
Abstract
Type-2 diabetes (T2D) and glucose metabolic imbalances have been linked to neurodegenerative diseases, including Parkinson’s disease (PD). To detect potential effects of different glucose levels on gene expression, by RNA-seq we analyzed the transcriptome of dermal fibroblasts from idiopathic PD (iPD) patients, LRRK2-associated PD (L2PD) patients, and healthy controls (total n = 21 cell lines), which were cultured at two different glucose concentrations (25 and 5 mM glucose). In PD patients we identified differentially expressed genes (DEGs) that were related to biological processes mainly involving the plasmatic cell membrane, the extracellular matrix, and also neuronal functions. Such pathway deregulation was largely similar in iPD or L2PD fibroblasts. Overall, the gene expression changes detected in this study were associated with PD independently of glucose concentration.
Similar content being viewed by others

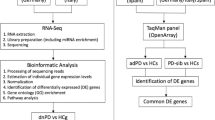
Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder associated with the progressive loss of dopaminergic neurons (DAn) in the substantia nigra. Although most patients are idiopathic PD cases (iPD) of unknown cause, around 5–10% encompass monogenic forms caused by pathogenic mutations. Among these, mutations in the leucine-rich repeat kinase 2 gene (LRRK2) causing LRRK2-associated PD (L2PD) are the most frequent cause of autosomal-dominant PD but also of sporadic cases by reduced penetrance1. Clinically, L2PD largely resembles iPD, suggesting that both PD types could share similar pathogenic mechanisms2. The most frequent LRRK2 G2019S mutation has been associated with cytoskeletal remodeling and abnormal vesicle trafficking resulting in shortening of the axonic and dendritic trees in cultured neurons3.
Imbalances of glucose metabolism and type-2 diabetes (T2D) have been linked to PD, eventually with conflicting results4,5. A recent meta‐analyses of observational studies and genetic data showed compelling evidence supporting an effect of diabetes on PD risk6. In fact, a certain degree of molecular pathway overlap between PD and T2D has been reported7,8. Yet, studies addressing specific effects of glucose levels in PD patients are needed. In this context, in the present study, we explored the transcriptome of PD by comprehensive RNA-seq analyses using primary skin fibroblasts from patients with iPD and monogenic L2PD, which were cultured at low and high-glucose concentrations (5 vs. 25 mM glucose). The goals of the study were to assess differential gene expression changes associated with PD using peripheral fibroblasts cells, compare the transcriptome of the iPD and L2PD disease forms, and explore the potential effects of different culture glucose levels on the PD transcriptome.
Results
Transcriptomic analysis in the discovery cohort
In the discovery phase, at a low-glucose concentration medium (5 mM Glucose), we found 636 DEGs associated with disease in the overall PD group compared to healthy controls (Fig. 1a, b). Segregating by subtype, we found 846 DEGs in iPD and 594 in L2PD (Supplementary Table 1). At high-glucose medium (25 mM Glucose), we identified 931 DEGs in the whole PD group with respect to controls and, by subtype, 1295 DEGs in iPD and 664 in L2PD. In addition, hierarchical clustering analysis of gene expression data confirmed a highly similar expression profile in all PD cases for which L2PD and iPD clustered together apart from controls in both low and high-glucose conditions (Fig. 2a, b).

Differentially expressed genes (DEGs) with a P-value <0.05 and a fold-change FC ≥ |2| are depicted in red (up-regulated) and blue (down-regulated). a Discovery cohort at 25 mM glucose. b Discovery cohort at 5 mM glucose. c Validation cohort at 25 mM glucose. d Validation cohort at 5 mM glucose. Volcano plots were done using default settings at the VolcaNoSer software. Annotated dots correspond to the ten DEG data points with the largest (Manhattan) distance from the origin above the significance thresholds indicated by the dashed line. VolcaNoSer: https://huygens.science.uva.nl/VolcaNoseR/.

a Discovery cohort at 25 mM glucose. b Discovery cohort at 5 mM glucose. c Validation cohort at 25 mM glucose. d Validation cohort at 5 mM glucose. Heatmaps of DEGs (P-value<0.05 and fold change FC ≥ |2|) were performed using default settings at the Clustvis software. e Venn diagram representation showing DEG overlap between PD vs. controls in the four different comparisons, where “A” corresponds to discovery 25 mM glucose, “B” discovery 5 mM glucose, “C” Validation 25 mM glucose, and “D” validation 5 mM glucose. Clustvis: https://biit.cs.ut.ee/clustvis/.
Transcriptomic analysis in the validation cohort
In addition, we used an independent set of additional iPD and L2PD patients to assess the validity of these results. Compared to controls, in the overall PD group, we again obtained similar transcriptomic changes with 405 DEGs associated with PD at low glucose (484 DEGs in iPD and 339 in L2PD), and 350 DEGs associated with PD at high-glucose (369 DEGs in iPD and 302 in L2PD) (Fig. 1c, d and Supplementary Table 2). As in the discovery set, hierarchical clustering analysis of DEGs at the validation phase also revealed similar expression patterns in iPD and L2PD with respect to controls both in high and low glucose (Fig. 2c, d)
Biological enrichment analysis
To assess the biological functions of the identified PD-associated DEGs at low and high-glucose concentration, we performed a functional gene enrichment analysis. In the discovery set, we observed a statistically significant association with biological processes mainly related to the plasmatic membrane, cell adhesion, extracellular matrix, and neuronal processes (Table 1). Despite the higher number of DEGs found in iPD with respect to L2PD which are potentially related to the higher number of iPD studied patients than L2PD, we found very similar affected biological terms in iPD or L2PD compared separately with controls (Supplementary Table 3). In the validation set, the biological enrichment analysis at low and high glucose again revealed biological terms mainly related to the extracellular matrix, cell adhesion, plasma membrane, and synapsis (Table 1 and Supplementary Table 3). Remarkably, despite the limited DEG overlap across all four comparisons (Fig. 1e) we found a highly consistent pathway deregulation (Table 1 and Supplementary Table 3).
Discussion
Here we report transcriptome alterations in primary skin fibroblast from a set of patients with iPD and monogenic L2PD for the first time. Akin to previous observations showing that L2PD is clinically undistinguishable from iPD1,2, here we found that the functional pathways altered at the transcriptome level in iPD and L2PD are also largely similar, thus indicating similarities at the molecular level between both disease forms. Yet, these results should be interpreted with caution considering the reduced number of samples when segregating PD patients into iPD and L2PD. Although we found a limited gene overlap among all four comparisons, we also found that biological pathways affected in PD patients, either L2PD or iPD, were very similar across the different cohorts and comparisons. These gene expression changes were independent of different glucose concentrations under the tested culture conditions.
The identified expression changes associated with PD in idiopathic and monogenic L2PD involved alterations related to the plasma membrane, extracellular matrix, cell adhesion, and neural processes. These results are in line with previous studies reporting deregulation in the cytoskeleton, cell adhesion, or extracellular matrix processes in various PD patients biospecimens such as blood cells9,10, iPSC-derived DAn11,12, or post-mortem substantia nigra13. Specifically, the previous transcriptomic study available in PD fibroblasts was done in patients carrying PRKN mutations14. Interestingly, PRKN-associated PD showed deregulation of cell adhesion processes and other terms that emerged specific for this disease form, such as aminoacid and folate metabolism suggesting the presence of additional alterations in this other PD monogenic. In addition, a remarkable finding is that given the peripheral non-neuronal type cell nature of skin fibroblasts, we found biological enrichment in terms related to neuron or synapsis in iPD and L2PD. These results can be interpreted given that neurons and non-neuronal peripheral cells such as fibroblasts share common molecular mechanisms involving cell adhesion and extracellular matrix leading to filopodia and cilia formation15, which mediate axon guidance and synaptic plasticity in a neural cell context16.
Overall, PD fibroblasts have been mostly used to investigate PD mitochondrial phenotypes until date17,18and to study cytoskeleton and cell adhesion regulatory mechanisms19. Based on our findings that the main expression changes detected in iPD and L2PD fibroblasts are related to cytoskeleton rather than mitochondria, additional functional studies investigating the interplay between gene expression deregulation and cytoskeleton-related processes are warranted in PD fibroblasts, most especially given the easy availability and robustness of this cell model. In addition, some of the top hits across all comparisons (Fig. 1) have been previously linked to PD. For example, the APOC4-APOC2 protein was significantly altered in plasma from PD patients20 and NOMO1 in isolated neurons from post-mortem sustantia nigra of PD patients21. Moreover, TUG1 was shown to modify neurodegeneration in a cell model of PD22, and IDO1 in a PD animal model23.
Previous studies suggested a role for glucose imbalances in PD pathology, including an association of T2D with PD, as reported in a recent comprehensive meta-analysis6. In PD fibroblasts, some studies reported alterations in the glycolytic activity and that this glycolytic capacity can be altered by the presence or absence of media glucose, although results are still conflicting and need further research (reviewed in ref. 24). In our study we did not observe an influence of different glucose concentrations on the biological processes related to PD, at least at the transcriptomic level and under the tested culture conditions. However, we cannot discard that glucose imbalances in the studied PD fibroblasts could affect other processes beyond gene expression like metabolic and mitochondrial plasticity25. Indeed, other studies described cell death effects of extracellular glucose levels in PD dopaminergic neurons26.
In summary, we identified similar biological processes altered in the transcriptome from iPD and G2019S L2PD related to cell adhesion processes and neural functions. These biological processes were associated with PD but did not seem to be influenced by the studied glucose concentrations in our cell model.
Methods
Subjects
Primary skin biopsies were obtained from iPD patients without a family history of disease and without LRRK2 mutations, L2PD patients carrying the G2019S mutation, and cultured fresh. The resulting dermal fibroblasts were expanded and cryopreserved in liquid N2 until use. In the discovery cohort, we screened seven PD patients (four iPD patients and three unrelated L2PD) and four healthy controls with no history of neurological disease. The validation cohort included five PD patients (three iPD and two unrelated L2PD) and five controls (Table 2). All subjects were Europeans of Spanish origin. We used Taqman assays (Thermo Fisher Sci.) on a Step One Plus Real-time PCR System (Thermo Fisher Sci.) to genotype LRRK2 G2019S (#C-63498123-10), and another TaqMan assay to genotype R1441G/C/H as previously described27. All 5 L2PD patients at discovery and validation were heterozygous G2019S carriers. Of these, 4 L2PD patients were familial cases with another affected family member, and 1 L2PD was the only affected family member. The studied LRRK2 mutations were absent in the iPD patients and healthy controls. No systematic sequencing was done to exclude additional mutations causing monogenic PD. The study was approved by the local ethics committee and conducted according to the Declaration of Helsinki. Written informed consent was obtained from all participating subjects. Patient data were encoded to protect confidentiality.
Cell culture
Fibroblasts from PD patients and controls were simultaneously cultured per duplicate in DMEM high-glucose (25 mM) (Thermofisher #41966029), and DMEM low-glucose (5 mM) (Thermofisher #31885023), supplemented with 10% calf serum (Gibco #10500064), and penicillin 100 U/mL / streptomycin 100 μg/mL (Sigma #15140122) at 37 °C and 10% CO2. Fibroblasts were seeded at cell passage numbers between 2 and 6 for discovery and 1–3 for validation, grown until reaching the first confluence and passed using trypsin/EDTA (Sigma #15090046). After the second confluence, fibroblasts were harvested, and dry pellets were stored at −80 °C until subsequent RNA isolation using the AllPrep DNA/RNA/Protein kit (Qiagen #80004). The total culture period was of 10–14 days depending on the cell line. During this period, cells were constantly maintained in their corresponding glucose concentration medium, with regular medium renovation every 2–3 days. Regarding glucose conditions, normal fasting glucose level in the blood for non-diabetics is 3.9–5.6 mmol/L, and accordingly, 5 mM glucose represents a normoglycaemic condition. In addition, 25 mM glucose has been usually chosen in the scientific literature to model hyperglycemia in different cell culture types28,29, including PD fibroblasts where hyperglycaemic associated changes were reported25.
mRNA library preparation and RNA-seq
Total RNA samples were quality controlled by Qubit® RNA BR Assay kit (Thermo Fisher Scientific) for quantity and RNA 6000 Nano Assay (Agilent) for integrity. Samples were analyzed by RNA-seq in three different array batches as follows, including discovery batches 2 (high glucose) and batch 3 (low glucose), and validation batch 3 (high and low glucose).
Batch_02: The RNASeq libraries were prepared from total RNA using the TruSeq™ RNA Sample Prep Kit v2 (Illumina) with minor modifications. Briefly, after poly-A based mRNA enrichment with oligo-dT magnetic beads and 0.5 μg of total RNA as the input material the mRNA was fragmented. After first and second-strand cDNA synthesis the double-stranded cDNA was end-repaired, 3´adenylated and the Illumina barcoded adapters were ligated. The ligation product was enriched by 10 cycles of PCR. The final library was validated on an Agilent 2100 Bioanalyzer with the DNA 7500 assay. Each library was sequenced on HiSeq2000 (Illumina) in paired-end mode with a read length of 2 × 76 bp using TruSeq SBS Kit v4 in a fraction of a sequencing v4 flow cell lane, following the manufacturer’s protocol. Image analysis, base calling, and quality scoring of the run were processed using the manufacturer’s software Real Time Analysis (RTA 1.18.61) and followed by generation of FASTQ sequence files by CASAVA.
Batch_03: The RNASeq libraries were prepared from total RNA using the TruSeq™ Stranded mRNA Library Prep Kit (Illumina). In brief, the mRNA was enriched with oligo-dT magnetic beads from total RNA (500 ng) and it was fragmented to 80–450 nt. The second strand cDNA synthesis was performed in the presence of dUTP to achieve the strand specificity. The blunt-ended double-stranded cDNA was 3´adenylated and Illumina single-index adapters were ligated. The ligation product was enriched with 15 PCR cycles. The final library was validated on an Agilent 2100 Bioanalyzer with the DNA 7500 assay. The libraries were sequenced on HiSeq2000 (Illumina) in paired-end mode with a read length of 2x76bp using TruSeq SBS Kit v4 in a fraction of a sequencing v4 flow cell lane, following the manufacturer’s protocol. Image analysis, base calling and quality scoring of the run were processed using the manufacturer’s software Real Time Analysis (RTA 1.18.66.3) and followed by the generation of FASTQ sequence files.
Batch_04: The RNASeq libraries were prepared with KAPA Stranded mRNA-Seq Illumina® Platforms Kit (Roche) following the manufacturer´s recommendations. Briefly, 500 ng of total RNA was used as the input material, the poly-A fraction was enriched with oligo-dT magnetic beads and the mRNA was fragmented. The strand specificity was achieved during the second strand synthesis performed in the presence of dUTP instead of dTTP. The blunt-ended double-stranded cDNA was 3´adenylated and Illumina platform compatible adaptors with unique dual indexes and unique molecular identifiers (Integrated DNA Technologies) were ligated. The ligation product was enriched with 15 PCR cycles and the final library was validated on an Agilent 2100 Bioanalyzer with the DNA 7500 assay. The libraries were sequenced on HiSeq4000 (Illumina) in a fraction of a HiSeq 4000 PE Cluster kit sequencing flow cell lane, following the manufacturer’s protocol for dual indexing. Image analysis, base calling, and quality scoring of the run were processed using the manufacturer’s software Real Time Analysis (RTA 2.7.7) and followed by the generation of FASTQ sequence files.
RNA-seq data processing and analysis
RNAseq reads were mapped against the human reference genome GRCh38 using STAR software version 2.5.1b30 with ENCODE parameters. Genes were quantified using RSEM version 1.2.2831with default parameters and annotation file from GENCODE version 24. Differential expression analysis was performed with DESeq2 v1.18. R package32 adjusting by sex and using the Wald test to compare the experimental conditions. We defined differentially expressed genes (DEGs) as those with P-value<0.05 and absolute fold-change difference |FC| > 2. To this end, we followed the cut-off consensus guidelines recommended for transcriptome studies in the clinical setting33,34 which have been specifically proposed for functional enrichment analysis35. Hierarchical clustering analyses were performed using the Clustvis software following the default standard settings (https://biit.cs.ut.ee/clustvis/). We computed Volcano plots showing fold-change and P-value distribution of all genes and DEG in the different comparisons (Fig. 1).
RT-qPCR validation
For technical validation of the RNA-seq study, we performed a real-time quantitative PCR (RT-qPCR) analysis. To this end, we selected 9 among the top-deregulated DEGs from the RNA-seq 25 mM glucose discovery analysis and quantified their expression by RT-qPCR. Briefly, fibroblast cDNA was synthesized from 2 μg of total RNA using a high-capacity reverse transcription kit according to the manufacturer’s protocol (Thermo Fisher Scientific Inc. #4368813); and diluted 1/10 in RNase-free water. For amplification, we used the Taqman assays below (Applied Biosystems) in a StepOnePlus Real-Time PCR System applying the thermal cycling conditions indicated by the manufacturer (Thermo Fisher Scientific Inc.). Optimal results were obtained using 12.5 μL of TaqMan Gene Expression Master Mix (Thermo Fisher Scientific Inc. #4369514), 1.25 μL of TaqMan Gene Expression Assay, 8.75 μL of RNase-free water, and 2.5 μL of the diluted template cDNA in a final reaction volume of 25 μL in 96-well plates. For relative quantification, we applied the comparative threshold cycle (CT) method (ΔΔCt) using the Dataassist 3.0 software (Applied biosystems). The internal references or housekeeping genes included: GAPDH (Hs02758991_g1), HPRT1 (Hs02800695_m1), and PPIA (Hs04194521_s1). The target genes included: EGR1 (Hs00152928_m1), IGF2BP3 (Hs00559907_g1), ITGA4 (Hs00168433_m1), ANK3 (Hs00241738_m1), ITGA10 (Hs00174623_m1), MFAP4 (Hs00412974_m1), MYL3 (Hs00264820_m1), and TMOD1 (Hs00984439_m1). Overall, we found a Pearson fold-change correlation coefficient of 0.95 (P-value=0.00019) between the RNA-seq and RT-qPCR (Supplementary Fig. 1).
Functional enrichment analysis
To determine whether DEGs in PD were enriched in particular gene ontology (GO) terms, specifically in Biological process and Cellular component terms, we used the GeneCodis 4.0 software (https://genecodis.genyo.es/).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
GEO data of the study and study details are deposited in the publicly available repositories GSE82340 and GSE167529.
References
Healy, D. G. et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 7, 583–590 (2008).
Tolosa, E., Vila, M., Klein, C. & Rascol, O. LRRK2 in Parkinson disease: challenges of clinical trials. Nat. Rev. Neurol. 16, 97–107 (2020).
Sánchez-Danés, A. et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 4, 380–395 (2012).
Palacios, N. et al. Obesity, diabetes, and risk of Parkinson’s disease. Mov. Disord. 26, 2253–2259 (2011).
Hogg, E. et al. High prevalence of undiagnosed insulin resistance in non-diabetic subjects with Parkinson’s disease. J. Parkinsons. Dis. 8, 259–265 (2018).
Chohan, H. et al. Type 2 diabetes as a determinant of Parkinson’s disease risk and progression. Mov. Disord. mds.28551. https://doi.org/10.1002/mds.28551 (2021).
Santiago, J. A. & Potashkin, J. A. Shared dysregulated pathways lead to Parkinson’s disease and diabetes. Trends Mol. Med. 19, 176–186 (2013).
Sánchez-Gómez, A. et al. Peripheral insulin and amylin levels in Parkinson’s disease. Park. Relat. Disord. 79, 91–96 (2020).
Mutez, E. et al. Transcriptional profile of Parkinson blood mononuclear cells with LRRK2 mutation. Neurobiol. Aging 32, 1839–1848 (2011).
Craig, D. W. et al. RNA sequencing of whole blood reveals early alterations in immune cells and gene expression in Parkinson’s disease. Nat. Aging 2021 18 1, 734–747 (2021).
Sim, H. et al. Quantitative proteomic analysis of primitive neural stem cells from lrrk2 g2019s-associated Parkinson’s disease patient-derived ipscs. Life 10, 1–19 (2020).
Fernandez-Santiago, R. et al. Aberrant epigenome in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. EMBO Mol. Med. 7, 1529–1546 (2015).
Grünblatt, E. et al. Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J. Neural Transm. 111, 1543–1573 (2004).
González-Casacuberta, I. et al. Transcriptional alterations in skin fibroblasts from Parkinson’s disease patients with parkin mutations. Neurobiol. Aging 65, 206–216 (2018).
Fischer, R. S., Lam, P. Y., Huttenlocher, A. & Waterman, C. M. Filopodia and focal adhesions: an integrated system driving branching morphogenesis in neuronal pathfinding and angiogenesis. Developmental Biol. 451, 86–95 (2019).
Chapman, M. A. Interactions between cell adhesion and the synaptic vesicle cycle in Parkinson’s disease. Med. Hypotheses 83, 203–207 (2014).
González-Casacuberta, I., Juárez-Flores, D. L., Morén, C. & Garrabou, G. Bioenergetics and autophagic imbalance in patients-derived cell models of Parkinsonas disease supports systemic dysfunction in neurodegeneration. Frontiers Neurosci 13, 894 (2019).
González-Casacuberta, I. et al. Mitochondrial and autophagic alterations in skin fibroblasts from Parkinson’s disease patients with Parkin mutations. Aging (Albany. NY) 11, 3750–3767 (2019).
Vergara, D. et al. Cytoskeletal alterations and biomechanical properties of parkin-mutant human primary fibroblasts. Cell Biochem. Biophys. 71, 1395–1404 (2015).
Hu, L. et al. Integrated metabolomics and proteomics analysis reveals plasma lipid metabolic disturbance in patients with Parkinson’s disease. Front. Mol. Neurosci. 13, 80 (2020).
A, Z. et al. Polyomic analyses of dopaminergic neurons isolated from human substantia nigra in Parkinson’s disease: an exploratory study. https://doi.org/10.21203/RS.3.RS-182873/V2 (2021).
Zhai, K., Liu, B. & Gao, L. Long-noncoding RNA TUG1 promotes Parkinson’s disease via modulating MiR-152-3p/PTEN pathway. Hum. Gene Ther. 31, 1274–1287 (2020).
Mazarei, G. & Leavitt, B. R. Indoleamine 2,3 dioxygenase as a potential therapeutic target in Huntington’s disease. J. Huntington’s Dis. 4, 109–118 (2015).
Bell, S. M. et al. Peripheral glycolysis in neurodegenerative diseases. Int. J. Mol. Sci. 21, 1–19 (2020).
Juárez-Flores, D. L. et al. Disrupted mitochondrial and metabolic plasticity underlie comorbidity between age-related and degenerative disorders as parkinson disease and type 2 diabetes mellitus. Antioxidants 9, 1063 (2020).
Yoon, S.-Y. & Oh, Y. J. Glucose levels in culture medium determine cell death mode in MPP + -treated dopaminergic neuronal cells. Exp. Neurobiol. 24, 197–205 (2015).
Gaig, C. et al. Screening for the LRRK2 G2019S and codon-1441 mutations in a pathological series of parkinsonian syndromes and frontotemporal lobar degeneration. J. Neurol. Sci. 270, 94–98 (2008).
Fiorello, M. L., Treweeke, A. T., Macfarlane, D. P. & Megson, I. L. The impact of glucose exposure on bioenergetics and function in a cultured endothelial cell model and the implications for cardiovascular health in diabetes. Sci. Rep. 10, 19547 (2020).
Humphries, T. L. R. et al. Par2-induced tissue factor synthesis by primary cultures of human kidney tubular epithelial cells is modified by glucose availability. Int. J. Mol. Sci. 22, 7532 (2021).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 1–16 (2011).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Patterson, T. A. et al. Performance comparison of one-color and two-color platforms within the MicroArray Quality Control (MAQC) project. Nat. Biotechnol. 24, 1140–1150 (2006).
Shi, L. et al. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 24, 1151–1161 (2006).
Zhang, L. et al. Investigating the concordance of Gene Ontology terms reveals the intra- and inter-platform reproducibility of enrichment analysis. BMC Bioinforma. 14, 143 (2013).
Acknowledgements
The authors thank the patients and their relatives for their participation in this study. This work was supported by CIBERNED / Instituto de Salud Carlos III (ISCIII) (grant PIE14/00061) (to M.-J.M.), and the Fondo de Investigaciones Sanitarias from ISCIII (grant PIE20/00259) (to M.E.). R.F.-S. was supported by a Jóvenes Investigadores (JIN) grant of the Spanish Ministry of Economy and Competitiveness (MINECO) and the Agencia Estatal de Investigación (AEI) (AEI/FEDER/UE) (grant #SAF2015-73508-JIN), and a Miguel Servet grant from the Instituto de Salud Carlos III (grant #CP19/00048). A.E-C. was funded by ISCIII /MINECO (PT17/0009/0019) and co-funded by FEDER. M.F. was funded through a María de Maeztu grant (#MDM-2017-0729). IDIBAPS receives funding from the CERCA Program from the Generalitat de Catalunya and the FEDER Program from the European Union.
Author information
Authors and Affiliations
Contributions
Conception, design, and organization: M.E., M.J.M., R.F.-S. Execution: R.F.-S., M.F., M.E., A.E.-C. Statistical analysis: A.E.-C. Manuscript preparation: M.E., R.F.-S., M.F., F.V., Y.C., M.J.M., A.E.-C., E.M., A.S.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fernández-Santiago, R., Esteve-Codina, A., Fernández, M. et al. Transcriptome analysis in LRRK2 and idiopathic Parkinson’s disease at different glucose levels. npj Parkinsons Dis. 7, 109 (2021). https://doi.org/10.1038/s41531-021-00255-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-021-00255-x
