
Abstract
Reducing the musculoskeletal deterioration that astronauts experience in microgravity requires countermeasures that can improve the effectiveness of otherwise rigorous and time-expensive exercise regimens in space. The ability of low-intensity vibrations (LIV) to activate force-responsive signaling pathways in cells suggests LIV as a potential countermeasure to improve cell responsiveness to subsequent mechanical challenge. Mechanoresponse of mesenchymal stem cells (MSC), which maintain bone-making osteoblasts, is in part controlled by the “mechanotransducer” protein YAP (Yes-associated protein), which is shuttled into the nucleus in response to cyto-mechanical forces. Here, using YAP nuclear shuttling as a measurement outcome, we tested the effect of 72 h of clinostat-induced simulated microgravity (SMG) and daily LIV application (LIVDT) on the YAP nuclear entry driven by either acute LIV (LIVAT) or Lysophosphohaditic acid (LPA), applied after the 72 h period. We hypothesized that SMG-induced impairment of acute YAP nuclear entry would be alleviated by the daily application of LIVDT. Results showed that while both acute LIVAT and LPA treatments increased nuclear YAP entry by 50 and 87% over the basal levels in SMG-treated MSCs, nuclear YAP levels of all SMG groups were significantly lower than non-SMG controls. LIVDT, applied in parallel to SMG, restored the SMG-driven decrease in basal nuclear YAP to control levels as well as increased the LPA-induced but not LIVAT-induced YAP nuclear entry over SMG only, counterparts. These cell-level observations suggest that daily LIV treatments are a feasible countermeasure for restoring basal nuclear YAP levels and increasing the YAP nuclear shuttling in MSCs under SMG.
Similar content being viewed by others


Introduction
The musculoskeletal deterioration that astronauts experience on long-term space missions and the resulting increase of traumatic physical injury risk is in part due to the reduction of mechanical loading on the musculoskeleton1. To alleviate the detrimental effects of unloading, astronauts undergo intensive regimens of running and resistance training in orbit2. Despite these efforts, astronauts lose an average bone density of 1% for each month they spend in space3. This loss necessitates new non-pharmacologic therapies, in addition, to exercise to keep musculoskeleton healthy during long-term space missions. In bone, tissue-level response to mechanical challenge is in part regulated by osteoblasts and osteocytes4. Both osteoblasts and osteocytes in turn share a common progenitor: the mesenchymal stem cell (MSC). Therefore, the growth and differentiation of MCSs in response to mechanical stimulation is required for the maintenance and repair of bone5. It is for this reason that the MSCs are a potential target for mechanical therapies aiming to alleviate bone loss in astronauts, injured service personnel with long periods of bed rest, and physically inactive aged individuals6.
To maintain healthy bone-making cell populations, MSCs rely on environmental mechanical signals inside the bone marrow niches and near bone surfaces. While the exact characteristics of the mechanical environment in which MSCs exist remain to be quantified, it is known that during habitual activities, our bones are subjected to combinations of complex loads including strain, fluid shear, and acceleration, each of which is inseparable7. For example, during moderate running, cortical bone can experience strains up to 2000 µε8,9, which also generates coupled fluid flow within canaliculi of up to 100 µm/s10. The interior of the bone is filled with bone marrow with viscosities in the range of 400–800 cP11. During moderate running, tibial accelerations are within the 2–5 g range12 (1 g = 9.81 m/s2), creating a complex loading at the bone marrow interface that depends on many factors including frequency, amplitude, and viscosity13. In silico studies reveal that when exposed to vibrations (0.1–2 g), marrow-filled trabecular compartments generate fluid shear stresses up to 2 Pa13,14, capable of driving bone cell functions15. Interestingly, while these high magnitude forces are only experienced a few times during the day, bones are bombarded by smaller mechanical signals arising from muscle contractions that generate bone strains ranging between 2 and 10 µε16.
Exogenous application of these small magnitude mechanical signals in the form of low-intensity vibrations (LIV) ranging between 0.1 and 2 g acceleration magnitudes and 20–200 Hz frequencies were shown to be effective in improving the tissue level bone and muscle indices17. The application of LIV has been shown to be effective in preclinical and clinical studies. Animal studies demonstrate that LIV increases trabecular bone density and volume18, enhance bone stiffness and strength19, and to slow bone loss caused by disuse20. Further, LIV enhanced muscle contractility21, strength22, and cross-sectional area23, showing that LIV signals are anabolic to skeletal muscle. Clinical studies support the beneficial effects of LIV where twice-daily exposure to LIV for one year increased bone mineral density in both postmenopausal24 and premenopausal women25, LIV resulted in enhanced muscle strength, size, and function in clinical trials25,26,27. Supporting the effectiveness of LIV in 3D in vivo environments, the application of LIV in both horizontal and vertical directions result in similar effects at the cell level in vitro28. At the cellular level, using both horizontal and vertical LIV systems, our group has reported that the application of LIV increases MSC contractility28, activates RhoA signaling29, and results in increased osteogenic differentiation as well as increased proliferation of MSCs30.
The cellular response relies on sensing and intracellular transduction of environmental information. This information is either coded in the extracellular matrix as growth factors or activates mechano-sensitive signaling cascades through dynamic environmental force gradients. Molecules that shuttle between cytoplasm and nucleus in response to mechanical forces such as βcatenin and Yes-associated protein (YAP), along with its orthologue TAZ, are widely recognized as molecular “transducers” of mechanical information17. YAP and its paralog TAZ have important overlapping but sometimes differing functions in stem cells. Focusing on the skeletal system, the interdependent functioning of YAP and TAZ is integral in skeletogenesis and bone regeneration. Not only does the deletion of both YAP and TAZ result in skeletal deficits31, but they also modulate the function and expression of the master osteogenic transcription factor Runx-2 in stem cells. For example, TAZ forms complexes with Runx-2 to increase its function32, and TAZ nuclear presence positively drives MSC osteogenesis32. YAP, on the other hand, maintains stem cell multipotentiality through repressing Runx-2 function33 and promoting the expression of Wnt inhibitory molecule Dkk-134. Further, we have shown that the absence of nuclear YAP amplifies osteogenesis in a Runx-2-dependent manner35. On the other hand, when YAP and TAZ enter into the nucleus, they both bind to their co-transcriptional activator TEAD and increases cell proliferation36,37. Therefore, while both YAP and TAZ act as pro-proliferative transcriptional co-factors for TEAD as well as playing differential role in osteogenesis, to function they need to shuttle from the cytoplasm into the cell nucleus. It has been reported that YAP nuclear entry is triggered by soluble or mechanical factors that increase F-actin contractilities such as Lysophosphatidic acid (LPA)38,39, increased substrate stiffness40, or substrate stretch ranging from 3 to 15%41,42, it is not known if LIV which we have shown to increase RhoA signaling29 and MSC contractility28 also trigger acute YAP nuclear entry.
The ability of molecular transducers such as β-catenin43, YAP, and TAZ to move between cytoplasm and nucleus has been associated with Linker of Nucleoskeleton and Cytoskeleton (LINC) complex function that physically couple cytoskeleton into nuclear lamina44 where polymerized F-actin binds to Nesprin (Nesprin-1 or Nesprin-2), spectrin repeat protein that pierces the nuclear envelope, connecting via its “KASH” (Klarsicht, ANC-1, Syne Homology) domain to intra-membrane leaflet Sun (Sun-1 and Sun-2) proteins45. When the LINC complex was de-functionalized via Nesprin deletion (siRNA), the strain was unable to impel YAP transport into the nucleus41. It has been further reported that pharmacologic inhibition of cytoskeletal contractility via cytochalasin D or disrupting nucleo-cytoskeletal connectivity via depletion of LINC complex function mutes the YAP nuclear entry triggered by either direct force application to nuclei via AFM or by increased substrate stiffness44.
The research aimed at studying the effects of microgravity at the cellular level often relies on simulated microgravity (SMG) devices designed to alter the gravitational conditions that cells experience by rotating on one or multiple axes at low speed46,47,48. Clinostats with a monolayer of cells is a microgravity model system49 and it has been used in a number of scientific studies to model aspects of microgravity since 70 s47,50,51,52. SMG decreases MSC proliferation53 and cytoskeletal contractility46,54,55. While SMG was reported to decrease nuclear TAZ levels52, the role of SMG on nuclear YAP levels is unknown.
As SMG effects are commonly associated with “unloading”, application of physical or soluble factors that induce cytoskeletal contractility are frequently used as countermeasures for SMG51,52. In this way, LIV was previously shown to improve decreased osteogenesis of preosteoblasts under SMG56. We have recently reported that SMG decreases cell proliferation and total YAP protein levels while daily LIV application can alleviate the decreased proliferation and YAP protein levels in MSCs57. This LIV effect was dependent on LINC complex function as depleting Sun-1 and Sun-2 element of the LINC complex muted the LIV response. Importantly, we have also reported that SMG decreases nuclear Lamina element LaminA/C as well as the Sun-2 element of the LINC complex, which was only partially recovered by daily LIV application. Sun-1 and Sun-2 were shown to have a role in maintaining nuclear YAP levels58 and Sun-2 was shown to interacts with F-actin cytoskeleton to maintain YAP nuclear entry in response to strain59. In this way, YAP-mechanosignaling may be altered by SMG due to alterations to the nuclear envelope. This suggests the possibility that LIV can be a feasible SMG countermeasure for increasing the YAP nuclear shuttling in response to subsequent mechanical challenges or soluble activators such as LPA.
Therefore, we tested whether SMG reduces basal nuclear YAP levels as well as YAP nuclear shuttling driven by either acute LIV (LIVAT) or Lysophosphohaditic acid (LPA), applied at the end of the 72 h period SMG period. We have further tested if daily LIV treatment (LIVDT), which was applied in parallel to SMG during the 72 h period alleviates SMG-driven effects on basal nuclear YAP levels as well as acute YAP nuclear shuttling driven by either acute LIVAT or LPA. We hypothesized that SMG-induced impairment of basal nuclear YAP levels as well as YAP nuclear entry in response to LIVAT and LPA would be alleviated by the daily application of LIVDT.
Results
Acute LIVAT application increases nuclear YAP levels
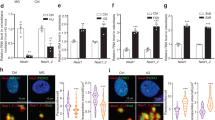
To quantify the acute YAP nuclear entry in response to LIV, MSCs were plated at a density of 5200 cells/cm2 and were allowed to attach for 24 hr. Following this, MSCs were subjected to treatment in two groups: control and acute LIV treatment regimen (LIVAT). The LIVAT regimen consisted of 5 × 20 min vibration periods separated by 1 hr in between each repetition at room temperature while control samples were treated identically (also taken out of the incubator) but were not vibrated. Immediately after LIVAT, samples were immunostained for YAP and DAPI. MATLAB was used to quantify the changes in the nuclear YAP levels. As shown in Fig. 1a, confocal images showed increased nuclear YAP following the LIVAT treatment. For this and all subsequent experiments, the average pixel intensities for all imaged nuclei per sample were normalized to the mean of all the nuclei for the control sample and then presented on a bar graph in order to compare average protein concentration in arbitrary units. Analysis of confocal images to quantify nuclear YAP intensity shown in Fig. 1b revealed a 32% increase in the average nuclear YAP levels in the LIVAT samples as compared to the control samples (p < 0.0001). We also used C2C12 myoblasts to confirm the LIVAT-induced YAP nuclear entry on a second cell line, quantitative analysis of confocal images showed a 40% increase of nuclear YAP in LIVAT samples compared to controls (Supplementary Fig. 1). As both LIV-induced focal adhesion signaling, initiated by focal adhesion kinase (FAK) phosphorylation at Tyr 397 residue29, and YAP nuclear entry in response to substrate strain41 requires intact LINC function, disabling LINC function via a dominant-negative overexpression of Nesprin KASH (Klarsicht, ANC-1, Syne homology) fragment both decreased basal nuclear YAP levels by 34% (p < 0.0001) and impeded the LIV-induced YAP nuclear entry when compared to empty plasmid (Supplementary Fig. 2). FAK phosphorylation at Tyr 397 residue (pFAK) was blocked via a FAK inhibitor (FAKi) PF573228 (3 µM) 1 h prior to LIVAT treatment as previously described29 and stained against DAPI and YAP (Supplementary Fig. 3a). FAKi inhibited the LIVAT-induced pFAK and decreased its basal levels (Supplementary Fig. 3b). As shown in Supplementary Fig. 3c, measuring nuclear YAP levels showed that LIVAT-induced YAP nuclear entry was not affected by FAKi when compared to DMSO-treated controls.

a MSCs were subjected to LIVAT and stained with DAPI (blue) and YAP (red). Confocal images indicated increased nuclear YAP levels following acute LIVAT applied as five 20 min vibration periods separated by 1 h. b Quantitative analysis of confocal images showed a 32% increase of nuclear YAP in LIVAT samples compared to controls. n > 400/grp, group comparison was made a Mann–Whitney U-test, ****p < 0.0001. Error bars represent standard deviation. Scale bar: 10 μm. Full statistical details were provided in Supplementary Table 4.
Basal nuclear YAP levels decreased by SMG were rescued by daily application of LIVDT
We next tested whether SMG decreases basal YAP levels and whether a daily LIV treatment regimen (LIVDT), applied in parallel with SMG, could alleviate decreased YAP in the nucleus. As we reported previously, LIVDT consisting of 2 × 20 min vibrations applied every 24 h during the 72 h period of SMG. This LIVDT regimen was effective at restoring MSC proliferation and whole-cell YAP levels when applied in conjunction with SMG57. MSCs were plated at a density of 1700 cells/cm2 in 9 cm2 tissue culture SlideFlasks (Nunc, #170920) and were allowed to attach for 24 h, after which point, the flasks were filled completely with growth medium, sealed, and subjected to 72 h of treatment followed by immunostaining for YAP and nuclear staining using DAPI. During the 72 h treatment period, MSCs were divided into three groups: control samples, SMG samples, which were subjected to the 72 h SMG alone, and SMG + LIVDT samples, which were subjected to both the 72 h SMG regimen and the daily LIVDT regimen. Representative images for YAP and DAPI stained images are shown in Fig. 2a. As depicted in Fig. 2b, the quantitative analysis of confocal images revealed a 42% decrease in the nuclear YAP intensity of the SMG group as compared to non-SMG controls (p < 0.0001). Compared to the SMG group, the LIVDT group increased nuclear YAP levels by 67% (p < 0.0001), and there was no significant difference between nuclear YAP levels of LIVDT-treated MSCs and non-SMG controls.

a MSCs were subjected to SMG, and SMG + LIVDT over 72 h period and stained with DAPI (blue) and YAP (red). b Quantitative analysis showed a 42% decrease in nuclear YAP levels in the SMG group compared to control levels. The SMG + LIVDT group showed a 67% increase in nuclear YAP when compared to the SMG group. There was no statistically significant difference between CTRL and SMG + LIVDT groups. n > 100/grp. Group comparisons were made via Kruskal–Wallis test followed by Tukey multiple comparison, ****p < 0.0001. Error bars represent standard deviation. Scale bar: 10 μm. Full statistical details were provided in Supplementary Table 5.
LIVAT-induced YAP nuclear entry decreased by SMG was not restored by daily LIVDT application
As SMG decreased basal nuclear YAP levels, we next tested whether SMG decreases LIVAT-induced YAP nuclear shuttling and whether LIVDT application alleviates this. A schematic of the experimental design is given in Fig. 3. MSCs were divided into six groups in which the CTRL, SMG, SMG + LIVDT groups were treated with ±LIVAT at the end of 72 h and nuclear YAP levels were measured. As shown in Fig. 4, SMG alone decreased basal nuclear YAP levels by 37% (p < 0.0001), which were restored back to control levels in the SMG + LIVDT group. As depicted in Fig. 4, +LIVAT increased nuclear YAP levels in the CTRL, SMG, and SMG + LIVDT groups by 50%, 69%, and 22%, respectively (p < 0.0001) while exhibiting the smallest increase in the SMG + LIVDT. As a result, final nuclear YAP levels in the SMG + LIVDT + LIVAT group were not significantly different from the SMG + LIVAT and 23% lower than the LIVAT group (p < 0.0001). Representative confocal images are presented in Supplementary Fig. 4.

MSCs were subcultured and plated in SlideFlasks and allowed to attach for 24 h before SlideFlasks were filled with culture medium and sealed. The treatment regimen for MSC’s involved 72 h of SMG (blue). LIVDT regimen consisted of one treatment cycle every 24 h during SMG treatment with each cycle consisting of 2 × 20 min LIV with an hour in between (yellow). LIVAT regimen was applied after 72 h SMG treatment period and consisted of 5 × 20 min LIV with an hour in between each (red). For LIV application, MSCs plated in SlideFlasks were placed in an LIV device constructed in the lab previous to this research. Vibrations were applied at peak magnitudes of 0.7 g at 90 Hz at room temperature. Control samples were treated the same but were not vibrated. For SMG application, MSCs plated in SlideFlasks were secured in lab custom-built clinostat inside the incubator. The clinostat subjected the MSCs to constant 15 RPM rotation simulated microgravity. After treatment, flasks were removed for immunofluorescence staining.

MSCs were subjected to either CTRL, SMG, SMG + LIVDT over 72 h period were subsequently treated with LIVAT. Quantitative analysis of confocal images showed that SMG alone decreased basal nuclear YAP levels by 37%, which were increased back to control levels in the SMG + LIVDT group. +LIVAT increased nuclear YAP levels in the CTRL, SMG, and SMG + LIVDT groups by 50%, 69%, and 22%, respectively. Nuclear YAP intensity in the SMG + LIVDT + LIVAT group remained not significantly different from the SMG + LIVAT and 23% smaller than the LIVAT group. n > 200/grp. Group comparisons were made via Kruskal–Wallis test followed by Tukey multiple comparison, ****p < 0.0001. Error bars represent standard deviation. Scale bar: 10 μm. Full statistical details were provided in Supplementary Table 6.
LPA treatment increases nuclear YAP levels
As the lack of difference between SMG + LIVAT and SMG + LIVDT + LIVAT treatment (Fig. 4) suggested that dosing cells daily with LIVDT may decrease cell responsiveness to LIV-AT, we sought an alternative soluble activator of YAP nuclear shuttling. LPA is frequently used for activating RhoA-mediated cytoskeletal response60,61. We have previously shown that the LPA effect on F-actin cytoskeleton and focal adhesion activation is additive with both LIV and substrate strain29. Since the usage of sealed culture flasks did not permit us to use substrate strain and LPA has been shown to induce nuclear YAP-shuttling in a number of cell types38,39,62,63, we utilized LPA as a soluble activator of YAP signaling. To test the effect of LPA on the acute YAP nuclear entry in MSCs, two LPA concentrations (50 and 100 µM) were compared against control samples. As shown in Fig. 5, nuclear YAP levels were almost doubled under a 2-h exposure to 50 µM LPA and 100 µM LPA treatments with 99 and 107% increases as compared to the control samples (p < 0.0001). Nuclear YAP levels for 50 µM LPA and 100 µM LPA treatments were not significantly different. Therefore, we chose to use 50 µM LPA treatment in the subsequent experiments.

a Representative confocal images of DAPI (blue) and YAP (red) stained MSCs with or without LPA treatment. MSCs were subjected to LPA addition at 50 and 100 µM concentrations. b Quantitative analysis of confocal images revealed a 99% and a 107% increase in the 50 µM LPA and 100 µM LPA treatments compared to DMSO-treated controls, respectively. Nuclear YAP levels for 50 µM LPA and 100 µM LPA treatments were not significantly different. n > 30/grp. Group comparisons were made via Kruskal–Wallis test followed by Tukey multiple comparison, ****p < 0.0001. Error bars represent standard deviation. Scale bar: 10 μm. Full statistical details were provided in Supplementary Table 7.
LPA-induced YAP nuclear entry decreased by SMG was alleviated by daily LIVDT application
After establishing that LPA induces YAP nuclear entry in MSCs, we next evaluated whether SMG decreases LPA-induced YAP nuclear shuttling and whether LIVDT application alleviates this. A 50 µM LPA dissolved in DMSO or DMSO as vehicle control was added to the samples at the end of the 72 h treatment of either CTRL, SMG, or SMG + LIVDT treatments. The CTRL group, SMG group, and SMG + LIVDT group were subjected to the same treatment as in the previous experiments and displayed similar results. As depicted in Fig. 6, +LPA increased nuclear YAP levels in the CTRL, SMG, and SMG + LIVDT groups by 105%, 67%, and 43% respectively (p < 0.0001). While final YAP nuclear levels in SMG + LIVDT + LPA remained 70% higher than SMG + LPA group (p < 0.0001), it remained 29% lower than the LPA group (p < 0.0001).

MSCs were subjected to SMG, and parallel SMG + LIVDT over 72 h period at the end of 72 h, samples were treated with either LPA (50 µM) or DMSO. Quantitative analysis of confocal images revealed that LPA addition increased nuclear YAP levels by 105%, 67%, and 43% in the CTRL, SMG, and SMG + LIVDT when compared to DMSO controls. When compared to nuclear YAP intensity of the LPA treatment alone, SMG + LPA and SMG + LIVDT + LPA samples were 55% and 29% lower, respectively. YAP nuclear levels in SMG + LIVDT + LPA remained 70% higher than SMG + LPA group. n > 100/grp. Group comparisons were made via Kruskal–Wallis test followed by Tukey multiple comparison, ****p < 0.0001. Error bars represent standard deviation. Scale bar: 10 μm. Full statistical details were provided in Supplementary Tables 8.
MSC stiffness and structure remain intact under SMG and SMG + LIVDT treatments
As YAP-mechanosignaling of SMG + LIVDT MSCs remained below control levels in response to both LIVAT and LPA, we quantified the effects of SMG and SMG + LIVDT on the cell stiffness, F-actin intensity, cell area, and nuclear area. AFM testing was used to quantify the elastic modulus of the nucleus by measuring load-displacement curves on top of the nucleus. AFM tests shown in Fig. 7a indicated a 21 and 27% stiffness decrease in the SMG or SMG + LIVDT groups but differences were not significant. Quantified from confocal images (Fig. 7b), mean F-actin intensities for all the cells in each imaging field were quantified by dividing the mean F-actin intensity to the number of nuclei in each imaging field. Shown in Fig. 7c, SMG and SMG + LIVDT-treated MSCs revealed 36% and 30% decreases in the mean F-actin intensity per cell, respectively, but the differences were not statistically significant. We have further quantified the nuclear area as a measure of cyto-mechanical forces on the nucleus64. As shown in Fig. 7d, analysis of the cross-sectional area of cell nuclei using DAPI stained images revealed no significant effect on average nuclear size by either SMG or combined SMG + LIVDT treatment compared to control levels.

MSCs were subjected to SMG and parallel SMG + LIVDT over a 72 h period. a Compared to CTRL samples, AFM measurement of the elastic moduli of SMG and SMG + LIVDT-treated MSCs revealed apparent decreases in elastic modules that were 21 and 27% below control levels, measured differences were not statistically significant. n = 10/grp. b Quantification of confocal images shows that c mean F-actin intensity of SMG and SMG + LIVDT-treated MSCs revealed a decrease of 36 and 30% below control levels, measured differences were not statistically significant. n = 15/grp. d No significant effects of either SMG or LIVDT treatment on the average nucleus size were found. n > 100/grp. Group comparisons were made via Kruskal–Wallis test followed by Tukey multiple comparison. Error bars represent standard deviation. Scale bar: 10 μm. Full statistical details were provided in Supplementary Tables 9–11.
Discussion
The mechanical forces that the bone and muscle cells are subjected to on Earth and in microgravity are complex and remain incompletely understood. At the same time, it is clear that these forces are required for healthy tissue growth and function. The complexity of these forces makes it difficult to design experiments that comprehensively simulate in vivo conditions. While, spheroid systems that provide in vivo like 3D growth conditions65, spheroid systems are harder to design in terms of fluid shear. Earlier studies that consider cell suspension systems66 indicated that very low fluid shear can be achieved if cell motion within the container can be avoided (i.e., low rpm, small particle size, high viscosity, etc). However, motion tracking studies with 3D microspheres with higher-than-water and lower-than water densities showed that while less dense scaffolds avoid repeated collisions with the bioreactor wall that impose a variety of confounding, non-quantifiable mechanical disruptions, both types generated fluid shear values ranging between 0.16 and 0.32 Pa67. As a result, rotated groups showed increased osteogenic differential compared to static ones. In contrast, rotation devices with adherent cells do not generate as high fluid shear values. For example, maximum fluid shear at flask walls during 15 rpm rotation was found to be 0.06 Pa68, which order or magnitude lower compared to microbead systems. These findings led us to adopt a monolayer SMG system. Another important discussion is the fluid shear generated by vibration stimuli. Sealed flasks would largely eliminate this fluid shear and sealing culture plates and flasks to avoid fluid motion is commonly utilized in both LIV69,70 and sMG-based studies48,53,71,72. Fluid shear generated therefore will be independent of fluid volume and is a result of deformations of a fluid-bounded well-bottom due to vertical accelerations73. These dynamic accelerations result in lateral fluid motion over-attached cells. We have reported that vertical 0.7 g, 90 Hz LIV in 6-well culture plates results in a peak velocity difference of 0.00004 m/s between fluid and well-bottom29, corresponding to peak fluid shear of 0.0008 Pa, well below of SMG-induced fluid shear. In this current study, we have utilized 10 cm2 culture flasks, which have a 5% more surface area compared to 6-well plates (9.5 cm2). While this difference in the area may result in larger peak deformations and thus generate larger fluid shear, we believe that using 10 cm2 flasks would be to not be radically different than 6-well culture plates in terms of fluid shear. We expect these fluid shear values to be inconsequential to LIV response as we previously tested the possible contribution of fluid shear to LIV vibration response74. Increasing fluid shear up to 2 Pa under 100 Hz horizontal vibration did not generate a statistically significant effect when compared to the LIV stimulus that generated 0.015 Pa fluid shear. As computational and ex vivo studies suggest that LIV may generate fluid shear forces between 0.5 and 2 Pa in vivo7. Our findings suggest that fluid shear does not contribute to the LIV vibration response. Therefore the in vitro experiments utilizing SMG and LIV treatments used in this study are limited in replicating the in vivo conditions and do not entirely correlate with the physiological behavior of these cells in vivo, the experiments presented here remain useful for testing cell behavior under these well-defined conditions.
Another limitation of the study includes our inability to apply SMG and LIV treatments simultaneously. When daily LIV was combined with SMG, the culture flasks needed to be removed from the clinostat SMG device in order to put them in the LIV device. This limitation is primarily due to our inability to dampen vibrations in the cell culture incubator. When we turn on the vibrating device inside the cell culture incubator, all the samples, including the controls also experience significant amounts of vibration. Further, vibrating the entire SMG system (i.e., simultaneous application) is a more complex problem due to (i) weight of the SMG setup necessitates a newer LIV device design and (ii) potential structural response of SMG device to 0.7 g, 90 Hz vibrations that would generate secondary vibrations were unknown. To avoid these confounding factors, we apply LIV on an isolated table while controls sit on a separate table with no vibration signal. While disruptions to SMG cannot be avoided, between the 20 min LIV sessions for the daily LIV treatment, the flasks were placed back into the clinostat such that the interruptions in SMG treatment were brief relative to the 72 h application period. This was not done for the acute LIV treatment as this treatment was designed to test acute YAP signaling activation after 72 h of SMG.
In this study, we focused on YAP-mechanosignaling of MSCs. The first experiments demonstrated that repeated LIVAT application over six hours was capable of stimulating YAP entry into the nucleus in both MSCs (Fig. 1) and in the C2C12 cell line (Supplementary Fig. 1). These findings suggest that similar to high magnitude substrate strains42 smaller mechanical signals such as LIV can be effective at increasing nuclear YAP levels. Further, in agreement with earlier reports utilizing uniaxial strain41, LIVAT-induced increase in nuclear YAP levels also required functional LINC complexes (Supplementary Fig. 2). Integrin related FAK signaling has been shown to promote YAP nuclear levels in the proliferative descendants of stem cells and that FAK inhibitor PF573228 decreased nuclear YAP in these cells75. Similarly, inhibiting integrin engagement via blocking FAK phosphorylation in Tyr 397 residue via FAKi also mutes the increase of GTP-bound RhoA levels in LIV-treated MSCs29. While we confirmed the loss of phosphorylation in Tyr 397 at both basal levels and in response to LIVAT (Supplementary Fig. 3b), FAKi treatment changed neither basal levels nor the LIVAT-induced increase in nuclear YAP (Supplementary Fig. 3c), suggesting a FAK independent mechanism. In these experiments, we did not compare LIVAT with strain because the application of 5–15% stretch onto sealed culture flasks was not technically possible without significantly altering experimental conditions. Instead, LPA addition served as the best option for applying a simple mechanical stimulation in order to evaluate the YAP mechanotransduction. LPA is a phospholipid derivative signaling molecule, which is capable of causing the simulation of a static transient stretch of a cell by activating GTPase Rho-mediated actin stress fiber creation, which results in increased contractility of the cytoskeleton30,60. The first experiments with LPA served to verify that the simulation of stretch via increased cytoskeleton contractility was capable of triggering YAP entry into the nucleus and the analysis methods utilized here were capable of detecting this response (Fig. 5).
The first SMG experiments confirmed a clear decrease of basal nuclear YAP levels. Interestingly, SMG-treated cells remained responsive, as both LIVAT and LPA treatments were able to increase the nuclear YAP levels at the end of the acute stimulation period (<6 h). However, final nuclear YAP levels in SMG-treated MSCs remained significantly lower when compared to non-SMG groups (Figs. 2, 4, and 6). These findings suggested that the YAP-mechanosignaling apparatus of MSCs, to some extent, was intact under SMG. When applied in parallel to SMG, daily LIVDT treatment was able to restore basal YAP levels in the cell nucleus (Figs. 2, 4, and 6) measured 24 h after the final LIVDT treatment. This increase in nuclear levels supported our earlier report that showed sustained recovery of MSC proliferation by LIVDT57. An interesting observation is that a closer look into Figs. 1b, 2b, and Supplementary Fig. S1 reveals that while all the sample sets fit into a general expectation of a “bell shaped” distribution, the shape of the LIVAT samples looks more unbalanced towards the positive end compared to others. The first interesting observation is that LIVDT groups do not show this shift probably because the imaging was conducted 24 h after the last LIVDT treatment to avoid any residual acute effects of LIVDT on the nuclear YAP levels. This timing should be sufficient as nuclear YAP levels have been shown to return to baseline within 12 h42. Therefore, in the lieu of our previous findings that LIVDT restores overall YAP levels decreased by SMG57, here we are seeing that nuclear YAP levels are also restored by LIVDT. As for the shape changes under LIVAT, there could be a number of reasons. First, it is possible that some cells are more responsive to LIVAT than others. More importantly, we apply LIVAT in five different bouts spanning a 6 h period, as YAP response to strain shown to be maximized around 6 h42 it may be possible that the shape is representative of the MSCs responding to LIVAT at different times. In this way, it is conceivable that shape might look different if a time point different than 6 h was selected. Ultimately, while shape may represent the dynamic and acute nature of the LIVAT response of YAP, the upward shift of the population intensity and the increased averages suggest increased nuclear YAP intensity in the cell nuclei under LIVAT treatment.
We next aimed to determine the effects of SMG and LIV on YAP signaling response to acute mechanical stimulus including the LIVAT treatment as well as the addition of soluble LPA. Interestingly, this increase in basal nuclear YAP levels under LIVDT was accompanied by a reduced MSC response to LIVAT treatment (Fig. 4). When SMG + LIVDT-treated MSCs were subjected to LIVAT, the increase in nuclear YAP from the non-LIVAT control was only 22% (p < 0.0001), which was small compared to the 77% increase seen in the SMG groups in response to LIVAT. As a result of this smaller increase in the SMG + LIVDT group, there was no measurable difference between SMG and SMG + LIVDT, samples that were subjected to LIVAT. It has been previously reported that an application of multiple LIV bouts separated by a refractory period is more effective at activating mechanosignaling pathways such as β-catenin76. It is possible that long-term application of LIVDT results in cell structural adaptations that serve to reduce MSC responsiveness to LIVAT treatment. To test this possibility, we replaced LIVAT with an LPA treatment. When LIVAT was replaced by LPA treatment (Fig. 6), the responsiveness of SMG + LIVDT-treated MSCs almost doubled to 43% (compared to 22% in response to LIVAT) and was significantly higher than the SMG + LPA group (p < 0.0001), suggesting that LIVDT increases the YAP-mechanosignaling in response to LPA.
Absolute nuclear YAP intensity in the SMG + LIVDT + LIVAT group, however, remained below the LIVAT group (p < 0.0001). Previously published findings using the same treatment protocols suggested that the total cellular YAP levels decreased by SMG were restored to control levels by daily LIV57. This indicates that the total availability of YAP protein was not responsible for this difference between the SMG + LIVDT + LIVAT and the LIVAT groups. In regards to other potential effects of SMG on the components of the mechanosignaling mechanism, one current prevailing hypothesis suggests a role for nuclear pore opening in response to cyto-mechanical forces44, which may be affected by changes in the nuclear stiffness. To test this possibility, we performed additional AFM and imaging experiments. While the AFM measured nuclear stiffness was 24% lower in the SMG and SMG + LIVDT groups on average, we were unable to identify any statistically significant effects of SMG or LIVDT treatment on nuclear stiffness. There was also slight F-actin intensity decreases in both the SMG and SMG + LIVDT groups, which were also not significant (Fig. 7). Similarly, cell and nuclear areas were not affected. While our results were not able to detect any changes in nuclear stiffness, considering the significant role that the nuclear membrane has as a mechanical structural component in the cell’s interpretation of mechanical stimulus77,78, more detailed future studies are needed to study the effects of SMG on the nuclear envelope and nuclear structure.
In summary, while the restoration of basal nuclear levels and improvement of LPA-induced YAP nuclear entry under daily LIVDT treatment identify LIV as a possible countermeasure to improve YAP nuclear import under simulated microgravity, future studies are required to understand why acute YAP nuclear entry in response LIVAT remains less responsive. Additionally, restoration of basal nuclear YAP under SMG + LIVDT may explain the previously observed increase in cell proliferation, the current study scope was limited to measuring basal YAP levels and YAP-shuttling only. Therefore, how these basal YAP levels and YAP-shuttling differences may affect MSC response to subsequent signaling, including proliferation and osteogenesis will be important to address in future studies to establish LIV as a viable countermeasure for unloading and microgravity.
Methods and materials
Cell culture
The primary mouse MSCs were isolated from multiple mice donors (8–10-wk-old male B6 mice)29,43,57. Briefly, these primary MSCs were extracted from 3 to 5 mice and all the cells were pooled together. Following the extraction protocol79, these MSCs were tested for adipogenic as well as osteogenic potential and subsequently frozen. For these experiments, MSCs between passages 6 and 9 were used. We have recently reported using the same protocol that these MSCs retain their multipotentiality beyond passage 15 as shown by unaltered expression of different lineage markers such as FGF-2. RUNX-2, PPARG, SOX-2, and SOX-280. C2C12 mouse myoblasts were derived from muscle satellite cells. MSCs were subcultured and plated in Iscove modified Dulbecco’s cell culture medium (IMDM, 12440053, Gibco) with 10% fetal calf serum (FCS, S11950H, Atlanta Biologicals) 10,000 unit/mL penicillin, and 10,000 μg/mL streptomycin (referred as 1% pen/strep). C2C12s were subcultured and plated in Dulbecco’s modified Eagle’s medium (DMEM, DML09, Caisson Laboratories) with 10% fetal calf serum (FCS, S11950H, Atlanta Biologicals) and 1% pen/strep. For each passage, stock cells were seeded in 55 cm2 culture dishes at a density of 1000 cells/cm2 and subcultured after an average of five population doublings at an average density of 32,000 cells/cm2. For experiments, MSCs were plated in SlideFlasks (Nunc, #170920) at a density of 5200 cells/cm2 for 1-day experiments and 1700 cells/cm2 for 3day experiments, while C2C12s were plated at a density of 10,000 cells/cm2. Experimental cells were plated and given 24 h to attach to the mounting surface prior to experiments. Cell passages for both MSCs and C2C12s used for experiments were limited to P7-P15. All methods were carried out in accordance with relevant guidelines and regulations of the Boise Institutional Animal Care and Use Committee and Institutional Biosafety Committee. All procedures were approved by the Boise State University Institutional Animal Care and Use Committee, and the Institutional Biosafety Committee.
Low-intensity vibrations treatment
SlideFlasks with plated MSCs were filled completely with the culture medium and placed in an LIV device designed and used in our lab (Fig. 3)57. LIV device subjected cells to low intensity 90 Hz lateral vibrations at 0.7 g at room temperature. MSCs were vibrated for 20 min intervals separated over time. The daily LIVDT regimen consisted of treatment cycles consisting of 2 × 20 min LIV with a 2 h refractory period in between, with one such treatment cycle applied per day over 3 days or 72 h. The acute LIVAT regimen consisted of 5 × 20 min LIV treatments with an hour in between each. Control samples were subject to the same growth conditions except they were not placed in the LIV device and were placed on a separate table surface entirely to isolate the cells from vibrations.
Simulated microgravity treatment
SlideFlasks (Nunc, #170920) with plated MSCs were filled completely with the culture medium and placed in a clinostat SMG device (Fig. 3). The clinostat shown is a redesign of a custom-made clinostat constructed in our lab with a new flask holder casing capable of holding SlideFlasks and fabricated with PTFE, which could be thoroughly sanitized by autoclaving The clinostat simulated microgravity by rotating the MSC’s in SlideFlasks at a constant 15 RPM, effectively rotating in one dimension the gravity vector acting on the sample and canceling out its effects as a result of the averaging out of the opposing gravity vectors over time. In all treatments, the clinostat was used to subject the MSCs to constant 15 RPM SMG for 72 h. When the LIVDT regimen was combined with this treatment, the design of the clinostat and design of the LIV device required that the SlideFlasks be removed from the clinostat and placed into the LIV device for the 20 min LIV treatment intervals. However, the SlideFlasks would be placed back into the clinostat for the full duration of the 2 h refractory periods. Additionally, when the LIVDT regimen was combined with this treatment, the samples that were subjected to control and SMG treatment were also taken out of the clinostat and placed on a separate table surface entirely to isolate the cells from vibrations. The LIVAT regimen, on the other hand, was only ever applied after SMG treatment and therefore the SlideFlasks were not placed back into the clinostat between LIV treatments.
Immunofluorescence staining and image analysis
Immediately after mechanical treatment, MSCs plated in Slideflasks were removed from treatment, and the SlideFlasks were disassembled in order to stain the MSCs on the slides (Fig. 3). The MSCs were fixed with 4% paraformaldehyde, then washed and permeabilized with 0.05% Triton X-100 in PBS, followed by immunostaining with YAP-specific antibody (YAP, D8H1X) Rabbit mAb, Cell Signaling Technologies #14074 at 1/100 dilution in PBS) and Alexa Fluor red secondary antibodies (Goat anti-Rabbit IgG (H + L) Cross-Adsorbed Secondary Antibody Alexa Fluor Plus 594, Thermo Fisher Scientific #A11037 at 1/500 dilution in PBS was used for all experiments prior to usage of LPA. Subsequently, Goat anti-Rabbit IgG (H + L) Cross-Adsorbed Secondary Antibody Alexa Fluor Plus 633, Thermo Fisher Scientific #A21070 at 1/500 dilution in PBS was used). Nuclear DNA was labeled via DAPI (VECTASHIELD HardSet Antifade Mounting Medium with DAPI, Vector Laboratories #H1500). Actin filaments were labeled via phalloidin stain (Phalloidin-iFluor 488 Reagent, Abcam #AB176753 at 1/500 dilution in PBS). Stained samples were imaged with a Leica TCS SP8 confocal microscope (×40x, HC PL APO CS2 Oil Immersion) prior to usage of LPA, after this point in time, the Leica machine became inoperable and therefore a Zeiss LSM 510 Meta Confocal Microscope (40x, HC PL APO CS2 Oil Immersion) was used. Exported images were used to quantify relative YAP levels within each nucleus (nuclear regions traced by DAPI stained nucleus) via a custom-made MATLAB program (The MathWorks, Natick, MA). DAPI images were analyzed using an edge-detection algorithm in order to determine the regions of cell nuclei for each cell. This algorithm used a thresholding method, which defined these regions based on experimentally determined threshold parameters, identification of regions based on 8-directional connectivity of image pixels as evaluated by the “bwlabel” function, and removal of regions substantially smaller than standard nucleus size by the “bwareaopen” function. The nuclear outline was then used as a mask to quantify the average pixel intensity of the YAP stain within the individual nuclei of each cell.
Sample sizes varied between experiments based on the number of images taken. Based on power calculations with 40% effects size and standard deviation of 50%—which was our average LIV or SMG effect on YAP nuclear levels during preliminary experiments, 25 samples is sufficient to reach 80% statistical power assuming 95 percentile confidence. In each major experiment, we imaged on average 100 to 200 nuclei to quantify YAP levels, this was an approximate range as we cannot really control how many nuclei we image in each field of view during microscopy sessions. All images from the individual trials were pooled and the final sample sizes were provided in the figure legends. We have also added Supplementary Tables S4–S14 to provide exact sample sizes and analysis details.
Atomic force microscopy
Bruker Dimension FastScan AFM was used for the collection of the atomic force measurements81. Tipless MLCT-D probes with a 0.03 N/m spring constant were functionalized with 10 µm diameter borosilicate glass beads for force collection. The AFM’s optical microscope was used to locate individual live MSCs plated on the SlideFlask slides with the flask section removed for access to the cells. The nucleus of each cell was tested with at least 3 s of rest between each test. In each test, three force-displacement curves were obtained (ramping rate: 2 µm/s over 2 µm total travel, 1 µm approach, 1 µm retract), which were analyzed using Nanoscope software with the implementation of a best-fit curve to a Hertzian (spherical) model (optimized such that R2 value was >0.95, or p < 0.05) to obtain elastic moduli of nuclear membrane of individual nuclei.
Western blotting
Western blotting was performed using standard gel electrophoresis procedure29,43,57,82. 20 μg of lysed cell protein from each sample was run on 10% polyacrylamide gels, transferred onto polyvinylidene difluoride (PVDF) membranes, blocked with 5%(w/v) milk for 1 h. After washing, the membranes were incubated overnight at 4 °C in a solution of primary antibodies diluted in 5% w/v bovine serum albumin (BSA), 1X tris-buffered saline (TBS), 0.1% tween. Protein bands were visualized via horseradish peroxidase-conjugated secondary antibodies (1:5000, Cell Signaling) and ECL plus chemiluminescence kit (Amersham Biosciences, Piscataway, NJ) and scanned using C-DiGit blot scanner (Licor, Lincoln, NE). All blots derive from the same experiment and were processed in parallel.
Statistical analysis
All data analysis results were displayed graphically based on the mean value with standard deviation. All the graphs were generated and analyzed using GraphPad Prism 8. Differences between treatments were not assumed to follow a Gaussian distribution. Therefore, group differences were identified via either non-parametric two-tailed Mann–Whitney U-test (Fig. 1a) or Kruskal–Wallis test followed by Tukey multiple comparisons (Figs. 2b, 4, 5, 6, 7, and Supplementary Figs. 1–3). p-values of < 0.05 were considered significant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data sets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Smith, S. M. et al. Calcium metabolism before, during, and after a 3-mo spaceflight: kinetic and biochemical changes. Am. J. Physiol. 277, R1–R10 (1999).
Greenleaf, J. E., Bulbulian, R., Bernauer, E. M., Haskell, W. L. & Moore, T. Exercise-training protocols for astronauts in microgravity. J. Appl. Physiol. 67, 2191–2204 (1989).
Vico, L. et al. Effects of long-term microgravity exposure on cancellous and cortical weight-bearing bones of cosmonauts. Lancet 355, 1607–1611 (2000).
Thompson, W. R., Rubin, C. T. & Rubin, J. Mechanical regulation of signaling pathways in bone. Gene 503, 179–193 (2012).
Ozcivici, E. et al. Mechanical signals as anabolic agents in bone. Nat. Rev. Rheumatol. 6, 50–59 (2010).
Rando, T. A. & Ambrosio, F. Regenerative rehabilitation: applied biophysics meets stem cell therapeutics. Cell Stem Cell 22, 306–309 (2018).
Chan, M. E., Uzer, G. & Rubin, C. The potential benefits and inherent risks of vibration as a non-drug therapy for the prevention and treatment of osteoporosis. Curr. Osteoporos. Rep. 1–9, https://doi.org/10.1007/s11914-012-0132-1 (2013).
Judex, S., Gross, T. S. & Zernicke, R. F. Strain gradients correlate with sites of exercise-induced bone-forming surfaces in the adult skeleton. J. Bone Miner. Res. 12, 1737–1745 (1997).
Rubin, C. T. & Lanyon, L. E. Dynamic strain similarity in vertebrates; an alternative to allometric limb bone scaling. J. Theor. Biol. 107, 321–327 (1984).
Price, C., Zhou, X. Z., Li, W. & Wang, L. Y. Real-time measurement of solute transport within the lacunar-canalicular system of mechanically loaded bone: direct evidence for load-induced fluid flow. J. Bone Miner. Res. 26, 277–285 (2011).
Gurkan, U. A. & Akkus, O. The mechanical environment of bone marrow: a review. Ann. Biomed. Eng. 36, 1978–1991 (2008).
Vainionpaa, A. et al. Intensity of exercise is associated with bone density change in premenopausal women. Osteoporos. Int. 17, 455–463 (2006).
Dickerson, D. A., Sander, E. A. & Nauman, E. A. Modeling the mechanical consequences of vibratory loading in the vertebral body: microscale effects. Biomech. Model. Mechanobiol. 7, 191–202 (2008).
Coughlin, T. R. & Niebur, G. L. Fluid shear stress in trabecular bone marrow due to low-magnitude high-frequency vibration. J. Biomech. 45, 2222–2229 (2012).
Riddle, R. C. & Donahue, H. J. From streaming potentials to shear stress: 25 years of bone cell mechanotransduction. J. Orthop. Res. 27, 143–149 (2009).
Fritton, S. P., McLeod, K. J. & Rubin, C. T. Quantifying the strain history of bone: spatial uniformity and self-similarity of low-magnitude strains. J. Biomech. 33, 317–325 (2000).
Pagnotti, G. M. et al. Combating osteoporosis and obesity with exercise: leveraging cell mechanosensitivity. Nat. Rev. Endocrinol. https://doi.org/10.1038/s41574-019-0170-1 (2019).
Rubin, C., Turner, A. S., Bain, S., Mallinckrodt, C. & McLeod, K. Anabolism. Low mechanical signals strengthen long bones. Nature 412, 603–604 (2001).
Rubin, C. et al. Quantity and quality of trabecular bone in the femur are enhanced by a strongly anabolic, noninvasive mechanical intervention. J. Bone Min. Res. 17, 349–357 (2002).
Rubin, C., Xu, G. & Judex, S. The anabolic activity of bone tissue, suppressed by disuse, is normalized by brief exposure to extremely low-magnitude mechanical stimuli. FASEB J. 15, 2225–2229 (2001).
McKeehen, J. N. et al. Adaptations of mouse skeletal muscle to low-intensity vibration training. Med. Sci. Sports Exerc. 45, 1051–1059 (2013).
Mettlach, G. et al. Enhancement of neuromuscular dynamics and strength behavior using extremely low magnitude mechanical signals in mice. J. Biomech. 47, 162–167 (2014).
Xie, L., Rubin, C. & Judex, S. Enhancement of the adolescent murine musculoskeletal system using low-level mechanical vibrations. J. Appl. Physiol. 104, 1056–1062 (2008).
Rubin, C. et al. Prevention of postmenopausal bone loss by a low-magnitude, high-frequency mechanical stimuli: a clinical trial assessing compliance, efficacy, and safety. J. Bone Miner. Res. 19, 343–351 (2004).
Gilsanz, V. et al. Low-level, high-frequency mechanical signals enhance musculoskeletal development of young women with low BMD. J. Bone Miner. Res. 21, 1464–1474 (2006).
Blottner, D. et al. Human skeletal muscle structure and function preserved by vibration muscle exercise following 55 days of bed rest. Eur. J. Appl. Physiol. 97, 261–271 (2006).
Muir, J., Kiel, D. P. & Rubin, C. T. Safety and severity of accelerations delivered from whole body vibration exercise devices to standing adults. J. Sci. Med. Sport 16, 526–531 (2013).
Pongkitwitoon, S., Uzer, G., Rubin, J. & Judex, S. Cytoskeletal configuration modulates mechanically induced changes in mesenchymal stem cell osteogenesis, morphology, and stiffness. Sci. Rep. 6, 34791 (2016).
Uzer, G. et al. Cell mechanosensitivity to extremely low-magnitude signals is enabled by a LINCed nucleus. Stem Cells 33, 2063–2076 (2015).
Uzer, G., Pongkitwitoon, S., Ete Chan, M. & Judex, S. Vibration induced osteogenic commitment of mesenchymal stem cells is enhanced by cytoskeletal remodeling but not fluid shear. J. Biomech. 46, 2296–2302 (2013).
Kegelman, C. D. et al. Skeletal cell YAP and TAZ combinatorially promote bone development. FASEB J. 32, 2706–2721 (2018).
Matsumoto, Y. et al. Reciprocal stabilization of ABL and TAZ regulates osteoblastogenesis through transcription factor RUNX2. J. Clin. Investig. 126, 4482–4496 (2016).
Zaidi, S. K. et al. Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. EMBO J. 23, 790–799 (2004).
Seo, E. et al. SOX2 regulates YAP1 to maintain stemness and determine cell fate in the osteo-adipo lineage. Cell Rep. 3, 2075–2087 (2013).
Sen, B. et al. Intranuclear actin regulates osteogenesis. Stem Cells. https://doi.org/10.1002/stem.2090 (2015).
Das, A., Fischer, R. S., Pan, D. & Waterman, C. M. YAP nuclear localization in the absence of cell-cell contact is mediated by a filamentous actin-dependent, myosin II- and phospho-YAP-independent pathway during extracellular matrix mechanosensing. J. Biol. Chem. 291, 6096–6110 (2016).
Yuan, Y. et al. YAP1/TAZ-TEAD transcriptional networks maintain skin homeostasis by regulating cell proliferation and limiting KLF4 activity. Nat. Commun. 11, 1472 (2020).
Ho, L. T. Y., Skiba, N., Ullmer, C. & Rao, P. V. Lysophosphatidic acid induces ECM production via activation of the mechanosensitive YAP/TAZ transcriptional pathway in trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 59, 1969–1984 (2018).
Cai, H. & Xu, Y. The role of LPA and YAP signaling in long-term migration of human ovarian cancer cells. Cell Commun. Signal. 11, 31 (2013).
Dupont, S. et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011).
Driscoll, T. P., Cosgrove, B. D., Heo, S.-J., Shurden, Z. E. & Mauck, R. L. Cytoskeletal to nuclear strain transfer regulates YAP signaling in mesenchymal stem cells. Biophys. J. 108, 2783–2793 (2015).
Benham-Pyle, B. W., Pruitt, B. L. & Nelson, W. J. Cell adhesion. Mechanical strain induces E-cadherin-dependent Yap1 and beta-catenin activation to drive cell cycle entry. Science 348, 1024–1027 (2015).
Uzer, G. et al. Sun-mediated mechanical LINC between nucleus and cytoskeleton regulates betacatenin nuclear access. J. Biomech. 74, 32–40 (2018).
Shiu, J.-Y., Aires, L., Lin, Z. & Vogel, V. Nanopillar force measurements reveal actin-cap-mediated YAP mechanotransduction. Nat. Cell Biol. 20, 262–271 (2018).
Stewart-Hutchinson, P. J., Hale, C. M., Wirtz, D. & Hodzic, D. Structural requirements for the assembly of LINC complexes and their function in cellular mechanical stiffness. Exp. Cell Res. 314, 1892–1905 (2008).
Janmaleki, M., Pachenari, M., Seyedpour, S. M., Shahghadami, R. & Sanati-Nezhad, A. Impact of simulated microgravity on cytoskeleton and viscoelastic properties of endothelial cell. Sci. Rep. 6, 32418 (2016).
Qian, A. R. et al. Fractal dimension as a measure of altered actin cytoskeleton in MC3T3-E1 cells under simulated microgravity using 3-D/2-D clinostats. Biomed. Eng. IEEE Trans. 59, 1374–1380 (2012).
Pardo, S. J. et al. Simulated microgravity using the random positioning machine inhibits differentiation and alters gene expression profiles of 2T3 preosteoblasts. Am. J. Physiol. Cell Physiol. 288, https://doi.org/10.1152/ajpcell.00222.2004 (2005).
Herranz, R. et al. Ground-based facilities for simulation of microgravity: organism-specific recommendations for their use, and recommended terminology. Astrobiology 13, 1–17 (2013).
Dedolph, R. R. & Dipert, M. H. The physical basis of gravity stimulus nullification by clinostat rotation. Plant Physiol. 47, 756–764 (1971).
Uddin, S. M. Z. & Qin, Y.-X. Enhancement of osteogenic differentiation and proliferation in human mesenchymal stem cells by a modified low intensity ultrasound stimulation under simulated microgravity. PLoS ONE 8, e73914 (2013).
Chen, Z., Luo, Q., Lin, C., Kuang, D. & Song, G. Simulated microgravity inhibits osteogenic differentiation of mesenchymal stem cells via depolymerizing F-actin to impede TAZ nuclear translocation. Sci. Rep. 6, 30322 (2016).
Dai, Z. Q., Wang, R., Ling, S. K., Wan, Y. M. & Li, Y. H. Simulated microgravity inhibits the proliferation and osteogenesis of rat bone marrow mesenchymal stem cells. Cell Prolif. 40, 671–684 (2007).
Shi, F. et al. Simulated microgravity promotes angiogenesis through RhoA-dependent rearrangement of the actin cytoskeleton. Cell. Physiol. Biochem. 41, 227–238 (2017).
Corydon, T. J. et al. Reduced expression of cytoskeletal and extracellular matrix genes in human adult retinal pigment epithelium cells exposed to simulated microgravity. Cell. Physiol. Biochem. 40, 1–17 (2016).
Patel, M. J. et al. Low magnitude and high frequency mechanical loading prevents decreased bone formation responses of 2T3 preosteoblasts. J. Cell. Biochem. 106, 306–316 (2009).
Touchstone, H. et al. Recovery of stem cell proliferation by low intensity vibration under simulated microgravity requires intact LINC complex npj. Microgravity 5, https://doi.org/10.1038/s41526-019-0072-5 (2019).
Aureille, J. et al. Nuclear envelope deformation controls cell cycle progression in response to mechanical force. EMBO Rep. 20, e48084 (2019).
Hoffman, L. M. et al. Mechanical stress triggers nuclear remodeling and the formation of transmembrane actin nuclear lines with associated nuclear pore complexes. Mol. Biol. Cell. https://doi.org/10.1091/mbc.E19-01-0027 (2020).
Riddick, N., Ohtani, K. & Surks, H. K. Targeting by myosin phosphatase-RhoA interacting protein mediates RhoA/ROCK regulation of myosin phosphatase. J. Cell. Biochem. 103, 1158–1170 (2008).
Jaganathan, B. G. et al. Rho inhibition induces migration of mesenchymal stromal cells. Stem Cells 25, 1966–1974 (2007).
Yasuda, D. et al. Lysophosphatidic acid–induced YAP/TAZ activation promotes developmental angiogenesis by repressing Notch ligand Dll4. J. Clin. Investig. 129, 4332–4349 (2019).
Hsueh, Y.-J. et al. Lysophosphatidic acid induces YAP-promoted proliferation of human corneal endothelial cells via PI3K and ROCK pathways. Mol. Ther. 2, https://doi.org/10.1038/mtm.2015.14 (2015).
Saeed, M. & Weihs, D. Finite element analysis reveals an important role for cell morphology in response to mechanical compression. Biomech. Model. Mechanobiol. https://doi.org/10.1007/s10237-019-01276-5 (2019).
Qiu, Q. Q., Ducheyne, P. & Ayyaswamy, P. S. 3D bone tissue engineered with bioactive microspheres in simulated microgravity. Vitr. Cell. Dev. Biol. Anim. 37, 157–165 (2001).
Klaus, D. M., Todd, P. & Schatz, A. Functional weightlessness during clinorotation of cell suspensions. Adv. Space Res. 21, 1315–1318 (1998).
Yu, X. J., Botchwey, E. A., Levine, E. M., Pollack, S. R. & Laurencin, C. T. Bioreactor-based bone tissue engineering: The influence of dynamic flow on osteoblast phenotypic expression and matrix mineralization. Proc. Natl. Acad. Sci. USA 101, 11203–11208 (2004).
Wuest, S. L., Stern, P., Casartelli, E. & Egli, M. Fluid dynamics appearing during simulated microgravity using random positioning machines. PLoS ONE 12, e0170826 (2017).
Bacabac, R. G. et al. Bone cell responses to high-frequency vibration stress: does the nucleus oscillate within the cytoplasm? FASEB J. 20, 858–864 (2006).
Lau, E. et al. Effect of low-magnitude, high-frequency vibration on osteogenic differentiation of rat mesenchymal stromal cells. J. Orthop. Res. 29, 1075–1080 (2011).
Gershovich, P. M., Gershovich, J. G. & Buravkova, L. B. Cytoskeleton structure and adhesion properties of human stromal precursors under conditions of simulated microgravity. Cell Tissue Biol. 3, 423–430 (2009).
Dai, Z. et al. Actin microfilament mediates osteoblast Cbfa1 responsiveness to BMP2 under simulated microgravity. PLoS ONE 8, e63661 (2013).
Dareing, D. W., Yi, D. & Thundat, T. Vibration response of microcantilevers bounded by a confined fluid. Ultramicroscopy 107, 1105–1110 (2007).
Uzer, G. et al. Separating fluid shear stress from acceleration during vibrations in vitro: identification of mechanical signals modulating the cellular response. Cell. Mol. Bioeng. 5, 266–276 (2012).
Hu, J. K.-H. et al. An FAK-YAP-mTOR signaling axis regulates stem cell-based tissue renewal in mice. Cell Stem Cell 21, 91–106.e106 (2017).
Sen, B. et al. Mechanical signal influence on mesenchymal stem cell fate is enhanced by incorporation of refractory periods into the loading regimen. J. Biomech. 44, 593–599 (2011).
De Magistris, P. & Antonin, W. The dynamic nature of the nuclear envelope. Curr. Biol. 28, R487–R497 (2018).
Czapiewski, R., Robson, M. I. & Schirmer, E. C. Anchoring a Leviathan: how the nuclear membrane tethers the genome. Front. Genet. 7, 82 (2016).
Peister, A. et al. Adult stem cells from bone marrow (MSCs) isolated from different strains of inbred mice vary in surface epitopes, rates of proliferation, and differentiation potential. Blood 103, 1662–1668 (2004).
Bas, G. et al. Low intensity vibrations augment mesenchymal stem cell proliferation and differentiation capacity during in vitro expansion. Sci. Rep. 10, 9369 (2020).
Newberg, J. et al. Isolated nuclei stiffen in response to low intensity vibration. J. Biomech. 110012, https://doi.org/10.1016/j.jbiomech.2020.110012 (2020).
Uzer, G. et al. Gap junctional communication in osteocytes is amplified by low intensity vibrations in vitro. PLoS ONE 9, e90840 (2014).
Acknowledgements
This study was supported by NASA ISGC NNX15AI04H, NIH grants R01AG059923, 5P2CHD086843–03, P20GM109095, P20GM103408, and NSF grants 1929188 & 2025505.
Author information
Authors and Affiliations
Contributions
M.T. experimental methods, data analysis/interpretation, manuscript writing, and final approval of the manuscript. K.W. data analysis and final approval of the manuscript. J.N. experimental methods and final approval of the manuscript. J.T.O. financial support and final approval of the manuscript. G.U. concept/design, financial support, data analysis/interpretation, manuscript writing, and final approval of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thompson, M., Woods, K., Newberg, J. et al. Low-intensity vibration restores nuclear YAP levels and acute YAP nuclear shuttling in mesenchymal stem cells subjected to simulated microgravity. npj Microgravity 6, 35 (2020). https://doi.org/10.1038/s41526-020-00125-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41526-020-00125-5
This article is cited by
-
The roles of Hippo/YAP signaling pathway in physical therapy
Cell Death Discovery (2024)
