels of cadaverine, ethanolamine, hydroxypropionic acid, isoleucine, leucine, and phenylalanine in PD94. In contrast, glutamic, pyroglutamic, and succinic acid levels are significantly reduced in PD patients. The authors speculated that the increased intestinal metabolite levels in PD may induce oxidative stress and promote inflammatory responses and α-synuclein aggregation in ENS95.
In contrast to intestinal amino acids, intestinal SCFAs (acetate, propionate, and butyrate) are decreased in PD across studies65,96,97,98. Since butyrate is involved in the formation of the mucus layer in the intestinal epithelium, the intestinal mucosa may not be sufficiently formed in PD. In IBD, butyrate administration prevents the reduction of tight junction proteins and inhibits activation of the NF-κB signaling pathway by binding to GPR109A99. Instead, propionate is metabolized in the liver and is therefore present at low concentrations in peripheral circulation100. Acetic acid is the most abundant SCFA in blood. Furthermore, acetic acid can cross the blood–brain barrier and reduce appetite via central homeostatic mechanisms101. Acetic and propionic acids in the gut stimulate GPR41 and GPR43 to release both PYY and GLP-1, affecting satiety and intestinal transit102. Butyric acid exerts its anti-inflammatory effect by binding to GPR41, GPR43, and GPR109A, as well as by inhibiting histone deacetylase, the mechanism of which has been elaborated further in this review. Furthermore, propionate is converted to glucose by the gluconeogenic pathway in the intestine, which causes satiety and reduces hepatic glucose production103,104. The involvement of SCFA in PD pathology is addressed in the next section.
The causal association of gut microbiota with PD
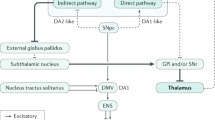
Based on the observations presented above, the following hypothesis was proposed for the gut–brain axis in PD (Fig. 1).

(Blue pathway) First, Akkermansia, which is increased in PD, degrades the intestinal mucus layer and increases intestinal permeability when dietary fibers are deficient. Thinning of the intestinal mucus layer allows toxic substances such as LPS and pesticides/herbicides to pass through the mucosal barrier, which triggers inflammation and oxidative stress in the intestine. Then, α-synuclein generated by the enteroendocrine cells and the intestinal neural plexus form insoluble fibrils. Abnormally aggregated α-synuclein fibrils ascend the vagal nerve and accumulate in the dorsal nucleus of the vagal nerve (the vagal nerve pathway). (Pink pathway) Second, Faecalibacterium, Roseburia, and Agathobacter, which are reduced in PD, are butyrate-producing bacteria. Butyrate is an energy source for colonic mucosal cells and is important for the maintenance of the intestinal epithelium and intestinal mucosa. Lack of SCFAs also causes thinning of the mucosal layer, and accelerates the vagal nerve pathway. SCFA also promotes the expression of Foxp3 (not shown) and the differentiation of naive T cells into regulatory T cells (Treg) by inhibiting histone deacetylases. LPS, pesticides, and herbicides additionally increase inflammatory cytokines. Peripheral inflammatory cytokines pass through the damaged blood–brain barrier (BBB) and trigger neuroinflammation. (Green pathway) Third, the lack of SCFAs also aggravates microglial activation, likely through reducing GLP-1, but the detailed mechanisms remain undetermined. (Mechanisms in DLB, not shown) Increased Collinsella and Ruminococcus torques in DLB, but not in PD, may mitigate neuroinflammation in the substantia nigra, possibly by producing secondary bile acids like ursodeoxycholate.
First, Akkermansia, whose abundance is increased in PD, degrades the intestinal mucus layer and increases intestinal permeability when dietary fiber is deficient105,106. Faecalibacterium and Roseburia, which are decreased in PD, produce butyrate, which is an energy source for colonic mucosal cells and is important to maintain the intestinal epithelium. Alterations in these bacteria may lead to thinning of the intestinal mucosa in PD and allow inflammatory substances such as LPS and even E. coli to pass through the mucosal layer64. Similarly, the abundance of Bacteroides and Verrucomicrobia are positively correlated with serum inflammatory cytokine levels in PD107. Furthermore, increased intestinal permeability also allows the enteric neural plexus to be exposed to toxins, such as pesticides and herbicides. Epidemiological studies have repeatedly demonstrated positive associations between pesticides/herbicides and PD108. Normal α-synuclein proteins generated in EECs and ENS are thus exposed to LPS, E. coli, and pesticides/herbicides, leading to the formation of abnormal α-synuclein fibrils, which may ascend the vagal nerve and accumulate in its dorsal nucleus.
In addition to PD, Akkermansia abundance also increases in multiple sclerosis109 and amyotrophic lateral sclerosis110. In contrast, its abundance is decreased in obesity, type 2 diabetes mellitus, autism, atopy, and IBD111. The paucity of Akkermansia in IBD may be accounted for by inflammatory thinning of the intestinal mucus layer. A high-fat diet decreases the intestinal mucus layer, and Akkermansia somehow rescues mucus layer thickness112. Increased Akkermansia levels may compensate for the thinned mucus layer triggered by a high-fat diet in obesity and diabetes. In contrast, a normal diet preserves the mucus layer in PD; however, an excessive abundance of Akkermansia may degrade the mucus layer.
Second, butyrate promotes the gene expression of FOXP3, and differentiates naive T cells into regulatory T (Treg) cells by inhibiting histone deacetylases113,114. Butyrate also acts on a G-protein-coupled receptor, GPR109a, expressed on dendritic cells and macrophages, and induces the differentiation of Treg cells and IL-10-producing T cells, leading to the expansion of Treg cells115. Butyrate also acts on GPR109a in the colonic epithelium and induces the production of IL-18, which also suppresses inflammation115. As a result, pro-inflammatory cytokine levels are elevated, and anti-inflammatory cytokines levels are suppressed in PD. The role of neuroinflammation in PD has been well-studied and reviewed18. For example, nonsteroidal anti-inflammatory drugs (NSAIDs) decrease the risk of PD116,117. Interestingly, ibuprofen, but not acetaminophen or aspirin, reduces the risk of PD by 35%118. Thus, a reduction in the abundance of butyrate-producing bacteria in PD may enhance neurodegeneration by failing to suppress neuroinflammation. In contrast to the protective effects of SCFAs against neuroinflammation in PD, the reduction in intestinal SCFAs owing to the depletion of gut microbiota by sanitization119 and antibiotics120 ameliorates motor deficits in rodent models of PD, whereas supplementation with SCFAs worsens them119. One possible reason that administration of SCFA worsened motor symptoms in a germ-free mouse model of PD119 was that microglia in germ-free mice was likely to be immature, because microglia requires SCFA to maturate and immature microglia cannot respond to endotoxin121. Administered SCFA was likely to have induced the maturation of microglia, which resulted in neuroinflammation and worsening of PD. In contrast to the germ-free mouse model of PD119, oral administration of butyrate protected the expression of α-synuclein in the colon and the substantia nigra, and prevented the loss of tyrosine hydroxylase-positive neurons in a rotenone-induced mouse model of PD122. Similarly, inulin, prebiotics yielding SCFA, mitigated microglial activation in mice123. More importantly, in PD patients, prebiotics yielding intestinal SCFA increased fecal and plasma SCFA levels, and decreased serum markers for intestinal and neuronal inflammations124.
Normal dopaminergic neurons, which are affected in Lewy body diseases, have multiple-branched, unmyelinated, and elongated neurites125. Energetic/metabolic burdens and subsequent oxidative stress are predicted to be high in neurons with multiple-branched neurites126. Thus, neurons in multiple brain regions are vulnerable to chronic neuroinflammation, and α-synuclein-expressing neurons may accumulate abnormal aggregates. This mechanism is likely to account for dopaminergic cell death in PD caused by decreased abundance of SCFA-producing bacteria. In contrast, in RBD, where Akkermansia abundance is increased but SCFA-producing bacterial abundance is not decreased, α-synuclein fibrils are formed in EECs and ENS and ascend the vagal nerve, but the lack of neuroinflammation is likely to suppress or delay the development of PD. Similarly, in DLB, neuroinflammation due to the lack of SCFAs is mitigated in the substantia nigra but not in the cortex by secondary bile acids generated by the increased abundance of Ruminococcus torques and Collinsella80. Indeed, intraperitoneal injection of LPS causes P2Y6 receptor-mediated activation of microglia and inflammatory neuronal loss in the substantia nigra but not in the cortex or hippocampus127. Thus, the mitigation of neuroinflammation is likely to be effective in the substantia nigra, but not in the cerebral cortex, where neuroinflammation may not critically aggravate neuronal cell death. DLB generally develops at the age of above 65 years128, and its average is higher than that of PD80,128. The lack of PD symptoms in older patients with DLB is consistent with the notion that dopaminergic neuronal cell death due to neuroinflammation at the substantia nigra is not critical in DLB. Similarly, the average age of RBD development is higher than75 or similar to129 that of PD. As stated above, the gut microbiota remains essentially unchanged over 2 years in both controls and PD66,70,74, and is independent of PD progression74. Therefore, the gut microbiota is likely to be a critical determinant of the nature and progression of brain pathology in patients with Lewy body diseases.
Early interventions for gut microbiota and their metabolites may potentially delay or mitigate the development and progression of non-motor, motor, and cognitive symptoms in Lewy body diseases.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
No datasets were generated or analyzed in this article.
References
Maeda, T. et al. Clinical manifestations of nonmotor symptoms in 1021 Japanese Parkinson’s disease patients from 35 medical centers. Parkinsonism Relat. Disord. 38, 54–60 (2017).
Abbott, R. D. et al. Frequency of bowel movements and the future risk of Parkinson’s disease. Neurology 57, 456–462 (2001).
Ito, M. et al. Drinking hydrogen water and intermittent hydrogen gas exposure, but not lactulose or continuous hydrogen gas exposure, prevent 6-hydorxydopamine-induced Parkinson’s disease in rats. Med. Gas. Res. 2, 15 (2012).
Braak, H. et al. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages). J. Neurol. 249, III/1–III/5 (2002).
Boeve, B. F. et al. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain 130, 2770–2788 (2007).
Hawkes, C. H., Del Tredici, K. & Braak, H. Parkinson’s disease: a dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 33, 599–614 (2007).
Cersosimo, M. G. & Benarroch, E. E. Pathological correlates of gastrointestinal dysfunction in Parkinson’s disease. Neurobiol. Dis. 46, 559–564 (2012).
Svensson, E. et al. Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 78, 522–529 (2015).
Liu, B. et al. Vagotomy and Parkinson disease: a Swedish register-based matched-cohort study. Neurology 88, 1996–2002 (2017).
Horsager, J. et al. Brain-first versus body-first Parkinson’s disease: a multimodal imaging case-control study. Brain 143, 3077–3088 (2020).
Zhu, F. et al. The risk of Parkinson’s disease in inflammatory bowel disease: A systematic review and meta-analysis. Dig. Liver Dis. 51, 38–42 (2019).
Peter, I. et al. Anti-tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol. 75, 939–946 (2018).
Park, S. et al. Patients with inflammatory bowel disease are at an increased risk of Parkinson’s disease: a South Korean nationwide population-based study. J. Clin. Med. 8, 1191 (2019).
Gibo, N. et al. Examination of abnormal alpha-synuclein aggregates in the enteric neural plexus in patients with ulcerative colitis. J. Gastrointestin. Liver Dis. 31, 290–300 (2022).
Franke, A. et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 42, 1118–1125 (2010).
Cookson, M. R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 11, 791–797 (2010).
Herrick, M. K. & Tansey, M. G. Is LRRK2 the missing link between inflammatory bowel disease and Parkinson’s disease? NPJ Parkinsons Dis. 7, 26 (2021).
Tansey, M. G. et al. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 22, 657–673 (2022).
Kim, S. et al. Transneuronal propagation of pathologic alpha-synuclein from the gut to the brain models Parkinson’s disease. Neuron 103, 627–641 (2019). e627.
Li, S. et al. Intestinal microbiota impact sepsis associated encephalopathy via the vagus nerve. Neurosci. Lett. 662, 98–104 (2018).
Killinger, B. A. et al. The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci. Transl. Med. 10, eaar5280 (2018).
Palacios, N., Hughes, K. C., Cereda, E., Schwarzschild, M. A. & Ascherio, A. Appendectomy and risk of Parkinson’s disease in two large prospective cohorts of men and women. Mov. Disord. 33, 1492–1496 (2018).
Gray, M. T., Munoz, D. G., Gray, D. A., Schlossmacher, M. G. & Woulfe, J. M. Alpha-synuclein in the appendiceal mucosa of neurologically intact subjects. Mov. Disord. 29, 991–998 (2014).
Forsythe, P., Bienenstock, J. & Kunze, W. A. Vagal pathways for microbiome-brain-gut axis communication. Adv. Exp. Med. Biol. 817, 115–133 (2014).
Stolzenberg, E. et al. A role for neuronal alpha-synuclein in gastrointestinal immunity. J. Innate Immun. 9, 456–463 (2017).
Chen, S. G. et al. Exposure to the functional bacterial amyloid protein curli enhances alpha-synuclein aggregation in aged Fischer 344 rats and Caenorhabditis elegans. Sci. Rep. 6, 34477 (2016).
Kelly, L. P. et al. Progression of intestinal permeability changes and alpha-synuclein expression in a mouse model of Parkinson’s disease. Mov. Disord. 29, 999–1009 (2014).
Kim, C. et al. Exposure to bacterial endotoxin generates a distinct strain of alpha-synuclein fibril. Sci. Rep. 6, 30891 (2016).
Chen, H. et al. Research on the premotor symptoms of Parkinson’s disease: clinical and etiological implications. Environ. Health Perspect. 121, 1245–1252 (2013).
Tanner, C. M. et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 119, 866–872 (2011).
Sherer, T. B., Kim, J. H., Betarbet, R. & Greenamyre, J. T. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp. Neurol. 179, 9–16 (2003).
Pan-Montojo, F. et al. Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS ONE 5, e8762 (2010).
Pan-Montojo, F. et al. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci. Rep. 2, 898 (2012).
Qualman, S. J., Haupt, H. M., Yang, P. & Hamilton, S. R. Esophageal Lewy bodies associated with ganglion cell loss in achalasia. Similarity to Parkinson’s disease. Gastroenterology 87, 848–856 (1984).
Wakabayashi, K., Takahashi, H., Ohama, E. & Ikuta, F. Parkinson’s disease: an immunohistochemical study of Lewy body-containing neurons in the enteric nervous system. Acta Neuropathol. 79, 581–583 (1990).
Kaelberer, M. M. et al. A gut-brain neural circuit for nutrient sensory transduction. Science 361, eaat5236 (2018).
Han, W. et al. A neural circuit for gut-induced reward. Cell 175, 665–678 (2018). e623.
Chandra, R., Hiniker, A., Kuo, Y. M., Nussbaum, R. L. & Liddle, R. A. alpha-Synuclein in gut endocrine cells and its implications for Parkinson’s disease. JCI Insight 2, e92295 (2017).
Shannon, K. M., Keshavarzian, A., Dodiya, H. B., Jakate, S. & Kordower, J. H. Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov. Disord. 27, 716–719 (2012).
Hilton, D. et al. Accumulation of alpha-synuclein in the bowel of patients in the pre-clinical phase of Parkinson’s disease. Acta Neuropathol. 127, 235–241 (2014).
Wakabayashi, K., Takahashi, H., Takeda, S., Ohama, E. & Ikuta, F. Parkinson’s disease: the presence of Lewy bodies in Auerbach’s and Meissner’s plexuses. Acta Neuropathol. 76, 217–221 (1988).
Beach, T. G. et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 119, 689–702 (2010).
Gjerloff, T. et al. Imaging acetylcholinesterase density in peripheral organs in Parkinson’s disease with 11C-donepezil PET. Brain 138, 653–663 (2015).
Fedorova, T. D. et al. Decreased intestinal acetylcholinesterase in early Parkinson disease: an (11)C-donepezil PET study. Neurology 88, 775–781 (2017).
Miki, Y. et al. Clinical availability of skin biopsy in the diagnosis of Parkinson’s disease. Neurosci. Lett. 469, 357–359 (2010).
Hopfner, F. et al. beta-adrenoreceptors and the risk of Parkinson’s disease. Lancet Neurol. 19, 247–254 (2020).
Hernan, M. A., Takkouche, B., Caamano-Isorna, F. & Gestal-Otero, J. J. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann. Neurol. 52, 276–284 (2002).
Tanner, C. M. et al. Smoking and Parkinson’s disease in twins. Neurology 58, 581–588 (2002).
Quik, M., Perez, X. A. & Bordia, T. Nicotine as a potential neuroprotective agent for Parkinson’s disease. Mov. Disord. 27, 947–957 (2012).
Prytz, H., Benoni, C. & Tagesson, C. Does smoking tighten the gut? Scand. J. Gastroenterol. 24, 1084–1088 (1989).
Wang, H. et al. Side-stream smoking reduces intestinal inflammation and increases expression of tight junction proteins. World J. Gastroenterol. 18, 2180–2187 (2012).
Benjamin, J. L. et al. Smokers with active Crohn’s disease have a clinically relevant dysbiosis of the gastrointestinal microbiota. Inflamm. Bowel Dis. 18, 1092–1100 (2012).
Biedermann, L. et al. Smoking cessation alters intestinal microbiota: insights from quantitative investigations on human fecal samples using FISH. Inflamm. Bowel Dis. 20, 1496–1501 (2014).
Bai, X. et al. Cigarette smoke promotes colorectal cancer through modulation of gut microbiota and related metabolites. Gut 71, 2439–2450 (2022).
Wirdefeldt, K., Adami, H. O., Cole, P., Trichopoulos, D. & Mandel, J. Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur. J. Epidemiol. 26, S1–S58 (2011).
Murakami, K., Okubo, H. & Sasaki, S. Dietary intake in relation to self-reported constipation among Japanese women aged 18-20 years. Eur. J. Clin. Nutr. 60, 650–657 (2006).
Prediger, R. D. Effects of caffeine in Parkinson’s disease: from neuroprotection to the management of motor and non-motor symptoms. J. Alzheimers Dis. 20, S205–S220 (2010).
Khokhlova, E. V. et al. Anti-inflammatory properties of intestinal Bifidobacterium strains isolated from healthy infants. Microbiol. Immunol. 56, 27–39 (2012).
Gan, J. et al. A survey of subjective constipation in Parkinson’s disease patients in shanghai and literature review. BMC Neurol. 18, 29 (2018).
Khalif, I. L., Quigley, E. M., Konovitch, E. A. & Maximova, I. D. Alterations in the colonic flora and intestinal permeability and evidence of immune activation in chronic constipation. Dig. Liver Dis. 37, 838–849 (2005).
Asnicar, F. et al. Blue poo: impact of gut transit time on the gut microbiome using a novel marker. Gut 70, 1665–1674 (2021).
Nishiwaki, H. et al. Meta-analysis of gut dysbiosis in Parkinson’s disease. Mov. Disord. 35, 1626–1635 (2020).
Hasegawa, S. et al. Intestinal dysbiosis and lowered serum lipopolysaccharide-binding protein in Parkinson’s disease. PLoS ONE 10, e0142164 (2015).
Forsyth, C. B. et al. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS ONE 6, e28032 (2011).
Unger, M. M. et al. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat. Disord. 32, 66–72 (2016).
Minato, T. et al. Progression of Parkinson’s disease is associated with gut dysbiosis: two-year follow-up study. PLoS ONE 12, e0187307 (2017).
Hopfner, F. et al. Gut microbiota in Parkinson disease in a northern German cohort. Brain Res. 1667, 41–45 (2017).
Bedarf, J. R. et al. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naive Parkinson’s disease patients. Genome Med. 9, 39 (2017).
Lin, A. et al. Gut microbiota in patients with Parkinson’s disease in southern China. Parkinsonism Relat. Disord. 53, 82–88 (2018).
Aho, V. T. E. et al. Gut microbiota in Parkinson’s disease: temporal stability and relations to disease progression. EBioMedicine 44, 691–707 (2019).
Romano, S. et al. Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. npj Parkinsons Dis. 7, 27 (2021).
Chen, S. J. et al. Association of fecal and plasma levels of short-chain fatty acids with gut microbiota and clinical severity in patients with Parkinson disease. Neurology 98, e848–e858 (2022).
Qian, Y. et al. Gut metagenomics-derived genes as potential biomarkers of Parkinson’s disease. Brain 143, 2474–2489 (2020).
Nishiwaki, H. et al. Short chain fatty acids-producing and mucin-degrading intestinal bacteria predict the progression of early Parkinson’s disease. npj Parkinsons Dis. 8, 65 (2022).
Nishiwaki, H. et al. Short-chain fatty acid-producing gut microbiota is decreased in Parkinson’s disease but not in rapid-eye-movement sleep behavior disorder. mSystems 5, e00797–20 (2020).
Heintz-Buschart, A. et al. The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov. Disord. 33, 88–98 (2018).
McKeith, I. G. et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology 89, 88–100 (2017).
Outeiro, T. F. et al. Dementia with Lewy bodies: an update and outlook. Mol. Neurodegener. 14, 5 (2019).
Gomperts, S. N. Lewy body dementias: dementia with Lewy bodies and Parkinson disease dementia. Continuum 22, 435–463 (2016).
Nishiwaki, H. et al. Gut microbiota in dementia with Lewy bodies. npj Parkinsons Dis. 8, 169 (2022).
Zhuang, Z. Q. et al. Gut microbiota is altered in patients with Alzheimer’s disease. J. Alzheimers Dis. 63, 1337–1346 (2018).
Haran, J. P. et al. Alzheimer’s disease microbiome is associated with dysregulation of the anti-inflammatory P-glycoprotein pathway. mBio 10, e00632–19 (2019).
Kobayashi, Y., Kuhara, T., Oki, M. & Xiao, J. Z. Effects of Bifidobacterium breve A1 on the cognitive function of older adults with memory complaints: a randomised, double-blind, placebo-controlled trial. Benef. Microbes 10, 511–520 (2019).
Png, C. W. et al. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am. J. Gastroenterol. 105, 2420–2428 (2010).
Chen, J. et al. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. 8, 43 (2016).
Lee, J. Y. et al. Contribution of the 7beta-hydroxysteroid dehydrogenase from Ruminococcus gnavus N53 to ursodeoxycholic acid formation in the human colon. J. Lipid Res. 54, 3062–3069 (2013).
Ko, W. K. et al. Anti-inflammatory effects of ursodeoxycholic acid by lipopolysaccharide-stimulated inflammatory responses in RAW 264.7 macrophages. PLoS ONE 12, e0180673 (2017).
Lapenna, D. et al. Antioxidant properties of ursodeoxycholic acid. Biochem. Pharmacol. 64, 1661–1667 (2002).
Mortiboys, H. et al. UDCA exerts beneficial effect on mitochondrial dysfunction in LRRK2(G2019S) carriers and in vivo. Neurology 85, 846–852 (2015).
Qi, H., Shen, D., Jiang, C., Wang, H. & Chang, M. Ursodeoxycholic acid protects dopaminergic neurons from oxidative stress via regulating mitochondrial function, autophagy, and apoptosis in MPTP/MPP(+)-induced Parkinson’s disease. Neurosci. Lett. 741, 135493 (2021).
Hirayama, M. & Ohno, K. Parkinson’s disease and gut microbiota. Ann. Nutr. Metab. 77, 28–35 (2021).
Agus, A., Clement, K. & Sokol, H. Gut microbiota-derived metabolites as central regulators in metabolic disorders. Gut 70, 1174–1182 (2021).
Yan, Z. et al. Alterations of gut microbiota and metabolome with Parkinson’s disease. Microb. Pathog. 160, 105187 (2021).
Vascellari, S. et al. Gut microbiota and metabolome alterations associated with Parkinson’s disease. mSystems 5, e00561–20 (2020).
Vascellari, S. et al. Clinical phenotypes of Parkinson’s disease associate with distinct gut microbiota and metabolome enterotypes. Biomolecules 11, 144 (2021).
Aho, V. T. E. et al. Relationships of gut microbiota, short-chain fatty acids, inflammation, and the gut barrier in Parkinson’s disease. Mol. Neurodegener. 16, 6 (2021).
Tan, A. H. et al. Gut microbial ecosystem in Parkinson disease: new clinicobiological insights from multi-omics. Ann. Neurol. 89, 546–559 (2021).
Chen, S. J. et al. Alteration of gut microbial metabolites in the systemic circulation of patients with Parkinson’s disease. J. Parkinsons Dis. 12, 1219–1230 (2022).
Chen, G. et al. Sodium butyrate inhibits inflammation and maintains epithelium barrier integrity in a TNBS-induced inflammatory bowel disease mice model. EBioMedicine 30, 317–325 (2018).
Brass, E. P. & Beyerinck, R. A. Effects of propionate and carnitine on the hepatic oxidation of short- and medium-chain-length fatty acids. Biochem. J. 250, 819–825 (1988).
Frost, G. et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 5, 3611 (2014).
D’Alessio, D. Intestinal hormones and regulation of satiety: the case for CCK, GLP-1, PYY, and Apo A-IV. J. Parenter. Enter. Nutr. 32, 567–568 (2008).
De Vadder, F. et al. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 156, 84–96 (2014).
Koh, A., De Vadder, F., Kovatcheva-Datchary, P. & Backhed, F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 165, 1332–1345 (2016).
Desai, M. S. et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 167, 1339–1353 (2016). e1321.
Earle, K. A. et al. Quantitative imaging of gut microbiota spatial organization. Cell Host Microbe 18, 478–488 (2015).
Lin, C. H. et al. Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J. Neuroinflammation 16, 129 (2019).
Islam, M. S. et al. Pesticides and Parkinson’s disease: current and future perspective. J. Chem. Neuroanat. 115, 101966 (2021).
Zoledziewska, M. The gut microbiota perspective for interventions in MS. Autoimmun. Rev. 18, 814–824 (2019).
Casani-Cubel, J. et al. The impact of microbiota on the pathogenesis of amyotrophic lateral sclerosis and the possible benefits of polyphenols. An overview. Metabolites 11, 120 (2021).
Derrien, M., Belzer, C. & de Vos, W. M. Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog. 106, 171–181 (2017).
Everard, A. et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl Acad. Sci. USA 110, 9066–9071 (2013).
Smith, P. M. et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 (2013).
Arpaia, N. et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455 (2013).
Singh, N. et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40, 128–139 (2014).
Casper, D., Yaparpalvi, U., Rempel, N. & Werner, P. Ibuprofen protects dopaminergic neurons against glutamate toxicity in vitro. Neurosci. Lett. 289, 201–204 (2000).
Chen, H. et al. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch. Neurol. 60, 1059–1064 (2003).
Chen, H. et al. Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann. Neurol. 58, 963–967 (2005).
Sampson, T. R. et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167, 1469–1480 (2016). e1412.
Bisht, R., Kaur, B., Gupta, H. & Prakash, A. Ceftriaxone mediated rescue of nigral oxidative damage and motor deficits in MPTP model of Parkinson’s disease in rats. Neurotoxicology 44, 71–79 (2014).
Erny, D. et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 18, 965–977 (2015).
Zhang, Y. et al. Sodium butyrate ameliorates gut dysfunction and motor deficits in a mouse model of Parkinson’s disease by regulating gut microbiota. Front. Aging Neurosci. 15, 1099018 (2023).
Caetano-Silva, M. E. et al. Inhibition of inflammatory microglia by dietary fiber and short-chain fatty acids. Sci. Rep. 13, 2819 (2023).
Hall, D. A. et al. An open label, non-randomized study assessing a prebiotic fiber intervention in a small cohort of Parkinson’s disease participants. Nat. Commun. 14, 926 (2023).
Braak, H. & Tredici, K. D. Neuroanatomy and pathology of sporadic Parkinson’s disease. Adv. Anat. Embryol. Cell Biol. 201, 1–119 (2009).
Uchihara, T. & Giasson, B. I. Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 131, 49–73 (2016).
Milde, S. et al. Inflammatory neuronal loss in the substantia nigra induced by systemic lipopolysaccharide is prevented by knockout of the P2Y(6) receptor in mice. J. Neuroinflammation 18, 225 (2021).
McKeith, I. G. Author response: diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology 90, 300–301 (2018).
Kashihara, K., Imamura, T. & Shinya, T. Cardiac 123I-MIBG uptake is reduced more markedly in patients with REM sleep behavior disorder than in those with early stage Parkinson’s disease. Parkinsonism Relat. Disord. 16, 252–255 (2010).
Acknowledgements
Studies performed in the authors’ laboratories were supported by Grants-in-Aid from the Japan Agency for Medical Research and Development (JP21gm1010002, JP22bm0804005, and JP22ek0109488), the Japan Society for the Promotion of Science (JP20H03561, JP22K17343, and JP22K15394), the Ministry of Health, Labor, and Welfare of Japan (20FC1036), the National Center of Neurology and Psychiatry (2–5), and the Hori Science and Arts Foundation.
Author information
Authors and Affiliations
Contributions
M.H. wrote the original draft. H.N., M.H., and K.O. scrutinized the content and references and made critical comments. All the authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics
Human studies performed in our laboratory were approved by the Ethical Review Committees of the Nagoya University Graduate School of Medicine (approval #2016-0151), Iwate Medical University (H28-123), Okayama Kyokuto Hospital (approval #kyoIR-2016002), and Fukuoka University School of Medicine (approval #2016M027).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hirayama, M., Nishiwaki, H., Hamaguchi, T. et al. Gastrointestinal disorders in Parkinson’s disease and other Lewy body diseases. npj Parkinsons Dis. 9, 71 (2023). https://doi.org/10.1038/s41531-023-00511-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-023-00511-2
This article is cited by
-
Gut microbiota, circulating cytokines and dementia: a Mendelian randomization study
Journal of Neuroinflammation (2024)
